
Abstract
Admixed American populations have different global proportions of European, Sub-Saharan African, and Native-American ancestry. However, individuals who display the same global ancestry could exhibit remarkable differences in the distribution of local ancestry blocks. We studied for the first time the distribution of local ancestry across the genome of 264 Brazilian admixed individuals, ascertained within the scope of the Brazilian Initiative on Precision Medicine. We found a decreased proportion of European ancestry together with an excess of Native-American ancestry on chromosome 8p23.1 and showed that this is due to haplotypes created by chromosomal inversion events. Furthermore, Brazilian non-inverted haplotypes were more similar to Native-American haplotypes than to European haplotypes, in contrast to what was found in other American admixed populations. We also identified signals of recent positive selection on chromosome 8p23.1, and one gene within this locus, PPP1R3B, is related to glycogenesis and has been associated with an increased risk of type 2 diabetes and obesity. These findings point to a selection event after admixture, which is still not entirely understood in recent admixture events.
Similar content being viewed by others

Introduction
Population structure due to genetic ancestry has been a significant confounding factor in genome-wide association studies (GWAS)1 since it can potentially lead to results that are not replicated among different populations2,3,4. To date, the majority of GWAS have been performed in European and Asian populations5,6 with a poor representation of other geographical areas and admixed populations. Even though ample GWAS reproducibility in medical genetic studies has been claimed7,8, it has also been shown that GWAS findings in European populations may not be transferable to admixed American cohorts due to the differences in population substructure, including a specific distribution of ancestry tracts in the genome caused by genetic drift9. In addition, admixed populations are likely to hide a larger number of genetic variants that have functional effects10, and local ancestry information could aid in identifying causal variants in complex traits11. Local ancestry has been inferred in different admixed American individuals, which showed ancestry-specific population substructures and differences in local ancestry proportions12,13,14,15,16,17,18.
Brazilian individuals encompass recent demographic admixture, including European, sub-Saharan African, and Native-American populations19,20,21. The proportion of global ancestries among the three populations is different between Brazilian and other admixed American populations13,22,23 and even among Brazilian individuals from different geographical regions19,20. However, local ancestry inference has been poorly explored in Brazilian individuals20, and since a specific demographic history can impact human populations differently, their study could potentially add relevant information about the distribution of local ancestry along the human genome. Furthermore, it has been established that ancestry proportions across different regions of the genome could present deviations, which can be due to the lack of methodological power to discriminate between two different ancestries in local ancestry inference, genetic drift after the admixture, or signals of recent natural selection24,25,26.
Therefore, our primary goals are to describe the distribution and to search for possible deviations of local ancestry blocks across the genome of Brazilian admixed individuals. Furthermore, if deviations in local ancestry are found beyond chance, we aim to explore the issue further by searching for signals of recent natural selection and the possibility of association with disease.
Results
Population structure and global ancestry inference
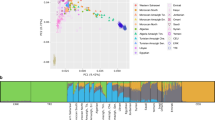
In a global PCA with the 1,000 Genomes Project (1 KGP) populations (Fig. 1a and Supplementary Fig. 1), BRS individuals were spread between Europeans, sub-Saharan Africans, and Native-Americans/East Asians, as are other admixed American populations, but were distributed mainly between Europeans and sub-Saharans rather than towards Native-Americans. Therefore, they appeared closer to Puerto Ricans and Colombians than to Peruvians and Mexicans; this is consistent with previous studies20,22. We observed similar population distributions when Native-American samples from Mao et al.27 are included in the PCA plot (Supplementary Fig. 1).

(a) The figure depicts a scatterplot showing the two first principal components for the BRS sample (black), Europeans (blue), sub-Saharan Africans (red), and admixed Americans (orange) from 1 KGP. (b) Global ancestry inference for ADMIXTURE K = 5 and K = 10, including the BRS and Europeans, sub-Saharan Africans and admixed Americans, where K = 10 had the lowest CV error estimate. Grades of blue, grades of red, and green indicate European, sub-Saharan African, and native American ancestry components, respectively.
We found the lowest cross-validation (CV) error at K = 10 in the global ancestry inference (0.45247; Supplementary Fig. 2), which shows the separation of subpopulations within continents (Fig. 1b and Supplementary Fig. 3). The component with the highest proportion in the BRS sample was the one most frequent in South Europeans (IBS, TSI; mean = 68.9%; SD = 14.4%), followed by the components most frequent in West Africans (ESN, YRI; mean = 7.7%; SD = 9.2%) and in Native-Americans (mean = 7.6%; SD = 5.8%). We show in the Supplementary Table 1 the remaining ancestry components, including SDs and 95% confidence intervals.
In order to avoid an excess of subpopulation variation in global and local ancestry comparison, we focused on global ancestry inference at K = 5, which identified the ancestry components from the five continental populations presented in the 1 KGP data (Fig. 1b, Supplementary Fig. 3). The BRS sample showed a high ancestry proportion of the European component (blue; mean = 76.9%; SD = 15.7%), followed by sub-Saharan African (red; mean = 13.8%; SD = 11.6%) and Native-American (green; mean = 7%; SD = 5.6%) components (Supplementary Table 1), which is consistent with previous studies19,20. We also observed that five individuals located between the European and East Asian clusters in PCA had an East Asian component in the admixture plots (Supplementary Fig. 3). Indeed, after this observation, we were able to retrieve some of the surnames of the volunteers, and four of them bore Japanese surnames.
To evaluate whether the BRS sample presents a geographical substructure based on the state of birth, we performed two tests: the analysis of molecular variance (AMOVA), which compared the genetic distance between the state of birth groups based on a set of 10,000 random SNPs across the genome and separately by chromosome; and the Kruskal–Wallis test which compared each EUR, AFR, and NAT proportions (ADMIXTURE K = 5) with the state of birth of individuals in the BRS sample. The AMOVA results from the set of 10,000 random SNPs showed that 99.98% of the variation component was observed within groups and 0.02% was observed among groups (total φ-statistics = 0.0002; p-value = 0.2327; Supplementary Table 2), indicating the absence of a population substructure, which is consistent with the lack of clusters observed in the PCA from the BRS sample based on the state of birth (Supplementary Fig. 4). The Kruskal-Wallis test showed EUR p-value = 0.0443, AFR p-value = 0.0567, and NAT p-value = 0.0701, which strengthens the hypothesis of the lack of substructure in the BRS sample based on the state of birth. However, previous studies have found some substructure in Brazilian populations20,28. We believe that our results differ from previous reports since most of our samples are from a different geographic region in Brazil, the state of São Paulo. In addition, we based our analysis of population substructure on the state of birth rather than ethnoracial self-classification28.
Local ancestry
Ancestry inference from ADMIXTURE at K = 5 and the average ancestry estimated over all the genome of RFMix from three reference panels, shows a high correlation (Pearson’s correlation ρ > 0.97), indicating consistency between the algorithms (Supplementary Fig. 5). We observed an average local ancestry proportion of 82.7% (SD = 2.2%) across the genome for the European component, 9.8% (SD = 1.3%) for the sub-Saharan African component, and 7.4% (SD = 1.9%) for the Native-American component.
