
Abstract
DNA polymerases often incorporate non-canonical nucleotide, i.e., ribonucleoside triphosphates into the genomic DNA. Aberrant accumulation of ribonucleotides in the genome causes various cellular abnormalities. Here, we show the possible role of human nucleotide excision repair (NER) and DNA polymerase η (Pol η) in processing of a single ribonucleotide embedded into DNA. We found that the reconstituted NER system can excise the oxidized ribonucleotide on the plasmid DNA. Taken together with the evidence that Pol η accurately bypasses a ribonucleotide, i.e., riboguanosine (rG) or its oxidized derivative (8-oxo-rG) in vitro, we further assessed the mutagenic potential of the embedded ribonucleotide in human cells lacking NER or Pol η. A single rG on the supF reporter gene predominantly induced large deletion mutations. An embedded 8-oxo-rG caused base substitution mutations at the 3′-neighboring base rather than large deletions in wild-type cells. The disruption of XPA, an essential factor for NER, or Pol η leads to the increased mutant frequency of 8-oxo-rG. Furthermore, the frequency of 8-oxo-rG-mediated large deletions was increased by the loss of Pol η, but not XPA. Collectively, our results suggest that base oxidation of the embedded ribonucleotide enables processing of the ribonucleotide via alternative DNA repair and damage tolerance pathways.
Similar content being viewed by others


Introduction
DNA replication is essential for maintaining genetic information in living organisms. DNA polymerases (Pols) specifically utilize the DNA precursors deoxynucleoside triphosphates (dNTPs) during replication; however, they often incorporate ribonucleoside triphosphates (rNTPs) into DNA because of the much higher concentration of rNTPs than that of dNTPs in the cellular nucleotide pool1,2. It has been suggested that millions of ribonucleotides are incorporated into the genome of eukaryotic cells3,4. The aberrant accumulation of ribonucleotides in the genome leads to Aicardi–Goutières syndrome (AGS), the severe autoimmune disease, and tumorigenesis5,6,7.
In eukaryotes, ribonucleotides embedded into DNA are primarily repaired by RNase H2-initiated ribonucleotide excision repair (RER)8. RNase H2 cleaves the 5′-side of the ribose sugar backbone, followed by excision by a flap endonuclease, strand displacement synthesis by Pol δ or ε, and nick sealing catalyzed by DNA ligase I. The inactivation of the canonical RER leads to various cellular abnormalities including the chronic activation of DNA damage response, aberrant innate immune activation, and epigenetic perturbations3,7,9,10,11,12. In the absence of RNase H2, the ribonucleotides are repaired by an alternative pathway involving DNA topoisomerase 1 (Top1). This Top1-mediated repair pathway is demonstrated to be associated with mutagenic consequences13,14,15. In yeast cells lacking RNase H2, an embedded ribonucleotide induces 2- to 5-bp deletion mutations at short tandem repeat sequences via Top1-mediated ribonucleotide processing14. However, whole-exome sequencing detected no such mutations in the genome of tumor cells derived from RNase H2 deficient mice5. The mutagenic potential of ribonucleotides embedded into DNA in mammalian cells remains to be elucidated.
Cellular DNA/RNA and its precursors are subject to oxidation from endogenous and exogenous sources of reactive oxygen species. The exposure of DNA to oxidative stress has been shown to promote the formation of a substantial amount of ribonucleotides and oxidized base damages in vitro and in vivo16. 7,8-Dihydro-8-oxo-riboguanosine-5′-triphosphate (8-oxo-rGTP), the oxidized form of rNTPs, can be generated in the nucleotide pool by the action of oxygen radicals17. 8-Oxo-rGTP is also utilized as the substrate during DNA synthesis by Pols18,19,20. Thus, the modified ribonucleotides could exist in DNA via the oxidation of the embedded ribonucleotide and/or the insertion of damaged rNTPs during DNA replication. Base oxidation in embedded ribonucleotide inhibits RNase H2-mediated excision repair21,22. Furthermore, the base excision repair (BER) pathway, involved in the repair of 7,8-dihydro-8-oxo-deoxyguanosine (8-oxo-dG), cannot excise the oxidized ribonucleotide 7,8-dihydro-8-oxo-riboguanosine (8-oxo-rG) in the DNA21,22. Therefore, a modified ribonucleotide would exhibit greater mutagenic potential compared with unmodified ribonucleotide in cells. Otherwise, an alternative repair pathway may participate in the repair of a damaged ribonucleotide. A possible mechanism is nucleotide excision repair (NER) involved in the removal of various helix-distorting DNA lesions from DNA23. A previous study has revealed that NER proteins derived from thermophilic eubacteria can excise ribonucleotides embedded into DNA24. However, another study has suggested that a ribonucleotide embedded into DNA is a poor substrate for NER function in vitro in both human and Escherichia coli (E. coli)25. Based on these findings, the involvement of NER in ribonucleotide removal in cells remains controversial.
Ribonucleotides that escape from repair can interfere with DNA replication. In vitro studies have revealed that single and multiple ribonucleotides in the template DNA influence DNA synthesis mediated by Pols in yeast and humans2,26. Furthermore, the presence of 8-oxo-rG in DNA strongly hinders the primer extension reaction catalyzed by human replicative Pol α21. To counteract these replication-blocking lesions, organisms possess specialized Pols that carry out translesion DNA synthesis (TLS) past DNA lesions. Pol η is one of TLS Pols that can bypass various DNA damages, including ultraviolet (UV) light-induced cyclobutane pyrimidine dimers27. Our in vitro experiments revealed that Pol η efficiently and accurately bypassed undamaged and damaged ribonucleotides (rG and 8-oxo-rG, respectively) in a more error-free manner compared with 8-oxo-dG21. Thus, it is of interest to investigate whether TLS Pols exert protective effects against the mutagenicity of incorporated ribonucleotides in cells.
