
Abstract
Respiratory syncytial virus (RSV) is a major cause of lower respiratory tract infections and hospital visits during infancy and childhood. Although risk factors for RSV infection have been identified, the role of microbial species in the respiratory tract is only partially known. We aimed to understand the impact of interactions between the nasal microbiome and host transcriptome on the severity and clinical outcomes of RSV infection. We used 16 S rRNA sequencing to characterize the nasal microbiome of infants with RSV infection. We used RNA sequencing to interrogate the transcriptome of CD4+ T cells obtained from the same set of infants. After dimension reduction through principal component (PC) analysis, we performed an integrative analysis to identify significant co-variation between microbial clade and gene expression PCs. We then employed LIONESS (Linear Interpolation to Obtain Network Estimates for Single Samples) to estimate the clade-gene association patterns for each infant. Our network-based integrative analysis identified several clade-gene associations significantly related to the severity of RSV infection. The microbial taxa with the highest loadings in the implicated clade PCs included Moraxella, Corynebacterium, Streptococcus, Haemophilus influenzae, and Staphylococcus. Interestingly, many of the genes with the highest loadings in the implicated gene PCs are encoded in mitochondrial DNA, while others are involved in the host immune response. This study on microbiome-transcriptome interactions provides insights into how the host immune system mounts a response against RSV and specific infectious agents in nasal microbiota.
Similar content being viewed by others


Introduction
Respiratory syncytial virus (RSV) is a major cause of lower respiratory tract infections and hospital visits during infancy and childhood1,2,3. Almost all children will have been infected with RSV by age two, with about 3% requiring hospitalization. A recent report estimated that in 2015 about 33.1 million children worldwide under age 5 had RSV-related acute lower respiratory infection; of those, approximately 10% required hospitalization with 59,000 in-hospital fatalities1. The incidence and mortality of RSV vary between geographic locations, with children from developing countries considered to be at higher risk4. In the United States, 20% of infants require outpatient treatment for RSV-related illnesses, with an associated economic burden of $1.9 billion5; around 3% of these cases are associated with serious bronchiolitis and viral pneumonia–the most common forms of severe RSV infection. In addition to major risk factors such as congenital or chronic cardiopulmonary disease, meta-analyses have identified additional risk factors for RSV, including preterm birth, low birth weight, siblings at home, day-care attendance, and maternal smoking6,7,8. Infants with RSV infection in early childhood are also at a higher risk of developing asthma and wheezing9,10,11. Although prophylactic treatments are available to prevent RSV infection in the most at-risk infants, attempts are ongoing to identify an effective and safe vaccine or small molecule drug to reduce the health burden of RSV.
Biomarker discovery through expression analysis is an important step in assessing disease severity and in distinguishing RSV from other common respiratory viruses12,13,14. Transcriptional profiles often reflect a host’s immune response to the virus, helping to explain disease progression and characterize its severity. Several studies have reported that T cell mediated response is crucial in clearing the viral load during RSV infection15,16. Recent reports also indicate that RSV infects CD4+ and CD8+ T cells and affects T cell function12,17,18, implicating T cells as potential biomarkers for RSV severity.
The microbiome in the respiratory tract is known to influence the course of acute infectious diseases19. The succession pattern of the nasal microbial community might influence host responses to RSV, thereby modulating inflammation and possibly disease severity. Indeed, several studies indicate that the nasal microbial composition affects the overall risk of developing respiratory tract infections20,21 and is associated with the severity of acute respiratory symptoms22.
Joint characterization of the nasal microbiome and host transcriptome may provide valuable insights into how viral infections influence the host response. However, the impact of the interactions between the nasal microbiome and the host transcriptome on the severity and clinical outcomes of RSV infection has not been fully understood. Recent studies have reported associations between nasal microbial compositions and whole blood gene expression22. In this work, we perform an integrative analysis to study associations between nasal microbial compositions and transcriptional profiles of bloodstream CD4+ T cells.
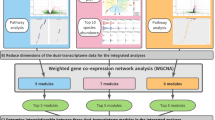
Network approaches provide an important framework for understanding complex relationships that influence human health23. In our study, we construct a network model that correlates microbial taxonomic profiles and host transcriptomic profiles. Examining this network in the context of RSV severity highlights important patterns of transcriptomic response related to immune processes and viral infection. An overview of our analysis approach is summarized in Fig. 1.