One challenge in our study design was to select an adequate Native-American reference for local ancestry inference. Therefore, we built two different Native-American references, as detailed in the Methods section: the first was based on individuals from the PEL population who presented more than 0.95 of Native-American ancestry proportion at K = 5 in ADMIXTURE, defined here as “Native-American Peruvians” (Supplementary Fig. 6). Based on this approach, we observed a decrease in the European ancestry proportion in the BRS sample on chromosome 8p23.1 (8092025 bp–11859740 bp; Fig. 2a) related to an excess of Native-American ancestry proportion, which was higher than 4.42 SDs from the genome-wide mean (Fig. 2e). The excess of Native-American ancestry in this genetic region was also observed in Colombian, Mexican, and Puerto Rican populations (Supplementary Figs 7–9). However, in a second approach using the pool of Aymaran, Nahuan, Mayan, and Quechuan individuals from Mao et al.27 as Native-American references, no signal of decreased European or an excess of Native-American ancestry on chromosome 8p23.1 was found in any of the samples, including BRS and Peruvian (Supplementary Figs 7–11).

(a,c,e) Local ancestry inference based on the 59 BRS individuals carrying the non-inverted haplotypes (NI/NI), showing each chromosome in the x-axis, and the proportion of European (EUR; grades of blue), sub-Saharan African (AFR; grades of red), and Native-American ancestries (NAT, grades of green) in the y-axis. Each dashed line indicates the 4.42 SD thresholds. (b,d,f) Tests of signals of positive selection based on study design described in Fig. S3, showing the proportion of FCS outliers in the y-axis, following the same colour pattern for each ancestry component. Each dashed line indicates the 99.5th percentile thresholds.
Chromosome 15cen-q11.2 (20116146 bp–20564575 bp) also showed a decreased European ancestry proportion (Fig. 2a) with an excess of sub-Saharan African (Supplementary Fig. 11) and Native-American Peruvian ancestries (Fig. 2e). Previous studies have shown a low accuracy of local ancestry inference in regions with complex patterns of LD and recombination rates, such as the centromeres, where abrupt deviations from reference panels can lead to a systematic bias in the ancestry inference24,25,26. We found nine SNPs located on the locus at 15cen-q11.2, which covered a region of 448.4 Kb of length divided into 456 tracts in RFMix inference. Among the 456 tracts, up to 260 presented posterior probabilities less than 0.9 (Supplementary Figs 12 and 13). This observation contrasts with what was found on chromosome 8p23.1 (1298 SNPs ranging 3.76 Mb, divided into 575 RFMix tracts), where we found only 76 tracts out of 575 with posterior probabilities less than 0.9 (Supplementary Figs 14 and 15). Therefore, the deviation on local ancestry detected on the locus at 15cen-q11.2 might be due to its location and the low number of SNPs, which could have led to a lack of power of RFMix in local ancestry inference in this genomic region.
Exploring the 8p23.1 region
To examine the inconsistency of Native-American ancestry estimation when using different references, we studied chromosome 8p23.1 in more detail. We observed that the upstream and downstream flanking regions of the locus on chromosome 8p23.1 were previously described as recombination hotspots and an inversion has been identified in this region (Fig. 3)29, which is consistent with the presence of long-range LD haplotypes30. Therefore, to discard false-positive results for the excess of Native-American ancestry on 8p23.1 based on inversions, we controlled the local ancestry inferences for the presence (I) or the absence (NI) of the inversion in both the target and reference populations by a haplotype-based method implemented in the invClust package in R software31. To do that, we analysed the distribution of NI, and I haplotypes on chromosome 8p23.1 in the BRS sample and other admixed American populations and compared NI, and I haplotypes with the ancestry identified on chromosome 8p23.1 (Fig. 4). As shown in Supplementary Table 3, we found 59, 103, and 55 BRS individuals with NI/NI, NI/I, and I/I genotypes, respectively.

(a) Zoom of chromosome 8p23.1, showing the Native-American (NAT) proportion with a solid green line (threshold: dashed green line), and the proportion of FCS outliers with a solid red line (threshold: dashed red line). Each grey dot represents the FCS value of each SNP (threshold: dashed grey line). (b) Standardised recombination ratio (SRR) in dark grey. The dashed line shows the threshold for recombination hotspots.

Stacked bar plot showing the proportion of the distribution of NI/NI (pink), NI/I (purple), and I/I (brown) genotypes according to the ancestry tract (x-axis), which was inferred for each admixed American population. The Native-American ancestry tracts from the 23 Peruvians (asterisk) were inferred based on Mao et al., 2007 as reference. I: inverted, and NI: non-inverted haplotypes.
The first local ancestry estimations were computed using the entire BRS sample; however, differences in the presence of the non-inversion or inversion segments in each individual could affect the population estimation. Therefore, we classified the BRS and the reference samples as homozygous for the non-inversion (NI/NI) as well as the inversion (I/I). Then, we recalculated the local ancestry distributions and deviations separately. Heterozygous individuals for the inversion (NI/I) were not included because of the inability to perform an accurate identification of the phased chromosome containing the inversion31,32. Consequently, it was impossible to match the genotype with the local ancestry estimations. The results show a deviation in local ancestry on chromosome 8p23.1 for the NI/NI genotype group, using both Native-American Peruvians and the pool of Native-American samples from Mao et al.27 as a reference. We observed an increase in the Native-American ancestry and a decrease in the European ancestry in the 8p23.1 region compared to the genome-wide estimations (Supplementary Fig. 16). We did not observe a deviation in the Native-American ancestry for the I/I genotype group on the 8p23.1 region when using the Native-American individuals from Mao et al.27 as a reference (we did not find I/I genotypes in the Native-American Peruvian reference). However, we observed a decrease in European ancestry and an increase in sub-Saharan ancestry compared to the genome-wide values in the same genomic region. We performed the same analysis for Colombian, Mexican, and Puerto Rican populations as well as for the 62 admixed Peruvians (using Mao et al.27 as a reference) (Supplementary Figs 17–20). Interestingly, in the Mexican sample, we observed the same pattern as in the BRS for both the NI/NI and I/I samples, using both Native-American Peruvians and Native-American individuals from Mao et al.27 as a reference. We also observed the deviation among admixed Peruvians who carry the NI/NI based on the analysis using Native-American individuals from Mao et al.27 as a reference (Supplementary Fig. 20).