In the current study, we investigated the possibility whether the reconstituted NER system can excise the ribonucleotide from the DNA. On the basis of in vitro analyses, we also examined the mutagenic potential of an embedded ribonucleotide in human lymphoblastoid cells lacking xeroderma pigmentosum group A (XPA), an essential factor for NER, and Pol η using a supF shuttle vector containing a single dG, rG, 8-oxo-rG, or 8-oxo-dG. Based on these biochemical and cell culture investigations, our findings provide new mechanistic insights underlying ribonucleotide-induced mutagenesis and the protective roles of DNA repair and tolerance processes in mammals.
Results
Reconstituted NER can excise an embedded oxidized ribonucleotide
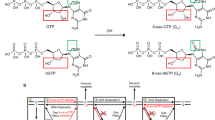
To test the possibility that NER is involved in the ribonucleotide removal from DNA, we analyzed whether the human cell-free NER system reconstituted with purified protein factors possesses the ribonucleotide excision activity against a single rG or 8-oxo-rG in the plasmid DNA (Fig. 1a). As shown in Fig. 1b, single rG was a poor substrate for human NER, a finding consistent with the previous report25. Conversely, 8-oxo-rG was recognized and excised by NER machinery. In addition, the incision activity of NER against 8-oxo-rG was greater than that against 8-oxo-dG (Fig. 1c). The ladder pattern of the excised products was also slightly different between 8-oxo-dG and 8-oxo-rG (Fig. 1b), which can be due to the difference in the sugar pucker conformation of deoxyribose and ribose that may affect the recognition and excision processes of NER28. Because the efficiency of NER in excising the ribonucleotide was lower compared with the canonical substrate for NER, i.e., UV-induced 6–4 photoproducts (Fig. 1b) (Supplementary Fig. S1), we further assessed the cellular impact of the NER deficiency on the mutagenic potential of an embedded ribonucleotide in the following section.

The dual incision activity of human nucleotide excision repair on duplex plasmid DNA containing a single ribonucleotide. (a) The DNA substrate used for the in vitro dual incision assay. The substrate plasmid harbored a single 6-4PP or dG, 8-oxo-dG, rG, or 8-oxo-rG at a defined position in the DNA sequence between PstI and HindIII sites of pBluescript II KS(+) as previously described53. (b) In vitro dual incision assays were performed using purified NER proteins and internally 32P-labeled DNA substrates containing a 6-4PP, dG, 8-oxo-dG, rG, or 8-oxo-rG as described in the Methods. DNA samples were subjected to 10% denaturing polyacrylamide gel electrophoresis followed by autoradiography. The full-length gel is shown in Supplementary Fig. S1. (c) Quantification of the products formed by NER-mediated dual incision reaction. Values are presented as mean ± S.E. of three independent experiments. Significant differences are indicated by asterisks (**P < 0.01, ****P < 0.0001 by two-way ANOVA).
Experimental design to determine the mutagenic potential of an embedded ribonucleotide in human cells
To examine the mutagenic potential of an embedded ribonucleotide in human cells, double-stranded plasmids containing rG or 8-oxo-rG were constructed as the model substrates. Ribonucleotides were inserted at position 112 (G112) on supF (Fig. 2a). The control plasmids harboring dG or 8-oxo-dG at the same position were also synthesized to clarify the effect of the ribose sugar backbone on mutations. The phenotype of cells used in this study was confirmed prior to the experiments; XPA−/− and POLH−/− cells were hypersensitive to UV exposure (Fig. 2b), as previously described29,30. The stable expression of XPA and POLH in each knock-out cells, i.e., “XPA−/− + XPA” and “POLH−/− + POLH”, rescued the phenotype, which was then utilized for the complementation experiment as described in the following section.

Analysis of ribonucleotide-induced mutagenesis in human lymphoblastoid cells. (a) Schematic diagram of the determination of mutant frequency and mutation spectrum using the supF shuttle vector. The closed circular double-stranded DNA, containing a single dG, rG, 8-oxo-rG, or 8-oxo-dG in supF, was transfected into WT, XPA−/−, and POLH−/− cells. After incubation for 48 h, the propagated plasmids were extracted from cells and introduced into KS40/pOF105 indicator strain. The mutant frequency and mutation spectrum were determined as described in the Methods. (b) Cell growth of WT (open circles), XPA−/− (closed triangles), POLH−/− (closed squares), XPA−/− + XPA (open triangles), and POLH−/− + POLH (open squares) cells after exposure to UVC light. Values are presented as mean ± S.E. of at least two independent experiments.
A single ribonucleotide predominantly induces large deletions in human cells
First, the plasmid DNA containing a single rG was transfected and replicated in wild-type (WT), XPA−/−, and POLH−/− cells. Plasmids replicated in cells were then recovered and introduced into KS40/pOF105 indicator strain, which was plated on titer and selection plates to calculate the frequencies of supF mutants. The mutant frequency induced by rG was comparable between the cells (3.9 ± 0.19, 3.6 ± 0.76, and 3.7 ± 0.056 × 10−3 in WT, XPA−/−, and POLH−/− cells, respectively), which was approximately four-fold higher than the control dG (0.88 ± 0.35, 0.79 ± 0.30, and 0.63 ± 0.12 × 10−3 in WT, XPA−/−, and POLH−/− cells, respectively) (Fig. 3a,b). We further analyzed the mutation spectrum induced by rG. Interestingly, large deletions were the predominant mutation (67%, 75%, and 82% in WT, XPA−/−, and POLH−/− cells, respectively) (Fig. 4b) (Supplementary Table S1). The length of deletions varied from 10 to 244 bp, with the majority of them being >100 bp in all cell lines (Supplementary Table S2). Notably, no sequence homology was found at each deletion junction. Because the frequency of large deletions observed was only approximately 10% in the control experiments involving dG (Fig. 4a) (Supplementary Table S1), large deletion mutations were specifically induced by rG embedded into the DNA.