An overview of the data and analyses performed in this study.
Results
Baseline characteristics of participants and data source
We analyzed data for 58 infants with RSV infection, each of whom had both CD4 + gene expression and nasal microbiome data. The RSV disease severity was measured by the Global Respiratory Severity Score (GRSS). This allowed us to divide infants into two groups based on severity: mild and severe (see Methods section). We emphasize that GRSS is a composite score that reflects the worst values during the entire illness, rather than a single time point, allowing us to address the problem of clinical variability among subjects. GRSS also recapitulates more frequently used factors such as viral load (which has been associated with RSV disease severity in several studies24,25) and need for hospitalization (Supplemental Fig. 1), thus acting as excellent surrogate for disease severity26. Among the infants, 23 had mild illness and 35 had severe illness. There was no difference in any demographic characteristics between mildly and severely ill subjects (Table 1). The data collection occurred in three stages (Fig. 2A).

(A) Overview of study timelines, including when each type of data was collected. All 58 infants included in this study had both transcriptomic and microbiome data collected for at least one time point. (B) Venn diagrams showing the overlap in the infants that had either transcriptomic or microbiome data collected during the acute visit (57 of the 58 infants), the overlap in the transcriptomic data collected at either the initial (Visit 1) or follow-up visit (Visit 3), and the overlap in the infants microbiome data collected at either the initial (Visit 1) or follow-up visit (Visit 2). Numbers are shown separately for mild and severe groups, with the total number in each category indicated below the Venn diagram. (C) Differential gene expression analysis comparing Visit 1 versus Visit 2 RNA-Seq samples yields 27 significantly differentially expressed genes (FDR < 0.05). The log2 of the expression levels of these genes are shown as boxplots combining mild and severe samples. The genes on the left side of the panel have higher mean expression during the acute visit and those on the right side of the panel have higher expression levels in the follow-up visit (D,E) The log2 expression levels for EZH1 comparing (D) data from infants with severe versus mild RSV infection and (E) data from the acute and follow-up visit. The reported FDR p-values are based on limma analysis. (F,G) Abundance profiles of nasal microbiota comparing groups of mild and severe infants at each of the two visits, including (F) observed diversity and (G) Shannon diversity index.
Transcriptome data analysis
We first characterized the gene expression data for these infants, as measured by RNA-sequencing (RNA-Seq) of CD4+ T cells collected at two distinct time points, with 46 samples collected during the acute visit (Visit 1) and 34 samples collected at follow-up day 12–16 (Visit 2). Out of 22 subjects who had RNA-Seq samples collected at both the acute and follow-up visit, 7 were mild and 15 were severe (Fig. 2B).
We used principal component analysis (PCA) to investigate the data structure in light of various measured clinical variables (Supplemental Fig. 2). This analysis indicated that enrollment season was a significant contributor to structure in our expression data. Therefore, we applied batch-correction to remove this signal from the data. We then performed differential expression analysis, comparing the expression levels of genes between the acute and follow-up visit. We identified 27 genes that were differentially expressed at a Benjamini-Hotchberg false discovery rate (FDR) less than 0.05 (Fig. 2C). Many of the genes that had higher expression levels in the acute visit are known to be important in mediating host immune response. This included IFITM1 and IFITM2, which are known to inhibit the infection and replication of respiratory syncytial virus27,28, IFI27, a known biomarker for RSV29, and IRF7, a gene associated with suppression of innate immunity response30. Other genes with known associations to RSV and higher expression in the acute visit included SOCS331, MX132, and the ISGylation pathway genes USP18 and ISG1533. Our analysis also identified several genes with higher expression in the follow-up visit, including MS4A1, which encodes the CD20 protein, and MCOLN1. CD20 + B cells are prominent in the lung tissue of infants with fatal RSV infection34,35 and MS4A1 is upregulated in infants after the administration of live attenuated influenza vaccine, indicating an association with immune system processes36. RSV activates innate immunity through the toll like receptor (TLR) pathway. MCOLN1 has been associated with TLR signaling through modulating viral pathogen-associated molecular pattern (PAMP) along with trafficking of single-stranded RNA (ssRNA) into lysosomes37,38. MCOLN1 also regulates autophagy39, as expected in the convalescence phase of an infection.