Haplotype bias evaluation by MDS
However, as stated above, it is important to note that the chromosome 8p23.1 region shows long-range LD haplotypes and is an inverted region, which could affect both the phasing and the local ancestry estimations30. Therefore, we performed multidimensional scaling (MDS) analysis based on the IBS values and Hudson’s Fst estimations on chromosome 8p23.1 to verify whether we had any bias in our previous analyses and to provide further proof of ancestry deviation by considering information unbiased by haplotype and LD information. The MDS plot from chromosome 8p23.1 in NI/NI individuals shows three clusters: one for European samples, one for sub-Saharan African samples, and one for Native-American samples (Supplementary Fig. 21a). All BRS NI/NI samples fall in the Native-American cluster, which is consistent with the observations in the local ancestry analyses. The MDS plot for I/I individuals also shows three clusters: one for European samples, one for sub-Saharan African samples, and one for the BRS and MXL individuals (Supplementary Fig. 22a). Unexpectedly, the Native-American individuals fall in the European cluster. These results, together with the fact that BRS and MXL do not cluster with the Europeans, agree with the local ancestry observations. However, the MDS pattern found in the 8p23.1 region could also be found in a set of random genomic regions. To investigate this issue, we estimated MDS among seven genomic regions at random across the genome with the same size of the 8p23.1 region (1883857 bp). The similarity between the MDS plot from the region at chromosome
8p23.1 and each of the MDS plots from the random genomic regions was evaluated by the Procrustes axis rotation analysis. We observed moderate Procrustes correlation values in the NI/NI samples (0.512–0.653; Supplementary Fig. 21b–h) and weak Procrustes correlation values in the I/I genotype (0.271–0.359; Supplementary Fig. 22b–h). Therefore, we suggest that the MDS pattern found in the 8p23.1 locus is not found in a set of random genomic regions.
Fst estimation on chromosome 8p23.1
Fst estimates on chromosome 8p23.1 show that the NI/NI genotypes from the BRS individuals are more similar to those from Native-American Peruvian and Native-American populations than to those from Sub-Saharan, Colombian, Puerto Rican, and European populations, in agreement with both the local ancestry and the MDS analyses (Supplementary Tables 4 and 6; Supplementary Fig. 23). For the I/I individuals, Fst on chromosome 8p23.1 shows a decreased similarity between BRS and Europeans, and also a decrease between BRS and Native-Americans (Supplementary Tables 5, 6; Supplementary Fig. 24), which it is in agreement with the clusters observed in the MDS. However, the sharp increase in Sub-Saharan African ancestry observed in the local ancestry is not seen in the Fst and is weakly supported by the MDS. This fact could be due to the lack of the appropriate sub-Saharan African proxies (according to slave disembarkation numbers, most sub-Saharan African ancestry in São Paulo and Southeast Brazil is expected to come from West-Central Africa (75%) and Southeast Africa (15%) (http://www.slavevoyages.org) regions, unsampled in the 1 KGP database33), or to a false positive in the local ancestry estimation18.
In addition, the evaluation of the DNA sequence of chromosome 8p23.1 based on phased haplotypes showed that the DNA sequence from the BRS samples with NI/NI genotype is more similar to the DNA sequence from Native-American PEL samples as reported by Mao et al. (2007), than to European and African samples (Supplementary Fig. 25). However, we could not observe the same results for samples with the I/I genotypes, probably due to the reduced number of individuals with the I/I genotype (Supplementary Fig. 26).
Therefore, the NI haplotypes found in the BRS sample are more similar to the NI haplotypes found in non-admixed Native-Americans than the NI haplotypes found in Europeans or some admixed Americans (such as Colombians or Puerto Ricans) although the European genome-wide ancestry found in BRS is much higher than the Native-American ancestry. The conclusions about I/I haplotypes are more challenging, due to the low frequency of the I allele in Native-American populations34; however, the lack of shared ancestry with European I haplotypes is clear. Therefore, our results depict a drop in the European ancestry in the region, both in the I and NI haplotypes, which in the case of the NI, is counterbalanced by an increase in the Native-American NI haplotypes.
Detection of positive selection
Local ancestry proportions more than 4.42 SDs from the genome-wide average could be indicative of positive selection18,24 since this deviation is equivalent to a statistical significance p-value < 1 × 10−5 from neutrality simulation tests24. To check whether the ancestry deviation detected on chromosome 8p23.1 might be related to a positive selection event, we tested our sample for positive selection by five neutrality statistics: intrapopulation absolute integrated haplotype scores (|iHS|, |ΔiHH|)35,36, interpopulation integrated haplotype score (|ΔiHHderived|)37, interpopulation extended haplotype homozygosity (XP-EHH)36, and population branch statistics (PBS)38 based on Hudson’s Fst39.
One tail p-values from the five different statistics were combined using the method proposed by Fisher18,40, defined as Fisher combined score (FCS). We assigned as outlier those SNPs within the 1% highest FCS value across the genome. In that way, we found 46 blocks of 100-SNPs with the highest proportion of outlier SNPs: 11 blocks identified as European ancestry; 19 as African; 12 as Native-American; one as European or African; one as African or Native-American; and two as European or Native-American (Supplementary Table 7). Among the Native-American blocks, we found the chromosome 8p23.1 locus (8962666 bp–9265464 bp) (Fig. 2f), which overlaps with the region found with an excess of local ancestry (Fig. 3). However, we did not observe the European and Native-American selection signal on chromosome 8p23.1 when the selection test is conducted separately for NI/NI and I/I individuals, although the selection signal maintains at the threshold line for NI/NI individuals (Supplementary Fig. 16). Although four of these tests are based on extended haplotype homozygosity35,36,37 and the structured haplotype on chromosome 8p23.1 could bias the result, they did not present signals of positive selection, separately. As shown in Fig. 3, the region within chromosome 8p23.1 with increased Native-American component and signals of positive selection spans 320.8 kb and includes two genes, PPP1R3B and ER1, and one noncoding RNA (LOC157273).
Discussion
Population genetics studies have led to a better understanding of human diseases in the context of human evolution, such as the heterozygous advantage phenomenon in sickle-cell anaemia41, cystic fibrosis42, and resistance to hepatitis C virus infection43. More recently, genome-wide studies have shown different risk allele frequencies from SNPs associated with the disease among populations worldwide3, as well as genotypes associated with target treatments in specific populations44,45. In this context, local ancestry information across the genome is likely to become relevant to determine whether a genetic variant is expected to be useful in precision medicine, especially for an admixed population. Although local ancestry information is available for admixed American populations9, it is still poorly explored in Brazilian individuals20. Therefore, in order to fulfil the lack of data, the present study provides information on ancestry tracts across a sample of Brazilian individuals.