Frequencies of supF mutants in a shuttle vector containing (a) dG, (b) rG, (c) 8-oxo-rG, and (d) 8-oxo-dG propagated in human cells. Data are expressed as mean ± S.E. of 2–5 independent experiments. **Significant difference between the assessed cells and WT cells; P < 0.01 (Dunnett’s Multiple Comparison Test).

Mutation spectrum of (a) dG, (b) rG, (c) 8-oxo-rG, and (d) 8-oxo-dG in supF plasmids propagated in wild-type, POLH−/−, and XPA−/− cells. The number of mutations were summarized in Supplementary Table S1.
Disruption of XPA or POLH enhances the mutagenic potential of an oxidized ribonucleotide
Next, we assessed whether base oxidation in a ribonucleotide affected its mutagenic potential in cells. The mutant frequency induced by 8-oxo-rG (3.0 ± 0.19 × 10−3) was not higher than that induced by rG (3.9 ± 0.19 × 10−3) and 8-oxo-dG (5.7 ± 0.15 × 10−3) in WT cells (Fig. 3b,c). In contrast with 8-oxo-dG that exclusively induced G:C to T:A transversion mutations at the position G112 (87%) (Fig. 4d) (Supplementary Table S1), 8-oxo-rG preferentially induced base-substitution mutations at a 3′-base neighboring the 8-oxo-rG site, i.e., GG112T to GGG (46%), followed by G:C to T:A (19%) and one-base deletions (14%) at the site of 8-oxo-rG (Fig. 4c). The frequency of large deletions induced by 8-oxo-rG was 7.9%, which was lower than that induced by rG (67%) (Fig. 4b). As some extent of G:C to T:A mutation (18%) was also induced by the control dG (Fig. 4a), mutations specifically induced by 8-oxo-rG were GG112T to GGG and one-base deletions, rather than large deletions and G:C to T:A transversion mutations. GG112T to GGG mutation was also observed with 8-oxo-dG in WT cells (6.7%) (Fig. 4d), suggesting that this mutation depends on the sequence context around G112.
To investigate the role of NER and TLS Pol on the mutagenic events caused by base oxidation in embedded ribonucleotide, we examined the mutant frequency and mutation spectrum of 8-oxo-rG in XPA−/− and POLH−/− cells. The mutant frequency was significantly increased in XPA−/− (4.5 ± 0.28 × 10−3) and POLH−/− (4.7 ± 0.22 × 10−3) compared with that in WT cells (3.0 ± 0.19 × 10−3) (Fig. 3c). The loss of XPA did not alter the mutation spectrum induced by 8-oxo-rG compared with that in WT cells (Fig. 4c) (Supplementary Table S1). In contrast, the loss of POLH resulted in an increased number of 8-oxo-rG-mediated large deletions (24%) in comparison with WT cells (7.9%). These deletions were mainly observed at a position 5′-upstream from the site of 8-oxo-rG (Supplementary Table S2). Accordingly, the number of GG112T to GGG mutation was decreased in POLH−/− (17%) as compared with WT cells (46%). To verify whether the increased mutagenic potential of 8-oxo-rG was due to the deficiency of XPA or POLH, we introduced the plasmid containing 8-oxo-rG into XPA−/− + XPA and POLH−/− + POLH cells. The frequency of the 8-oxo-rG-induced supF mutant was 3.6 ± 0.11 × 10−3 and 3.2 ± 0.11 × 10−3 in XPA−/− + XPA and POLH−/− + POLH cells, respectively, which was comparable with that in WT cells (3.0 ± 0.19 × 10−3) (Fig. 3c). The expression of POLH in POLH−/− cells also decreased the frequency of large deletions (7.9%) (Fig. 4c) (Supplementary Tables S1 and S2). It is noted that the disruption of XPA or POLH did not alter the mutant frequency of 8-oxo-dG (5.7 ± 0.15 × 10−3, 5.2 ± 0.17 × 10−3 and 5.4 ± 0.10 × 10−3 in WT, XPA−/−, and POLH−/− cells, respectively) (Fig. 3d). The predominant mutation induced by 8-oxo-dG was G:C to T:A transversion mutations in all cells (87%, 71%, and 67% in WT, XPA−/−, and POLH−/− cells, respectively) (Fig. 4d) (Supplementary Table S1). The overall mutation spectrum was slightly altered in POLH−/− cells; the number of GG112T to GGG was increased (Table S1), which might be because of the TLS event across 8-oxo-dG by other Pols in the absence of Pol η.
Discussion
The absence of RER leads to the accumulation of ribonucleotides in the genome, which is associated with increased genomic instability31. Although several studies have reported the importance of RER for maintaining genome integrity in higher eukaryotes3,32, the mutational specificity of embedded ribonucleotides remains largely unclear in mammals. To visualize mutations induced by a ribonucleotide that is readily repaired by RER in the genome, we employed the shuttle vector system that can rapidly propagate in cells. Our results suggest that an embedded rG can cause the deleterious mutations in human cells. On the other hand, its oxidative derivative 8-oxo-rG did not display the enhanced mutagenic potential compared with rG and 8-oxo-dG, which can be explained by the alternative repair and damage tolerance pathways based on in vitro investigations using the reconstituted NER and Pol η.