We also evaluated if any genes changed their expression levels in response to RSV infection severity. We found 600 genes differentially expressed at a nominal p-value significance of less than 0.05. Out of these, the most significantly differentially-expressed gene was EZH1 (p-value = 2.3 × 10−6; FDR = 0.026) (Fig. 2D,E). EZH1 was previously included in a biosignature proposed as a molecular diagnosis tool for RSV infection40. Interestingly, both EZH1 and IFITM have been implicated in immune response signaling indicating resolving infection27.
Because we were only able to identify a handful of genes, primarily related to immune response, as significantly differentially-expressed in these same infants, we also investigated whether differences between severe and mild RSV infection could be captured using other methods. In particular, we examined whether gene expression differences between mild and severe RSV infection might be better captured through changes in variability rather than in mean expression levels. We evaluated the differential variability in gene expression levels between mild versus severe samples using the F-test and identified 641 (5.5%) genes that were differentially variable at an FDR significance threshold of 0.05. Gene Ontology enrichment analysis41 identified a number of biological processes nominally associated with these genes (Supplemental Fig. 3). Many of these were associated with mitochondrial activity such as mitochondrial gene expression and translation. Mitochondria are important for viral suppression of the innate immunity30.
Microbiome data analysis
We next characterized microbiome data from nasal swabs obtained by 16 S rRNA sequencing. This included data collected at two time points, with 51 samples collected during the acute visit (Visit 1) and 40 collected during a one-month follow-up visit (Visit 3); 23 of these samples were from infants with mild RSV infection and 35 were from infants with severe RSV infection (Fig. 2B).
We used both PCA and Principal Coordinate Analysis (PCoA) with various dissimilarity metrics, including rooted-Jensen–Shannon divergence (rJSD)42 as well as weighted and unweighted Unifrac distance43, to reduce dimensionality and visualize the microbiome data (Supplemental Fig. 4)44. We also applied MaAsLin (Multivariate association Analysis with Linear modeling) to test for significant relationships between microbial taxa and clinical outcome, after adjusting for sex, race, and enrollment season45. At a nominal p-value of 0.05, this analysis identified one operational taxonomic unit (OTU) – H. influenza – as positively associated with severity, and two OTUs – Ralstonia and Streptococcus – as negatively associated with severity. We also assessed the influence of visit and infection severity on the microbiome composition by using two measures to quantify the α-diversity: (1) observed number of operational taxonomic units (OTUs), i.e., the OTU richness and (2) the Shannon index46. We found that both α-diversity measures have higher values for infants with severe infection compared to those with mild infection during the acute visit (Visit 1), as well as higher values in the acute as compared to the post-acute visit (Visit 3) for infants with severe infection (Fig. 2F,G). In particular, the Shannon diversity is significantly different between severe versus mild samples in Visit 1 (p-value = 0.011), and between the Visit 1 and Visit 3 samples among infants with severe RSV infection (p-value = 0.0057). The infection score was estimated during Visit 1 (the acute phase). Severity of infection generally decreases over time, potentially explaining why we observe that the α-diversity decreases between the Visit 1 and Visit 3 samples.
Integrative analysis of transcriptome and microbiome data
Although it has been suggested that host gene expression is influenced by the microbiome, the biological mechanisms that may facilitate these types of interactions are largely unknown47. Integrative analysis of microbiome and transcriptome data could help us understand the relationships between host gene expression, microbial composition, and disease pathogenesis. Therefore, we applied a network-based approach, which systematically evaluates the interdependence of multiple biological entities instead of looking at each one independently, to identify microbiome—transcriptomic relationships important for RSV severity.
We focused on the 40 infants from Visit 1 that had paired transcriptome and microbiome data (see Fig. 2B) and performed dimension reduction on these data using PCA (Fig. 3A). This analysis identified 13 gene principal components (gPCs) and 10 clade principal components (cPCs) that explained 95% of the variance in each of their respective data (Fig. 3B).