Our results showed that the BRS sample is composed of a high proportion of European ancestry followed by sub-Saharan African and Native-American ancestry, showing remarkable variability among BRS individuals. At a genome-wide level, BRS sample is closer to Puerto Ricans and Colombians than to Peruvian and Mexican populations, which is consistent with a large amount of European ancestry detected in other Brazilian samples from different geographic regions19,20,22,28. This result is also consistent with previous findings in other admixed American populations, such as in Mexico, Puerto Rico, Peru, Colombia, Ecuador, Chile, and Argentina13,22,23,46, including ancestry estimations by different haplotype-based methods47,48
Average genome-wide local ancestry showed similar results compared to global ancestry inference in the BRS sample, which indicates consistency between the two ancestry inferences. The local ancestry screening along the genome shows that BRS samples have decreased European ancestry tracts on chromosome 8p23.1, followed by an excess of Native-American ancestry tracts in this same region. This locus is known to present inversion events, which can generate long-range LD regions and could lead to a possible confounding of local ancestry inference30,49. Therefore, we further focused our analyses on both homozygous NI/NI and I/I genotype only since we cannot define the ancestry tract for each haplotype from the NI/I individuals. This approach considerably reduced the sample size for analyses performed independently in I and NI individuals.
Brazilian NI/NI samples have extreme lower values of European ancestry in the 8p23.1 region compared to average values along the chromosome 8 and genome-wide. These observations point to the predominance of Native-American origin rather than European of NI haplotypes in the Brazilian population. The Brazilian I/I sample also shows lower values of European ancestry but followed by an increase in sub-Saharan African ancestry. Together, these results point to the presence of selection pressure against the European haplotypes in this genomic region in favour of the Native-American ancestry tracts for the NI haplotypes, and probably the sub-Saharan African tracks for the I haplotypes.
Results in other admixed American populations show variability. Regarding NI/NI individuals, Mexican and Peruvian populations have more similarity on chromosome 8p23.1 with Native-Americans than with European populations, while Colombian and Puerto Rican populations have more similarity with European populations. Therefore, genomic ancestry distribution results cannot be extrapolated to different populations, and differences on 8p23.1 ancestry among populations point to different demographic histories and, more importantly, different selective pressures in the different populations.
Salm et al. showed that NI/I and I/I genotypes are rare in Native-American populations due to demographic events50, and their study suggests that a serial founder model of migration from Africa explains the demographic distribution of the chromosomal 8p23.1 inversion. We did not find evidence in favour or against this demographic explanation for the I and NI allelic frequency distributions in a pre-Columbian expansion. However, the increase of Native-American component on 8p23.1 in Brazilian, Mexican, and Peruvian samples is difficult to explain by demographic events alone, even considering extreme genetic drift, and it points to a selection event after admixture24,25,26.
Therefore, our findings suggest that either the inversion event occurred more than once, or that genetic drift has produced a high divergence between European and Native-American haplotypes in this genomic region before the European arrival in the Americas. Finally, we suggest that the European NI haplotypes on chromosome 8p23.1 were disadvantaged in favour of the Native-American NI haplotypes in the context of new heterogeneous genomic diversity landscapes and changing environmental conditions which arose after post-Columbian admixture events in the Americas, in Brazil and Mexico. Selection after admixture has been described in archaic admixture between modern humans and Neanderthals51,52, but it is not entirely understood in more recent admixture events53.
Deviation in local ancestry genomic distribution has also been found on chromosome 6p22 in African-American14,54, and on chromosome 8p23.1 in Mexican populations55. However, follow-up studies have depicted a critical view of these results, reporting the need for higher thresholds than p > 10−5 and the risk of errors in local ancestry inferences24, pointing to long-range LD as the possible origin of biased local ancestry predictions30. Moreover, they have highlighted the need for parallel analysis with convergent results to have enough evidence of a selection event after an admixture process. Taking these perspectives into account, we have corroborated our local ancestry deviation, reinforcing it with non-haplotype-dependent analyses and selection tests leading to the conclusion of selected Native-American ancestry in the 8p23 region in admixed Brazilians. Therefore, we consider this finding as one of the first pieces of substantial evidence of selection after admixture in admixed Americans, beyond the HLA region56,57,58.
The selection test revealed signs of positive selection on chromosome 8p23.1, in comparison with Peruvians and Native-Americans. Interesting, the PPP1R3B gene found within the candidate region encodes a regulatory subunit 3B of the phosphatase-1 protein. PPP1R3B plays an essential role in glycogen synthesis, and a previous study showed that PPP1R3B overexpression reduces hypoglycaemia after long-term fasting59. Indeed, the main clinical phenotypes associated with metabolic disorders, taking admixture into account, are related to type 2 diabetes, insulin secretion, body mass index, obesity, and adiposity10,59,60,61,62. In this way, we can hypothesise that during the Brazilian population admixture process after 1500 CE, some PPP1R3B haplotypes from the Native-American ancestry gene pool could have been advantageous, due to increased fat and glucose storage under a restrictive diet environment, such as severe famine periods63, which increased the frequency of the Native-American PPP1R3B haplotypes in the Brazilian population. Because of the long-range LD haplotype, due to inversion events, the locus on chromosome 8p23.1 from Native-American ancestry also had the frequency increased by PPP1R3B hitchhiking. Future gene expression studies considering genetic ancestry should confirm or disprove this hypothesis.
In conclusion, we found an excess of Native-American haplotypes in the BRS sample at a locus on chromosome 8p23.1, and the presence of different European and Native-American haplotypes, due to long-range LD effects, helped us to understand further the local ancestry deviation. This unusual local ancestry deviation, which could be due to a recent adaptation after the admixture, was further studied, and the hypothesis proposed was supported by additional selection tests. The study of admixed samples allowed us to identify a process of adaptation in a region where the role of selection and demography was unclear. Our results also emphasize the need to study additional admixed American populations independently, avoiding extrapolated conclusions from one to another, since the admixture process can show relevant differences, as we have demonstrated in the present work. Moreover, we propose that individual local ancestry information can be used to obtain relevant information about human health, assisting in the identification of populations that may be at higher risk for specific disorders, which can, in turn, be applied in the recent efforts to develop and implement precision medicine.
Methods
Subjects
We analysed 264 healthy individuals (159 females; 105 males) ascertained at the University of Campinas hospital (UNICAMP, Campinas, São Paulo, Brazil) within the scope of the Brazilian Initiative on Precision Medicine Project (BIPMed; http://bipmed.org). We defined them as the Brazilian-São Paulo sample (BRS). Among them, 202 had information about their state of birth (Supplementary Fig. 27). This study was approved by the Research Ethics Committee of the University of Campinas (UNICAMP, Campinas, São Paulo, Brazil), and we obtained informed consent from all participants who signed a consent form before entering the study. All methods were performed in accordance with the relevant guidelines and regulations.
We compared the BRS sample with two publicly available datasets: the first included sequencing data from 2504 unrelated individuals from 26 worldwide populations from the 1000 Genomes Project phase 3 (1 KGP)33 (GRCh37/hg19 assembly; ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/) (Supplementary Table 8), and the second including genotyped data from 43 Native-American individuals (25 Aymarans, 10 Nahuans, 6 Mayans, and 2 Quechuans) previously studied by Mao et al.27 and Martin et al.9.