It has been controversial whether NER can recognize a small lesion such as 8-oxo-dG- and ribonucleotide-containing DNA as the substrate24,25,30,33,34,35. The in vitro NER reconstitution experiments in this study implicated that 8-oxo-rG is the preferable substrate for human NER compared with rG and 8-oxo-dG in the plasmid DNA (Fig. 1c). Further, our results revealed that the loss of the NER factor XPA does not alter the mutant frequency and mutation spectra induced by rG and 8-oxo-dG, but significantly enhanced the mutagenic potential of 8-oxo-rG (Fig. 3). Hence, these observations suggest that the oxidation of guanine base in ribonucleotide enables ribonucleotide removal by human NER.
In a previous in vitro study, both replicative and TLS Pols bypassed rG in an error-free manner21. Conversely, the oxidation of a ribonucleotide, i.e., 8-oxo-rG, on the template DNA strongly inhibited DNA synthesis by Pols and contributed to miscoding events. Because 8-oxo-rG also could not be repaired by either RER or BER21,22, we hypothesized that such modification of the ribonucleotide enhances the mutagenic potential in cells. Contrary to the initial hypothesis, it seems that 8-oxo-rG did not display greater mutagenic potential than rG in WT cells (Fig. 3b,c). The frequency of large deletions induced by rG (67%) was much higher than that induced by 8-oxo-rG (7.9%) (Fig. 4b,c). These observations may be attributed to the recognition of the damaged ribonucleotide by the alternate repair and/or damage tolerance pathways. While 8-oxo-rG is the replication blocking lesion, Pol η rapidly and accurately bypasses the lesion21. Along with the in vitro study, our results suggest that the oxidized ribonucleotide on the template DNA is recognized by the DNA damage tolerance pathway and Pol η performs TLS in cells. Interestingly, the proportion of GG112T to GGG mutation caused by 8-oxo-rG was decreased in POLH−/− cells (Fig. 4c), indicating that such mutation is induced during Pol η-mediated TLS across 8-oxo-rG. Although the structural impact of the presence of 8-oxo-rG in DNA is still unclear, the mutation could be because of the sequence specific misalignment induced by 8-oxo-rG. For example, the nucleotide incorporated opposite T113 is encoded by 5′-neighboring 8-oxo-rG through the misalignment-mediated nucleotide insertion followed by realignment of the template DNA during TLS. It has been suggested that such mutations due to misalignment can be induced by DNA damage at the specific sequence context36. While the loss of POLH resulted in a three-fold increase in the frequency of large deletion mutations induced by 8-oxo-rG (Fig. 4c), the loss of XPA did not enhance the frequency of such deletions despite the increase in the frequency of mutations (Figs 3c and 4c). Based on these observations, ribonucleotide-induced large deletions may occur in conjunction with DNA synthesis. Our results also imply that an embedded ribonucleotide causes mutations not only in tandem repeat sequences but also in other genomic loci.
In contrast with findings of the current study, previous studies have reported that such large deletion mutations at the non-repetitive sequences were rare in the genome of the yeast strain lacking RER13,14,15. This difference may be because of a distinct repair mechanism activated during the mutagenic processing of the ribonucleotide. During Top1-mediated ribonucleotide processing, Top1 incises the 3′-side of the ribonucleotide and forms a Top1–DNA cleavage complex (Top1cc) between the catalytic tyrosine residue and the 3′-phosphate of the ribonucleotide14,37. Top1cc is further reversed by the nucleophilic attack of 2′-OH of the ribonucleotide to generate a 2′,3′-cyclic phosphate and to release Top1 from the complex. The subsequent processing of the released DNA by Top1 can lead to DNA double strand breaks (DSBs)38. Possible repair pathways following DSBs formation include homologous recombination (HR) and non-homologous end-joining (NHEJ). The predominant DSB repair pathway is NHEJ throughout the cell cycle in mammalian cells39, whereas HR is prevalent especially during the G2/S phase in yeast40. This pathway choice may explain the differential mutagenic property of embedded ribonucleotides between yeast and mammalian cells. Interestingly, large deletions induced by rG and 8-oxo-rG were often extended from the site of the ribonucleotide (G112) to a position 5ʹ-upstream in supF (Supplementary Table S2). These mutations could result from the Top1-mediated processing of a ribonucleotide in the following manner. The incision of a ribonucleotide by Top1 generates Top1cc. Upon the generation of a 2′,3′-cyclic phosphate-terminated nick and the release of Top1, one possible scenario is the formation of a second Top1cc 5′-upstream of the nick, leaving a gap between 3′-Top1cc and 5′-OH41,42. Because Top1 is coupled with DNA replication43, the generation of such a gap or nick during replication can lead to DSBs, in which the 3′-end of the strand contains Top1cc. In the present study, the deletion mutations observed toward the 5′ side of supF may have occurred via the processing of such complex DSBs, e.g., the resection of the dirty terminus by NHEJ. Reportedly, even a single DNA damage, e.g., cyclobutane pyrimidine dimer, abasic site, and DSB is sufficient to induce large deletion mutations in the episomal and genomic DNA in mammalian cells44,45,46. Our results provide a further insight into the mechanism of ribonucleotide-induced genomic instability and its suppression by alternative factors involving DNA repair and damage tolerance.
Figure 5 shows the probable model of ribonucleotide-induced mutagenesis and the mechanism for resolving a damaged ribonucleotide in DNA. In summary, unrepaired ribonucleotides in DNA can cause severe genomic instability via the formation of deleterious mutations in mammals. Our findings also suggest that NER and Pol η are employed the back-up pathways for suppressing ribonucleotide-induced mutagenesis under circumstances where embedded ribonucleotides cannot be properly resolved. These findings can deepen our understanding of the mechanism underlying genome instability caused by the genomic ribonucleotide accumulation.