(A) Principal component analysis was performed on Visit 1 samples for which we had both transcriptome and microbiome data to reduce the transcriptome into gPCs (top) and the microbiome into cPCs (bottom). (B) Cumulative distribution of the amount of variance explained by the top gPCs and cPCs. The vertical and horizontal lines indicate the number of PCs which explain 95% of variance and which were included in our network analysis. (C) The coordinates of the gene and clade principal components across the 40 analyzed samples from Visit 1. (D) Spearman correlation across the loadings of the top 13 gPCs and the top 10 cPCs.
The loadings of each cPC and gPC represent a pattern of highly correlated microbial and transcriptional abundances, respectively (Fig. 3C). In order to relate these patterns and identify associations between highly varying genes and clades, we calculated the Spearman correlation between the loadings of the top gPCs and cPCs (Fig. 3D). We find several gPC—cPC pairs that are highly correlated, such as gPC5 and cPC4 (most positively correlated; ρ = 0.48), and gPC4 and cPC7 (most negatively correlated; ρ = −0.43). Interestingly, these relationships are not limited to the top few PCs, which are normally the primary focus of dimension reduction analysis. Instead, these relationships highlight that prominent patterns in microbiome data may be associated with more subtle patterns in gene expression, and vice versa.
Integration of microbiome/transcriptome relationships with clinical characteristics
To interpret these results, we considered this correlation matrix as edges in a bipartite graph, where nodes are gPCs and cPCs. This network framework can help us identify important transcriptomic-microbiomic (gPC—cPC) relationships. However, since this network was derived using information from all samples, by itself it is unable to shed light on which of these relationships might be associated with differences in the various phenotypic or clinical properties of the input samples, including disease severity.
To overcome this limitation, we applied LIONESS48,49, which employs a jackknife approach to reverse engineer a set of sample-specific networks, to our gPC/cPC correlation network. This allowed us to construct separate correlation networks between gPCs and cPCs for each of the infants and to analyze gPC—cPC relationships in light of clinical information for these infants.
The weights of edges across these networks are shown in Fig. 4A. We compared the distribution of the sample-specific edge-weights between the mild and severe groups and identified six edges which were nominally significant with a p-value less than 0.05: gPC1 with cPC2, gPC8 with cPC8 and cPC4, gPC3 with cPC10, gPC13 with cPC9, and gPC12 with cPC1 (Fig. 4B).

(A) The weights of edges predicted for each of the 40 sample-specific networks obtained from the LIONESS analysis. Columns are grouped based on each infant’s infection severity, sex, and race. Rows are sorted based on the significance of edges. (B) The significance of each edge as defined from multivariate linear analysis comparing edge-weights between two groups, mild and severe, and corrected for sex and race. The top significant edges with nominal p-value less than 0.05 are labeled.
To assess the robustness of this result, we performed a sensitivity analysis by randomly selecting 10 samples each from mild and severe groups and repeating this analysis 1000 times. This analysis allows us to assess the robustness of our results given our relatively low sample number as well as the disparity between the number of mild and severe samples. If an edge is robustly different between the severe and mild groups, we will recover that association repeatedly across the randomizations. We found that the top edges obtained above were frequently (at least 20% of the time compared to 5% expected by chance) identified as significant with the same direction of effect (Supplemental Fig. 5).
Microbiome/transcriptome relationships identify clades that may impact the host immune response
Six clade principal components, cPC2, cPC8, cPC10, cPC9, cPC1, and cPC4 (ordered by significance) were identified as differentially-associated with gPCs in the context of RSV severity based on our network analysis. To understand why these cPCs might have a different relationship with the host transcriptome in the context of RSV severity, we identified their associated top OTUs (Table 2). We find that genus like Streptococcus, Corynebacterium, Alloiococcus, Haemophilus influenzae, and Staphylococcus are among the top OTUs in cPC2, Ralstonia, Corynebacterium, Neisseriaceae (family, genus not known), and Pseudomonas are among the top OTUs in cPC8, and Corynebacterium, Ralstonia, Alloiococcus, and Staphylococcus are among the top OTUs in cPC10. Some of these same OTUs were also identified as OTUs that discriminate between severe and mild RSV infection in our MaAsLin analysis.