SNP genotyping and initial data processing
The genomic DNA of the BRS sample was obtained from peripheral blood using the phenol-chloroform procedure64. The quantification and quality of each DNA sample were evaluated by Qubit® 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and Epoch 2 microplate spectrophotometer (BioTek Instruments Inc., Winooski, VT, USA). Genotype calling was performed by Genome-Wide Human SNP Array 6.0 platform (Affymetrix Inc, Santa Clara, CA) in the Laboratory of Multiuser Equipment facility at UNICAMP. We called the genotypes from fluorescent signals by CLRMM algorithm65, which were converted to PLINK format66 by in-house scripts.
We used PLINK 1.966 software to perform SNP and individual filtering.67 Among the 906,600 genotyped SNPs, we removed 1,428 unmapped, 37,149 non-autosomal, 134,006 ambiguous (SNPs with G/C or A/T alleles), and 128,380 SNPs in Hardy–Weinberg disequilibrium (p-value < 0.01)67. We also checked for SNP and individual missing data > 2%, but all SNPs and individuals passed this filter because the genotype call rate by the CRLMM algorithm was 100%65. After genotype filtering, 605,637 SNPs remained (Supplementary Table 9). We performed the same SNP filtering procedure in the Native-American sample, and 719,472 SNPs remained (Supplementary Table 9).
Since we analysed an admixed population, an excess of heterozygote SNPs could be due to recent population substructure, and SNPs with Hardy-Weinberg disequilibrium could be maintained23. However, Hardy-Weinberg disequilibrium filtering is performed to remove possible genotyping errors67 and, although it can eliminate signals of selection; it reduces the possibility of finding false-positive results. For this reason, we aimed to be conservative in terms of excluding genotyping errors, including a high p-value threshold, which guarantees that the signals of selection we found are not produced by systematic genotyping errors.
We evaluated the heterozygosity rate for each individual to search for inbreeding (low heterozygosity rates) or sample contamination (high heterozygosity rates)67. The mean heterozygosity rate of the BRS sample was 0.317 (SD = 0.012). We removed 11 individuals with heterozygosity rate higher (n = 5) or lower (n = 6) than two SDs from the mean. We also removed eight individuals from pairs of individuals who presented identical-by-state alleles > 0.85, which could indicate duplicated samples66,67. Next, we excluded individuals from identical-by-descent and genomic relatedness matrix estimations > 0.125, which is the expected genomic relatedness for third-degree relatives. To maximise the sample size, we used a greedy algorithm implemented in PLINK 1.9 software. After individual filtering, 222 individuals remained suitable for further analysis (Supplementary Table 10). We did not perform individual filtering in the Native-American sample in order to analyse the same 43 individuals evaluated previously by Martin et al.9.
Merging of datasets
First, we merged BRS with 1 KGP, removing 5750 SNPs that did not match between the two datasets. After merging, we removed 3,612 SNPs with a minor allele frequency (MAF) < 0.01. To avoid bias in population structure, MDS, and global ancestry inference, we removed 291,734 SNPs in linkage disequilibrium (LD) by the following parameters: window size = 50 SNPs, shift step = 5 SNPs, and r2 = 0.567. In the end, we had 304,541 SNPs for the population structure and global ancestry inference and 596,275 SNPs for local ancestry inference (Supplementary Table 9). Next, we generated a second dataset by merging the Native-American sample with the BRS + 1 KGP dataset, and we kept 605,637 SNPs common to both datasets. After MAF and LD pruning, 294,823 SNPs remained for the population structure and global ancestry inference, and 570,323 SNPs remained for local ancestry inference (Supplementary Table 9). The merging process and the following filters were performed by PLINK 1.9 software.
Population structure and global ancestry inference
We compared the BRS sample and the 26 populations from 1 KGP using principal component analysis (PCA) in PLINK v1.9 software. Global ancestry was inferred by unsupervised ADMIXTURE maximum likelihood approach68 from K = 2 to K = 13 ancestral components. We also performed three different runs with different random seeds for each K to observe the consistency of estimates. The BRS sample has a sample size two times larger than other 1 KGP populations, which could lead to bias in admixture results69; therefore, we divided the BRS sample into two random subsamples (n = 111) and ran ADMIXTURE separately for each subsample.
Analysis of molecular variance
Brazilian individuals could present different ancestries based on their geographical location. Therefore, we evaluated possible clustering based on available state of birth information by PCA. In addition, we evaluated whether the state of birth groups are genetically similar by the Analysis of Molecular Variance (AMOVA) using poppr.amova70 and Kruskal-Wallis test based on EUR, AFR, and NAT ancestry proportions (ADMIXTURE K = 5). Kruskal-Wallis p-values were corrected by Bonferroni adjustment under three comparisons. The analyses were performed by an in-house script developed in R software.
Local ancestry inference
We phased the SNPs without LD pruning with SHAPEIT2 v2.r387 software using the default parameters71. After phasing, we converted the output data from SHAPEIT2 to the input files required by RFMix v.1.5.4 software32, following a pipeline reported by Martin et al.9 (https://github.com/armartin/ancestry_pipeline).
We estimated the Native-American local ancestry from two approaches. The first approach was to build a reference by selecting 23 individuals from the PEL population (Peruvian individuals from 1 KGP) who presented more than 0.95 of Native-American ancestry proportion at K = 5 in ADMIXTURE (18 individuals had > 0.99 Native-American ancestries). To avoid biases due to unbalanced reference panel sizes32, we selected 23 random individuals among Europeans (CEU, GBR, IBS, and TSI) and 23 among Africans (ESN, GWD, LWK, MSL, and YRI) (Supplementary Fig. 6a). The second approach was to build the Native-American reference based on the 43 Native-American samples from Mao et al.27 (25 Aymarans, 10 Nahuans, 6 Mayans, and 2 Quechuans), 43 random individuals among Europeans, and 43 among Africans to evaluate the consistency of local ancestry inference using the first approach (Supplementary Fig. 6b).
We ran RFMix in the PopPhased mode with a minimum window size of 0.2 cM, one EM iteration, and node size 5, maintaining the reference panel after the initial inference step and saving the forwards–backwards probabilities. We developed an in-house R script (https://www.r-project.org/) to analyse the proportion of European, African, or Native-American ancestry among BRS individuals for each SNP across the genome. We inferred local ancestry and estimated the proportion of SNPs associated with each ancestry across the genome for Colombian (CLM), Mexican (MXL), and Puerto Rican (PUR) populations from 1 KGP to compare results with the BRS sample. We also analysed the local ancestry of all PEL individuals in the second local ancestry approach, where we used Native-American individuals from Mao et al.27 as a Native-American reference.
Identification of haplotype inversions
Since our results show evidence of haplotype inversions, we identified non-inverted (NI) and inverted (I) haplotypes by a haplotype-based method developed by Cáceres and González (invClust package in R)31. We set invClust parameters as follows: two multidimensional scaling components; method = 1, which performed an EM algorithm for three genotypes; 1,000,000 iterations for the EM algorithm; and 10e−6 for EM convergence tolerance.