A possible model of the processing of an embedded ribonucleotide and ribonucleotide-induced mutagenesis. 8-Oxo-rG could be generated by the incorporation of oxidized ribonucleoside triphosphates into DNA or by the oxidation of the embedded ribonucleotides. RNase H2 and BER cannot remove 8-oxo-rG21,22. NER is involved in the excision of such a damaged ribonucleotide. Alternatively, Pol η can bypass 8-oxo-rG to suppress the mutagenic events induced by the embedded ribonucleotide. The unrepaired ribonucleotide is subject to Top1-mediated processing, leading to the deleterious mutations.
Methods
Cell culture
Human lymphoblastoid TSCER2 cells, derived from TK6 cell line47, were cultured in RPMI-1640 medium (Nacalai Tesque) supplemented with 200 μg/mL sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin, and 10% (v/v) heat-inactivated fetal bovine serum at 37 °C in an atmosphere of 5% CO2 and 100% humidity. XPA−/− and POLH−/− cells were established as described previously29.
Protein preparation
Expression and purification of recombinant NER proteins, Flag-XPC/RAD23B-His48, CETN249, TFIIH Core750, Flag-XPA, ERCC1-His/XPF, and RPA51, were achieved as previously described. For the preparation of XPG-Flag, High-Five cells (grown in twenty 150-mm culture dishes) were infected with a recombinant baculovirus. After incubation at 27 °C for 72 h, the infected cells were collected and washed with ice-cold phosphate-buffered saline. The cell pellet was resuspended in an 8-fold volume of extraction buffer [25 mM Tris-HCl (pH 7.5), 5 mM NaCl, 0.1 mM EDTA, 1.5 mM MgCl2, 0.1% Nonidet P-40, 20% glycerol, 1 mM dithiothreitol (DTT), 0.25 mM PMSF, protease inhibitor cocktail (Complete: Roche)]. After incubation on ice for 30 min, the cells were disrupted using Dounce homogenization (20 strokes of pestle A). The cell lysate was then adjusted at 0.3 M NaCl and further incubated on ice for 60 min. A clear extract obtained after centrifugation at 35,000 rpm for 30 min (Beckman 50.2Ti rotor) was loaded onto a HiPrep 16/10 Heparin FF column (GE Healthcare) equilibrated with buffer PET [20 mM sodium phosphate (pH 7.8), 1 mM EDTA, 10% glycerol, 0.01% Triton X-100, 1 mM DTT, 0.25 mM PMSF] containing 0.1 M NaCl. After extensively washing with the same buffer, bound proteins were eluted with buffer PET containing 1 M NaCl. The eluate was then loaded onto an anti-Flag M2 agarose column (1 cm diameter × 5 cm long), which had been equilibrated with buffer PT [20 mM sodium phosphate (pH 7.8), 0.15 M NaCl, 10% glycerol, 0.01% Triton X-100, 1 mM 2-mercaptoethanol, 0.25 mM PMSF). After an extensive wash with the same buffer, the column was inverted and bound proteins were eluted with 10 ml of buffer PT containing 0.1 mg/ml Flag peptide (Sigma). The eluate fractions were dialyzed against buffer A [25 mM Hepes-NaOH (pH 7.5), 0.1 M NaCl, 10% glycerol, 1 mM 2-mercaptoethanol, 0.25 mM PMSF] and loaded onto a HiTrap Q HP column (5 ml, GE healthcare), which was previously equilibrated with the same buffer. Bound proteins were then eluted using a linear NaCl gradient (0.1 to 1 M) in buffer A. The fractions containing XPG-Flag were dialyzed against buffer PET containing 0.1 M NaCl, and loaded onto a Mono S PC 1.6/5 column (GE Healthcare) equilibrated with the same buffer. XPG-Flag was eluted using a linear NaCl gradient (0.1 to 1 M) in buffer PET. The eluted XPG-Flag was dialyzed against buffer PET containing 0.1 M NaCl.
In vitro dual incision assay
The oligonucleotide containing 6-4PP was prepared as described previously52. The oligonucleotides containing dG, 8-oxo-dG, rG, or 8-oxo-rG were synthesized and purified by Tsukuba Oligo Service Co., Ltd. (Ibaraki, Japan). The oligonucleotides were purified by polyacrylamide gel electrophoresis. After the purification step, the homogeneity and no detectable degradation of the oligonucleotides were confirmed by the same method prior to use. The internally 32P-labeled DNA substrates, containing a site-specific lesion, were prepared as previously described53. In vitro dual incision assays were performed basically according to the previously described procedures54. Briefly, the DNA substrates (~1 × 106 cpm) were incubated at 30 °C for the indicated time in 15-μl reactions, each containing 40 mM Hepes-KOH (pH 7.8), 70 mM NaCl, 0.5 mM DTT, 5 mM MgCl2, 0.5 mM EDTA, 2 mM ATP, acetylated bovine serum albumin (Ambion: 5.4 μg), Flag-XPA (87.5 ng), ERCC1-His/XPF (15 ng), RPA (100 ng), XPG-Flag (60 ng), TFIIH Core7 (250 ng), Flag-XPC/RAD23B-His (10 ng), and CETN2 (10 ng). The purified DNA samples were subjected to 10% denaturing polyacrylamide gel electrophoresis and were detected using the Typhoon FLA9500 (GE Healthcare) and analyzed with the ImageQuant TL software (GE Healthcare).