The gene principal components that were identified as differentially-associated with cPCs in the context of RSV severity included gPC1, gPC8, gPC3, gPC13, and gPC12. We identified the top genes from these gPCs based on their loadings (Table 3). Interestingly, we found that 10 of top 20 genes in gPC1 are mitochondrial genes. We also found IFITM1 or IFITM2 in the top loadings of gPC3. These two genes are also significantly differentially-expressed between the acute and post-acute visit, but not between infants with severe versus mild RSV infection (see Fig. 2C). CCR7 (Chemokine receptor type 7), a top gene in gPC1, gPC8, and gPC3, and ILR7 (interleukin 7 receptor) from gPC8 and gPC12 have been found to be downregulated in RSV patients50. Other genes present in the loadings of multiple gene PCs included L-ribosomal proteins (RPL family), which are involved in various pathophysiological process, and SELL (L-Selectin), a cell surface lectin mostly expressed in leukocytes, which is down-regulated in RSV infection. SELL also plays a key role in the recruitment of neutrophils to roll along the endothelium to the infected tissue51,52.
We performed Gene Ontology (GO) enrichment analysis on the top 100 genes associated with each of these gPCs. For gPC1, top significant terms included “immune system process”, “immune response”, “viral process”, “T cell activation”, and “leukocyte activation”. Terms associated with gPC8 included “viral gene expression”, “viral transcription”, and “multi-organism process”. Terms associated with gPC3 were associated with signaling pathways like “cytokine-mediated signaling”, “type I interferon”, “innate immune response”, “defense response”, “response to other organism”, “viral genome replication”, “defense response”, and “regulation of viral genome replication” (Fig. 5). Similar functions were identified for gPC12 and gPC13 (Supplemental Fig. 6). All of these GO terms are consistent with viral infection and immune response in infected infants. The association of these viral and immune processes with the taxa identified in the associated cPCs allows us to hypothesize that certain microbial species may modulate the immune response in the host CD4+ T cells.

GO term enrichment analysis for the top 100 genes based on the loadings of (A) gPC1, (B) gPC8, and (C) gPC3. These bubble plots include the 25 most significant GO terms identified in each analysis based on the FDR significance. All terms shown are significant at an FDR < 0.05. Size of each bubble indicates the number of genes annotated to the respective GO term and the color indicates the percentage of the top 100 genes annotated to that term. The top 25 GO terms enriched in the other identified gPCs are shown in Supplemental Fig. 6A,B and all GO terms enriched at an FDR < 0.05 are shown in Supplemental Fig. 6C–G.
Discussion
Our microbiome data analysis indicated that taxonomic diversity is highest in infants with severe infection during the acute visit, and decreases over time. Moreover, using MaAsLin and at a nominal p-value of 0.05, we identified one OTU (H. influenza) that is positively associated with RSV severity, and two OTUs (Ralstonia and Streptococcus) that are negatively associated with RSV severity. These results are partially consistent with previous observations that infants within H. influenzae-enriched clusters mount a distinct host inflammatory response characterized by the overexpression of genes related to toll-like receptor signaling and neutrophil recruitment and activation22. Interestingly, our subsequent network analysis implicated many of the same OTUs as MaAsLin did. Top OTUs in the network-identified clade PCs included Streptococcus, Corynebacterium, Alloiococcus, H. influenzae, Staphylococcus, Moraxella, Ralstonia, and Pseudomonas. These are consistent with a similar study analyzing the microbiome from nasopharyngeal bacterial swabs alongside whole blood transcriptomic data collected from RSV infected infants22. In addition, among these identified OTUs, incidence of co-infection of Streptococcus, Haemophilus influenza, and Moraxella with RSV has been reported in studies of nasopharyngeal aspirate samples from RSV infected infants53,54,55. During acute respiratory illness most infants show stable colonization of these microbes with Alloiococcus or Moraxella21.