Fst estimation and multidimensional scaling analysis
In order to evaluate which haplotype from the 1 KGP populations has a high similarity with the BRS sample, we separated the NI/NI and I/I genotypes from all populations and estimated Hudson’s Fst39,72,73 across all chromosome 8 by a 1 Mb window. To complement the Fst information, we calculated and plotted multidimensional scaling (MDS) values from IBS estimations for each individual, using the R software, in the 8p23.1 and additional seven genomic regions at random across the genome with the same size of the 8p23.1 region (1883857 bp). To evaluate the similarity between the MDS plot from 8p23.1 and each MDS plot from the random genomic region, we performed Procrustes axis rotation. We compared the Procrustes results from the MDS plots by estimating the Procrustes correlation test under 10,000 permutations. All Procrustes analyses were performed by the vegan package in R74.
Haplotype analysis by DNA sequence
The SHAPEIT software was used to convert the phased haplotypes to VCF files, separating individuals with the NI/NI and the I/I genotype and by continental populations. The VCF files were converted to DNA sequence by read.vcfR and vcfR2DNAbin from the vcfR R package75. The DNA sequences were plotted by image.DNAbin function from the ape R package76.
Positive selection test
We tested our sample for positive selection by five neutrality statistics: intrapopulation absolute integrated haplotype scores (|iHS|, |ΔiHH|)35,36, interpopulation integrated haplotype score (|ΔiHHderived|)37, interpopulation extended haplotype homozygosity (XP-EHH)36, and population branch statistics (PBS)38 based on Hudson’s Fst39. Integrated haplotype scores and extended haplotype homozygosity were estimated by rehh package in R software35,36,57. PBS and Hudson’s Fst were calculated by an in-house script in R software based on previously reported formulae38,39. Interpopulation statistics require a reference population and PBS statistics for an outgroup population. Therefore, the sample we used as a reference population for the scans of positive selection in the BRS group were 404 European (CEU, GBR, IBS, TSI), 504 sub-Saharan African (ESN, GWD, LWK, MSL, YRI), and the 23 Native-American Peruvian (PEL) individuals of the 1 KGP for the first local ancestry approach, and the 43 Native-American individuals27 for the second local ancestry approach. PBS was evaluated using 96 Punjabi from Lahore, Pakistan (PJL) as outgroup population. We performed the same positive selection test for Colombian, Mexican, and Puerto Rican populations, to compare results with our sample, and also changed the Native-American reference panel (Supplementary Fig. 28).
The ancestral and derived allele assignation followed the alignment from 1 KGP data. We combined the one tail p-values of the different statistics by the method proposed by Fisher18,40 and showed in the following formulae:
where FCS is Fisher combined score, k is the number of combined statistics, and pi the empirical p-value, i.e., the genomic rank of the ith statistics divided by the total number of SNPs18,40. Since small values of pi lead to large FCS values, we defined outlier SNPs as those with values of FCS higher than 99% of the SNP FCS values across the genome (i.e., the 1% highest FCS values). To search for regions with a signal of positive selection, we split the genome into 100-SNP blocks. Then, we estimated the proportion of outliers within each block, and if a block was found to present a proportion of outliers higher than the 99.5th percentile (the highest 0.5%) among the proportions of all blocks, it was defined as a region under positive selection18,40. Although FCS violates the assumption of dependency among empirical p-values, it has a chi-square distribution with 2k degrees of freedom under neutrality40. Previous studies have also shown that combining different neutrality tests increases the power to detect signals of positive selection37,40.
References
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nature Reviews Genetics 11, 459–463, https://doi.org/10.1038/nrg2813 (2010).
Marchini, J., Cardon, L. R., Phillips, M. S. & Donnelly, P. The effects of human population structure on large genetic association studies. Nat Genet 36, 512–517, https://doi.org/10.1038/ng1337 (2004).
Myles, S., Davison, D., Barrett, J., Stoneking, M. & Timpson, N. Worldwide population differentiation at disease-associated SNPs. BMC medical genomics 1, 22–22, https://doi.org/10.1186/1755-8794-1-22 (2008).
Moonesinghe, R. et al. Estimating the contribution of genetic variants to difference in incidence of disease between population groups. European journal of human genetics: EJHG 20, 831–836, https://doi.org/10.1038/ejhg.2012.15 (2012).
Need, A. C. & Goldstein, D. B. Next generation disparities in human genomics: concerns and remedies. Trends Genet 25, 489–494, https://doi.org/10.1016/j.tig.2009.09.012 (2009).
Popejoy, A. B. & Fullerton, S. M. Genomics is failing on diversity. Nature 538, 161–164, https://doi.org/10.1038/538161a (2016).
Marigorta, U. M. & Navarro, A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet 9, e1003566, https://doi.org/10.1371/journal.pgen.1003566 (2013).
Marigorta, U. M., Rodríguez, J. A., Gibson, G. & Navarro, A. Replicability and Prediction: Lessons and Challenges from GWAS. Trends Genet 34, 504–517, https://doi.org/10.1016/j.tig.2018.03.005 (2018).
Martin, A. R. et al. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. The American Journal of Human Genetics 100, 1–15, https://doi.org/10.1016/j.ajhg.2017.03.004 (2017).
Goetz, L. H., Uribe-Bruce, L., Quarless, D., Libiger, O. & Schork, N. J. Admixture and clinical phenotypic variation. Human Heredity 77, 73–86, https://doi.org/10.1159/000362233 (2014).
Zhang, J. & Stram, D. O. The Role of Local Ancestry Adjustment in Association Studies Using Admixed Populations. Genetic Epidemiology 38, 502–515, https://doi.org/10.1002/gepi.21835 (2014).
Ko, A. et al. Amerindian-specific regions under selection harbour new lipid variants in Latinos. Nature communications 5, 3983–3983, https://doi.org/10.1038/ncomms4983 (2014).
Homburger, J. R. et al. Genomic Insights into the Ancestry and Demographic History of South America. PLoS Genetics 11, https://doi.org/10.1371/journal.pgen.1005602 (2015).
Gravel, S. et al. Reconstructing Native American Migrations from Whole-Genome and Whole-Exome Data. PLoS Genetics 9, https://doi.org/10.1371/journal.pgen.1004023 (2013).
Moreno-Estrada, A. et al. Reconstructing the Population Genetic History of the Caribbean. PLoS Genetics 9, https://doi.org/10.1371/journal.pgen.1003925 (2013).
Deng, L., Ruiz-linares, A., Xu, S. & Wang, S. Ancestry variation and footprints of natural selection along the genome in Latin American populations. Scientific Reports 6, 1–7, https://doi.org/10.1038/srep21766 (2016).
Browning, S. R. et al. Local Ancestry Inference in a Large US-Based Hispanic/Latino Study: Hispanic Community Health Study/Study of Latinos (HCHS/SOL). G3 (Bethesda, Md.) 6, 1525–1534, https://doi.org/10.1534/g3.116.028779 (2016).