Establishment of POLH and XPA-complemented cells
The XPA-expressing cell clone (XPA−/− + XPA) was established as previously reported30. To construct the POLH expression plasmid, the coding region of POLH was amplified from the cDNA of TK6 cells by PCR with primers 5′-GAGCTCGGATCCTGAAAAATGGCTACTGGACAGGATC-3′ and 5′-CAGCGGGTTTAAACCTAATGTGTTAATGGCTTAAAAAATGATTCCAATG-3′. The resulting product was digested with BamHI and PmeI and cloned into the BamHI/PmeI sites of pEF6/myc-His (ThermoFisher Scientific). The plasmid obtained was named pEF-POLH. POLH−/− cells (1 × 106) were transfected with 10 µg of ScaI-linearized pEF-POLH using a NEPA21 Super Electroporator (Nepa Gene Co., Ltd.) according to the manufacturer’s instructions. After incubating for 48 h in RPMI-1640 medium, cells were plated into 96-microwell plates in the presence of 10 µg/ml blasticidin S. Blasticidin S-resistant clones expressing POLH were then separated. The XPA- and POLH-expressing clones were then subjected to UV irradiation and supF forward mutation assay.
UV irradiation
Cells in the exponential phase were washed and resuspended with RPMI-1640 medium without phenol red prior to the irradiation. Moreover, cells (2.5 × 106) suspended in 5 ml medium were placed in a 100 mm petri dish and were exposed to UVC light. After irradiation, cells were cultured for 3 days. The relative growth rate was determined by counting cells over the 3 day period using Coulter Counter Z2 (Beckman).
supF forward mutation assay
The position 112 of the supF gene was selected as the site for embedding rG, 8-oxo-rG, or 8-oxo-dG according to the previous study55. The 5′-phosphorylated 21-mer primer (5′-CGACTTCGAAGNTTCGAATCC-3′, where N represents dG, rG, 8-oxo-rG, or 8-oxo-dG) was synthesized and provided by Tsukuba Oligo Service Co., Ltd. (Ibaraki, Japan). Each primer was annealed with pMY189 single-stranded DNA that was prepared using the VCSM13 helper phage. The closed circular double-stranded DNA, containing a single dG, rG, 8-oxo-rG, or 8-oxo-dG, was then synthesized and purified with ultracentrifugation as previously described56.
This closed circular double-stranded DNA (500 ng) was introduced into 2 × 106 cells using NEPA21 Super Electroporator (Nepa Gene Co., Ltd.) according to the manufacturer’s instructions. After incubating for 48 h, the replicated plasmid was isolated from the cells and digested with DpnI to remove the unreplicated plasmids57. The resulting plasmid was introduced into the E. coli KS40/pOF105 indicator strain. To select supF mutants, the transformed cells were plated on LB plates containing nalidixic acid (Nal, 50 μg/ml), streptomycin (Sm, 100 μg/ml), ampicillin (Amp, 150 µg/ml), chloramphenicol (Cm, 30 μg/ml), X-Gal (80 μg/ml), and IPTG (23.8 μg/ml). For determining the total number of transformants, the transformed cells were plated onto LB plates containing Amp (150 μg/ml) and Cm (30 μg/ml). The frequency of supF mutants and mutation spectrum was determined as described previously56.
References
Traut, T. W. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140, 1–22 (1994).
Nick McElhinny, S. A. et al. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proceedings of the National Academy of Sciences of the United States of America 107, 4949–4954, https://doi.org/10.1073/pnas.0914857107 (2010).
Reijns, M. A. et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022, https://doi.org/10.1016/j.cell.2012.04.011 (2012).
Williams, J. S. et al. Topoisomerase 1-mediated removal of ribonucleotides from nascent leading-strand DNA. Molecular cell 49, 1010–1015, https://doi.org/10.1016/j.molcel.2012.12.021 (2013).
Aden, K. et al. Epithelial RNase H2 Maintains Genome Integrity and Prevents Intestinal Tumorigenesis in Mice. Gastroenterology 156, 145–159 e119, https://doi.org/10.1053/j.gastro.2018.09.047 (2019).
Crow, Y. J. et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nature genetics 38, 910–916, https://doi.org/10.1038/ng1842 (2006).
Hiller, B. et al. Ribonucleotide Excision Repair Is Essential to Prevent Squamous Cell Carcinoma of the Skin. Cancer research 78, 5917–5926, https://doi.org/10.1158/0008-5472.CAN-18-1099 (2018).
Sparks, J. L. et al. RNase H2-initiated ribonucleotide excision repair. Molecular cell 47, 980–986, https://doi.org/10.1016/j.molcel.2012.06.035 (2012).
Lim, Y. W., Sanz, L. A., Xu, X., Hartono, S. R. & Chedin, F. Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutieres syndrome. eLife 4, https://doi.org/10.7554/eLife.08007 (2015).
Gunther, C. et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. The Journal of clinical investigation 125, 413–424, https://doi.org/10.1172/JCI78001 (2015).
Pizzi, S. et al. Reduction of hRNase H2 activity in Aicardi-Goutieres syndrome cells leads to replication stress and genome instability. Human molecular genetics 24, 649–658, https://doi.org/10.1093/hmg/ddu485 (2015).
Mackenzie, K. J. et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. The EMBO journal 35, 831–844, https://doi.org/10.15252/embj.201593339 (2016).
Nick McElhinny, S. A. et al. Genome instability due to ribonucleotide incorporation into DNA. Nature chemical biology 6, 774–781, https://doi.org/10.1038/nchembio.424 (2010).
Kim, N. et al. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332, 1561–1564, https://doi.org/10.1126/science.1205016 (2011).
Clark, A. B., Lujan, S. A., Kissling, G. E. & Kunkel, T. A. Mismatch repair-independent tandem repeat sequence instability resulting from ribonucleotide incorporation by DNA polymerase epsilon. DNA repair 10, 476–482, https://doi.org/10.1016/j.dnarep.2011.02.001 (2011).
Randerath, K. et al. Formation of ribonucleotides in DNA modified by oxidative damage in vitro and in vivo. Characterization by 32P-postlabeling. Mutation research 275, 355–366, https://doi.org/10.1016/0921-8734(92)90038-q (1992).
Hayakawa, H. et al. Metabolic fate of oxidized guanine ribonucleotides in mammalian cells. Biochemistry 38, 3610–3614, https://doi.org/10.1021/bi982361l (1999).
Sastre-Moreno, G., Sanchez, A., Esteban, V. & Blanco, L. ATP insertion opposite 8-oxo-deoxyguanosine by Pol4 mediates error-free tolerance in Schizosaccharomyces pombe. Nucleic acids research 42, 9821–9837, https://doi.org/10.1093/nar/gku711 (2014).
Ordonez, H. & Shuman, S. Mycobacterium smegmatis DinB2 misincorporates deoxyribonucleotides and ribonucleotides during templated synthesis and lesion bypass. Nucleic acids research 42, 12722–12734, https://doi.org/10.1093/nar/gku1027 (2014).
Cilli, P., Minoprio, A., Bossa, C., Bignami, M. & Mazzei, F. Formation and Repair of Mismatches Containing Ribonucleotides and Oxidized Bases at Repeated DNA Sequences. The Journal of biological chemistry 290, 26259–26269, https://doi.org/10.1074/jbc.M115.679209 (2015).
Sassa, A. et al. Impact of Ribonucleotide Backbone on Translesion Synthesis and Repair of 7,8-Dihydro-8-oxoguanine. The Journal of biological chemistry 291, 24314–24323, https://doi.org/10.1074/jbc.M116.738732 (2016).
Malfatti, M. C. et al. Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2. Nucleic acids research 45, 11193–11212, https://doi.org/10.1093/nar/gkx723 (2017).
Kusakabe, M. et al. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ 41, 2, https://doi.org/10.1186/s41021-019-0119-6 (2019).
Vaisman, A. et al. Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucleotide excision repair. PLoS genetics 9, e1003878, https://doi.org/10.1371/journal.pgen.1003878 (2013).
Lindsey-Boltz, L. A., Kemp, M. G., Hu, J. & Sancar, A. Analysis of Ribonucleotide Removal from DNA by Human Nucleotide Excision Repair. The Journal of biological chemistry 290, 29801–29807, https://doi.org/10.1074/jbc.M115.695254 (2015).
Clausen, A. R., Zhang, S., Burgers, P. M., Lee, M. Y. & Kunkel, T. A. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase delta. DNA repair 12, 121–127, https://doi.org/10.1016/j.dnarep.2012.11.006 (2013).
Masutani, C. et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399, 700–704, https://doi.org/10.1038/21447 (1999).
DeRose, E. F., Perera, L., Murray, M. S., Kunkel, T. A. & London, R. E. Solution structure of the Dickerson DNA dodecamer containing a single ribonucleotide. Biochemistry 51, 2407–2416, https://doi.org/10.1021/bi201710q (2012).
Mohiuddin, M. et al. SUMOylation of PCNA by PIAS1 and PIAS4 promotes template switch in the chicken and human B cell lines. Proceedings of the National Academy of Sciences of the United States of America 115, 12793–12798, https://doi.org/10.1073/pnas.1716349115 (2018).
Sassa, A., Kamoshita, N., Kanemaru, Y., Honma, M. & Yasui, M. Xeroderma Pigmentosum Group A Suppresses Mutagenesis Caused by Clustered Oxidative DNA Adducts in the Human Genome. PLoS One 10, e0142218, https://doi.org/10.1371/journal.pone.0142218 (2015).
Sassa, A., Yasui, M. & Honma, M. Current perspectives on mechanisms of ribonucleotide incorporation and processing in mammalian DNA. Genes Environ 41, 3, https://doi.org/10.1186/s41021-019-0118-7 (2019).
Hiller, B. et al. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. The Journal of experimental medicine 209, 1419–1426, https://doi.org/10.1084/jem.20120876 (2012).
Reardon, J. T., Bessho, T., Kung, H. C., Bolton, P. H. & Sancar, A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proceedings of the National Academy of Sciences of the United States of America 94, 9463–9468, https://doi.org/10.1073/pnas.94.17.9463 (1997).
Dianov, G., Bischoff, C., Piotrowski, J. & Bohr, V. A. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. The Journal of biological chemistry 273, 33811–33816, https://doi.org/10.1074/jbc.273.50.33811 (1998).
Cai, Y., Geacintov, N. E. & Broyde, S. Ribonucleotides as nucleotide excision repair substrates. DNA repair 13, 55–60, https://doi.org/10.1016/j.dnarep.2013.10.010 (2014).
Fang, H. & Taylor, J. S. Serial analysis of mutation spectra (SAMS): a new approach for the determination of mutation spectra of site-specific DNA damage and their sequence dependence. Nucleic Acids Res 36, 6004–6012, https://doi.org/10.1093/nar/gkn595 (2008).
Sekiguchi, J. & Shuman, S. Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Molecular cell 1, 89–97 (1997).
Huang, S. N., Williams, J. S., Arana, M. E., Kunkel, T. A. & Pommier, Y. Topoisomerase I-mediated cleavage at unrepaired ribonucleotides generates DNA double-strand breaks. The EMBO journal 36, 361–373, https://doi.org/10.15252/embj.201592426 (2017).
Mao, Z., Bozzella, M., Seluanov, A. & Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7, 2902–2906, https://doi.org/10.4161/cc.7.18.6679 (2008).
Mathiasen, D. P. & Lisby, M. Cell cycle regulation of homologous recombination in Saccharomyces cerevisiae. FEMS Microbiol Rev 38, 172–184, https://doi.org/10.1111/1574-6976.12066 (2014).