Patin, E. et al. Dispersals and genetic adaptation of Bantu-speaking populations in Africa and North America. Science 356, 543–546, https://doi.org/10.1126/science.aal1988 (2017).
Rodrigues de Moura, R. et al. Meta-analysis of Brazilian genetic admixture and comparison with other Latin America countries. American Journal of Human Biology 27, 674–680, https://doi.org/10.1002/ajhb.22714 (2015).
Kehdy, F. S. G. et al. Origin and dynamics of admixture in Brazilians and its effect on the pattern of deleterious mutations. Proceedings of the National Academy of Sciences 112, 8696–8701, https://doi.org/10.1073/pnas.1504447112 (2015).
Mychaleckyj, J. C. et al. Genome-wide analysis in brazilians reveals highly differentiated native American genome regions. Molecular Biology and Evolution 34, 559–574, https://doi.org/10.1093/molbev/msw249 (2017).
Ruiz-Linares, A. et al. Admixture in Latin America: Geographic Structure, Phenotypic Diversity and Self-Perception of Ancestry Based on 7,342 Individuals. PLoS Genetics 10, e1004572–e1004572, https://doi.org/10.1371/journal.pgen.1004572 (2014).
Adhikari, K., Mendoza-Revilla, J., Chacón-Duque, J. C., Fuentes-Guajardo, M. & Ruiz-Linares, A. Admixture in Latin America. Current Opinion in Genetics and Development 41, 106–114, https://doi.org/10.1016/j.gde.2016.09.003 (2016).
Bhatia, G. et al. Genome-wide Scan of 29,141 African Americans Finds No Evidence of Directional Selection since Admixture. The American Journal of Human Genetics 95, 437–444, https://doi.org/10.1016/j.ajhg.2014.08.011 (2014).
Pasaniuc, B. et al. Analysis of Latino populations from GALA and MEC studies reveals genomic loci with biased local ancestry estimation. Bioinformatics 29, 1407–1415, https://doi.org/10.1093/bioinformatics/btt166 (2013).
Baran, Y. et al. Fast and accurate inference of local ancestry in Latino populations. Bioinformatics 28, 1359–1367, https://doi.org/10.1093/bioinformatics/bts144 (2012).
Mao, X. et al. A Genomewide Admixture Mapping Panel for Hispanic/Latino Populations. The American Journal of Human Genetics 80, 1171–1178, https://doi.org/10.1086/518564 (2007).
Lima-Costa, M. F. et al. Genomic ancestry and ethnoracial self-classification based on 5,871 community-dwelling Brazilians (The Epigen Initiative). Sci Rep 5, 9812, https://doi.org/10.1038/srep09812 (2015).
Kong, A. et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103, https://doi.org/10.1038/nature09525 (2010).
Price, A. L. et al. Long-Range LD Can Confound Genome Scans in Admixed Populations. American Journal of Human Genetics 83, 132–135, https://doi.org/10.1016/j.ajhg.2008.06.005 (2008).
Cáceres, A. & González, J. R. Following the footprints of polymorphic inversions on SNP data: from detection to association tests. Nucleic acids research 43, e53–e53, https://doi.org/10.1093/nar/gkv073 (2015).
Maples, B. K., Gravel, S., Kenny, E. E. & Bustamante, C. D. RFMix: A discriminative modeling approach for rapid and robust local-ancestry inference. American Journal of Human Genetics 93, 278–288, https://doi.org/10.1016/j.ajhg.2013.06.020 (2013).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74, https://doi.org/10.1038/nature15393 (2015).
Alves, J. M., Chikhi, L., Amorim, A. & Lopes, A. M. The 8p23 Inversion polymorphism determines local recombination heterogeneity across human populations. Genome Biology and Evolution 6, 921–930, https://doi.org/10.1093/gbe/evu064 (2014).
Voight, B. F., Kudaravalli, S., Wen, X. & Pritchard, J. K. A map of recent positive selection in the human genome. PLoS Biology 4, 0446–0458, https://doi.org/10.1371/journal.pbio.0040072 (2006).
Sabeti, P. C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918, https://doi.org/10.1038/nature06250 (2007).
Grossman, S. R. et al. A Composite of Multiple Signals Distinguishes Causal Variants in Regions of Positive Selection. Science 327, 883–886, https://doi.org/10.1126/science.1183863 (2010).
Yi, X. et al. Sequencing of 50 Human Exomes Reveals Adaptation to High Altitude. Science 329, 75–78, https://doi.org/10.1126/science.1190371 (2010).
Bhatia, G., Patterson, N., Sankararaman, S. & Price, A. L. Estimating and interpreting FST: The impact of rare variants. Genome Research 23, 1514–1521, https://doi.org/10.1101/gr.154831.113 (2013).
Deschamps, M. et al. Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. American Journal of Human Genetics 98, 5–21, https://doi.org/10.1016/j.ajhg.2015.11.014 (2016).
Wiesenfeld, S. L. Sickle-cell trait in human biological and cultural evolution. Development of agriculture causing increased malaria is bound to gene-pool changes causing malaria reduction. Science 157, 1134–1140 (1967).
Danks, D. M., Allan, J. & Anderson, C. M. A genetic study of fibrocystic disease of the pancreas. Annals of Human Genetics 28, 323–356, https://doi.org/10.1111/j.1469-1809.1965.tb00489.x (1965).
Hraber, P., Kuiken, C. & Yusim, K. Evidence for human leukocyte antigen heterozygote advantage against hepatitis C virus infection. Hepatology 46, 1713–1721, https://doi.org/10.1002/hep.21889 (2007).
Nyakutira, C. et al. High prevalence of the CYP2B6 516G–>T(*6) variant and effect on the population pharmacokinetics of efavirenz in HIV/AIDS outpatients in Zimbabwe. Eur J Clin Pharmacol 64, 357–365, https://doi.org/10.1007/s00228-007-0412-3 (2008).
Nordling, L. How the genomics revolution could finally help Africa. Nature 544, 20–22, https://doi.org/10.1038/544020a (2017).
Eyheramendy, S., Martinez, F. I., Manevy, F., Vial, C. & Repetto, G. M. Genetic structure characterization of Chileans reflects historical immigration patterns. Nature. Communications 6, 6472–6472, https://doi.org/10.1038/ncomms7472 (2015).
Montinaro, F. et al. Unravelling the hidden ancestry of American admixed populations. Nat Commun 6, 6596, https://doi.org/10.1038/ncomms7596 (2015).
Chacón-Duque, J. C. et al. Latin Americans show wide-spread Converso ancestry and imprint of local Native ancestry on physical appearance. Nat Commun 9, 5388, https://doi.org/10.1038/s41467-018-07748-z (2018).
Tian, C. et al. Analysis and application of European genetic substructure using 300 K SNP information. PLoS Genetics 4, 0029–0039, https://doi.org/10.1371/journal.pgen.0040004 (2008).