Sparks, J. L. & Burgers, P. M. Error-free and mutagenic processing of topoisomerase 1-provoked damage at genomic ribonucleotides. The EMBO journal 34, 1259–1269, https://doi.org/10.15252/embj.201490868 (2015).
Huang, S. Y., Ghosh, S. & Pommier, Y. Topoisomerase I alone is sufficient to produce short DNA deletions and can also reverse nicks at ribonucleotide sites. The Journal of biological chemistry 290, 14068–14076, https://doi.org/10.1074/jbc.M115.653345 (2015).
Pommier, Y., Sun, Y., Huang, S. N. & Nitiss, J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nature reviews. Molecular cell biology 17, 703–721, https://doi.org/10.1038/nrm.2016.111 (2016).
Kamiya, H., Iwai, S. & Kasai, H. The (6-4) photoproduct of thymine-thymine induces targeted substitution mutations in mammalian cells. Nucleic acids research 26, 2611–2617, https://doi.org/10.1093/nar/26.11.2611 (1998).
Suzuki, T., Katayama, Y., Komatsu, Y. & Kamiya, H. Analysis of large deletion mutations induced by abasic site analog in human cells. Genes Environ 40, 24, https://doi.org/10.1186/s41021-018-0110-7 (2018).
Honma, M. et al. Non-homologous end-joining for repairing I-SceI-induced DNA double strand breaks in human cells. DNA repair 6, 781–788, https://doi.org/10.1016/j.dnarep.2007.01.004 (2007).
Honma, M. et al. Deletion, rearrangement, and gene conversion; genetic consequences of chromosomal double-strand breaks in human cells. Environ Mol Mutagen 42, 288–298, https://doi.org/10.1002/em.10201 (2003).
Yasuda, G. et al. In vivo destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation. Molecular and cellular biology 27, 6606–6614, https://doi.org/10.1128/MCB.02166-06 (2007).
Nishi, R. et al. Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Molecular and cellular biology 25, 5664–5674, https://doi.org/10.1128/MCB.25.13.5664-5674.2005 (2005).
Li, C. L. et al. Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Molecular cell 59, 1025–1034, https://doi.org/10.1016/j.molcel.2015.08.012 (2015).
Sugasawa, K. et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121, 387–400, https://doi.org/10.1016/j.cell.2005.02.035 (2005).
Iwai, S., Shimizu, M., Kamiya, H. & Ohtsuka, E. Synthesis of a phosphoramidite coupling unit of the pyrimidine (6-4) pyrimidone photoproduct and its incorporation into oligodeoxynucleotides. Journal of the American Chemical Society 118, 7642–7643, https://doi.org/10.1021/ja9603158 (1996).
Sugasawa, K. et al. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev 15, 507–521, https://doi.org/10.1101/gad.866301 (2001).
Sugasawa, K., Akagi, J., Nishi, R., Iwai, S. & Hanaoka, F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Molecular cell 36, 642–653, https://doi.org/10.1016/j.molcel.2009.09.035 (2009).
Suzuki, T., Harashima, H. & Kamiya, H. Effects of base excision repair proteins on mutagenesis by 8-oxo-7,8-dihydroguanine (8-hydroxyguanine) paired with cytosine and adenine. DNA repair 9, 542–550, https://doi.org/10.1016/j.dnarep.2010.02.004 (2010).
Sassa, A. et al. In vivo evidence that phenylalanine 171 acts as a molecular brake for translesion DNA synthesis across benzo[a]pyrene DNA adducts by human DNA polymerase kappa. DNA repair 15, 21–28, https://doi.org/10.1016/j.dnarep.2013.12.008 (2014).
Stary, A. & Sarasin, A. Simian virus 40 (SV40) large T antigen-dependent amplification of an Epstein-Barr virus-SV40 hybrid shuttle vector integrated into the human HeLa cell genome. J Gen Virol 73(Pt 7), 1679–1685, https://doi.org/10.1099/0022-1317-73-7-1679 (1992).
Acknowledgements
We thank Dr. Tomonari Matsuda (Kyoto University) for providing us with pMY189 and Dr. Tatsuo Nunoshiba (International Christian University) for providing us with KS40/pOF105. We are grateful to Dr. Samuel H. Wilson (NIEHS, National Institutes of Health) and Dr. Masayuki Yokoi (Kobe University) for helpful comments and discussions. We wish to thank Dr. Takeshi Yasuda (National Institute of Radiological Sciences) for allowing us to use the UV irradiator. We thank Enago (www.enago.jp) for English-language review. This work was supported by the joint research program of the Biosignal Research Center, Kobe University [291002 to A.S.] and JSPS KAKENHI [19K12339 and 16K16195 to A.S., 25281022 to M.Y. and 16H02953 to H.S.]. This work was also supported by the grants from the Takeda Science Foundation [to K.U. and H.S.] and Mitsubishi research foundation [to H.S.].
Author information
Authors and Affiliations
Contributions
A.S. and H.T. designed the research, A.S., H.T., A.T., K.H., M.S., M.T., H.S., S.T., K.S., M.Y., M.H. and K.U. discussed the study and set up the experiments. A.S., H.T., A.T., K.H., M.S. and M.T. performed the research and analyzed data. A.S. and H.T. wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sassa, A., Tada, H., Takeishi, A. et al. Processing of a single ribonucleotide embedded into DNA by human nucleotide excision repair and DNA polymerase η. Sci Rep 9, 13910 (2019). https://doi.org/10.1038/s41598-019-50421-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50421-8
This article is cited by
-
RNA: a double-edged sword in genome maintenance
Nature Reviews Genetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