Salm, M. P. A. et al. The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Research 22, 1144–1153, https://doi.org/10.1101/gr.126037.111 (2012).
Sankararaman, S. et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507, 354–357, https://doi.org/10.1038/nature12961 (2014).
Kuhlwilm, M. et al. Ancient gene flow from early modern humans into Eastern Neanderthals. Nature 530, 429–433, https://doi.org/10.1038/nature16544 (2016).
Laso-Jadart, R. et al. The Genetic Legacy of the Indian Ocean Slave Trade: Recent Admixture and Post-admixture Selection in the Makranis of Pakistan. Am J Hum Genet 101, 977–984, https://doi.org/10.1016/j.ajhg.2017.09.025 (2017).
Bhatia, G. et al. Genome-wide Comparison of African-Ancestry Populations from CARe and Other Cohorts Reveals Signals of Natural Selection. The American Journal of Human Genetics 89, 368–381, https://doi.org/10.1016/j.ajhg.2011.07.025 (2011).
Guan, Y. Detecting structure of haplotypes and local ancestry. Genetics 196, 625–642, https://doi.org/10.1534/genetics.113.160697 (2014).
Meyer, D., C Aguiar, V. R., Bitarello, B. D., C Brandt, D. Y. & Nunes, K. A genomic perspective on HLA evolution. Immunogenetics 70, 5–27, https://doi.org/10.1007/s00251-017-1017-3 (2018).
Tang, H. et al. Recent Genetic Selection in the Ancestral Admixture of Puerto Ricans. The American Journal of Human Genetics 81, 626–633, https://doi.org/10.1086/520769 (2007).
Zhou, Q., Zhao, L. & Guan, Y. Strong Selection at MHC in Mexicans since Admixture. PLoS Genet 12, e1005847, https://doi.org/10.1371/journal.pgen.1005847 (2016).
Mehta, M. B. et al. Hepatic protein phosphatase 1 regulatory subunit 3B (Ppp1r3b) promotes hepatic glycogen synthesis and thereby regulates fasting energy homeostasis. J Biol Chem 292, 10444–10454, https://doi.org/10.1074/jbc.M116.766329 (2017).
Dunn, J. S. et al. Examination of PPP1R3B as a candidate gene for the type 2 diabetes and MODY loci on chromosome 8p23. Annals of Human Genetics 70, 587–593, https://doi.org/10.1111/j.1469-1809.2005.00248.x (2006).
Hayes, M. G. et al. Identification of HKDC1 and BACE2 as Genes Influencing Glycemic Traits During Pregnancy Through Genome-Wide Association Studies. Diabetes 62, 3282–3291, https://doi.org/10.2337/db12-1692 (2013).
Flores, Y. N. et al. Association between PNPLA3 (rs738409), LYPLAL1 (rs12137855), PPP1R3B (rs4240624), GCKR (rs780094), and elevated transaminase levels in overweight/obese Mexican adults. Molecular Biology Reports 43, 1359–1369, https://doi.org/10.1007/s11033-016-4058-z (2016).
Davis, M. Late Victorian holocausts: El Niño famines and the making of the third world (Verso, 2001).
Maniatis, T., Fritsch, E. F. & Sambrook, J. Molecular cloning: a laboratory manual (Cold Spring Harbor Laboratory, 1982).
Scharpf, R. B., Irizarry, R. A., Ritchie, M. E., Carvalho, B. & Ruczinski, I. Using the R Package crlmm for Genotyping and Copy Number Estimation. Journal of statistical software 40, 1–32, https://doi.org/10.1016/j.str.2010.08.012.Structure (2011).
Purcell, S. et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics 81, 559–575, https://doi.org/10.1086/519795 (2007).
Anderson, C. A. et al. Data quality control in genetic case-control association studies. Nat Protoc. 5, 1564–1573, https://doi.org/10.1038/nprot.2010.116 (2010).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Research 19, 1655–1664, https://doi.org/10.1101/gr.094052.109 (2009).
Puechmaille, S. J. The program structure does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Molecular Ecology Resources 16, 608–627, https://doi.org/10.1111/1755-0998.12512 (2016).
Excoffier, L., Smouse, P. E. & Quattro, J. M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491 (1992).
O’Connell, J. et al. A General Approach for Haplotype Phasing across the Full Spectrum of Relatedness. PLoS Genetics 10, https://doi.org/10.1371/journal.pgen.1004234 (2014).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genetics 2, 2074–2093, https://doi.org/10.1371/journal.pgen.0020190 (2006).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–909, https://doi.org/10.1038/ng1847 (2006).
Peres-Neto, P. R. & Jackson, D. A. How well do multivariate data sets match? The advantages of a Procrustean superimposition approach over the Mantel test. Oecologia 129, 169–178, https://doi.org/10.1007/s004420100720 (2001).
Knaus, B. J. & Grünwald, N. J. vcfr: a package to manipulate and visualize variant call format data in R. Mol Ecol Resour 17, 44–53, https://doi.org/10.1111/1755-0998.12549 (2017).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528, https://doi.org/10.1093/bioinformatics/bty633 (2019).
Acknowledgements
We are grateful to all DNA donors for their helpful cooperation. This study was supported by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, Brazil) grants BEPE-PD #2016/14423-9, and CEPID-BRAINN #2013/07559-3 IL-C and F.C. were also supported by grants from CNPq (Conselho Nacional de Pesquisa, Brazil). A.M.S., L.R.A. and D.C. were also supported by the Agència de Gestió d’Ajuts Universitaris i de la Recerca (Generalitat de Catalunya) grant 2014SGR866 and ‘Unidad de Excelencia María de Maeztu’ funded by MINECO (MDM-2014-0370).
Author information
Authors and Affiliations
Contributions
R.S. contributed with the study design, conceptualisation, data acquisition, and statistical analysis; A.M.S. and L.A.R. contributed to population genetics analysis; F.R.T., T.K.A. and M.L.S. contributed to data acquisition and genotyping; C.S.R. and B.S.C. contributed with genotype processing and public availability of data; and F.C., I.L.C. and D.C. contributed with project conceptualisation and served as principal investigators. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Secolin, R., Mas-Sandoval, A., Arauna, L.R. et al. Distribution of local ancestry and evidence of adaptation in admixed populations. Sci Rep 9, 13900 (2019). https://doi.org/10.1038/s41598-019-50362-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50362-2
This article is cited by
-
Putative role of HLA polymorphism among a Brazilian HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) population
Scientific Reports (2023)
-
Multivariate statistical approach and machine learning for the evaluation of biogeographical ancestry inference in the forensic field
Scientific Reports (2022)
-
Genetic variability in COVID-19-related genes in the Brazilian population
Human Genome Variation (2021)
-
The Brazilian Initiative on Precision Medicine (BIPMed): fostering genomic data-sharing of underrepresented populations
npj Genomic Medicine (2020)
-
Prospective avenues for human population genomics and disease mapping in southern Africa
Molecular Genetics and Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