Our transcriptome analysis found that genes involved in immune response, such as IFITM1 and IFITM2, decreased in expression after the acute visit, while EZH1, which has also been implicated in immune response signaling27, was expressed at lower levels in infants with severe RSV infection. Interferons are widely expressed and are among the key genes responsible for mediating immune response27,56. In particular, the IFN-inducible protein is localized in the cell membrane and endocytic vesicle and is important in restricting viral entry to the cell57,58,59. In27, IFITM genes have been shown to potentially inhibit RSV infection by interfering with virus entry and subsequent viral multiplication. Despite these promising findings, we note that standard differential expression only identified one gene as significantly (FDR < 0.05) differentially expressed based on RSV infection severity –EZH1 with an FDR of 0.026.
In this paper, we have proposed and applied a novel approach for integrating data from multiple omics platforms, in our case the host transcriptome and nasal microbiome, in order to extract meaningful associations. Our method constructs infant-specific correlation networks between the transcriptome and microbiome in reduced dimensions using PCA. Linear modeling of these interactions, accounting for various confounding factors, allowed us to identify associations between top gene PCs and clade PCs that differ between infants with mild versus severe RSV infection. This allowed us to better understand the etiology of RSV infection and its impact on disease severity by highlighting key associations between active components in the transcriptome and dominant constituents of the microbial composition.
The functional enrichment analysis of the top genes in the network-identified gene PCs indicated that the top-loading genes are highly enriched in pathways related to immune response and viral infection. These top genes included IFITM1 and IFITM2, which were significantly expressed between the acute and post-acute visit, but not between infants with severe versus mild infection, and CCR7, which has been found to be downregulated in RSV patients50. Interestingly, many of the top genes are encoded in mitochondrial DNA and included members of NADH dehydrogenase (MT-ND4L and MT-ND5), members of cytochrome c oxidase (MT-CO1, MT-CO2, and MT-CO3), and members of ATP synthase (MT-ATP6 and MT-ATP8). Mitochondrial function can be modulated by viruses60,61 and mitochondrial genes play a key role in the host immune response62,63. These genes are also important for viral suppression of natural immunity in RSV infection30.
Importantly, our analysis indicates that the association of some clade PCs with gene PCs is context-dependent and a function of RSV infection severity. Further, the enrichment of immune and viral pathways in the network-identified gene PCs implies that specific microbial taxa in the nasal microbiome may impact host immune response, potentially mediating RSV infection severity. In this context, the identification of mitochondrial genes in our integrative analysis is an especially intriguing finding. The relationship between mitochondria and the microbiome is only beginning to emerge64,65, and has not been previously described in the context of RSV severity. However, interestingly, the role of microbiota has been noted in the immune response to influenza66.
We also point out that the sign of the identified associations (negative versus positive correlations) may imply that both adverse as well as cooperative relationships exist between certain bacterial species and immune and viral defense response. For example, clade PC4 and clade PC8 both have positive associations with the gene PC8, indicated a potentially multifactorial host response. However, in the context of this analysis, we note that it is impossible to establish whether the microbiome-transcriptome associations we identified are indeed causal, and, if so, their direction of effect (whether the microbiome influences the transcriptome, vice versa, or both). Without data-collection in infants prior to RSV infection, we can only hypothesize as to whether targeting these associations might help to prevent or minimize the impact of RSV infection.
Finally, it is also important to note that, although highly intriguing, the associations identified in our network analysis were only nominally significant. This likely reflects heterogeneity in the disease as well as a limitation in statistical power due to sample size. However, we point out that the network analysis uncovered very similar microbial taxa as MaAsLin did, lending confidence to our results. Furthermore, highly plausible biological pathways related to viral processes and immune response were implicated in RSV severity using this integrative analysis. These pathways were not found using more standard approaches, such as differential-expression analysis in the context of infection severity, and despite the fact that we did observe some immune genes differentially-expressed between the acute and post-acute visits. We believe these results illustrate the strength of using an integrative analysis approach to bring novel insights into the disease.
This study has several limitations. First, the number of samples with simultaneous collection of transcriptome and microbiome data was small. Hence most of the statistical analysis with interesting results, although nominally significant, did not show significance after adjusted for multiple hypothesis testing. Second, there may have been other host factors such as maternal or household smoking history, breast-feeding status, number of siblings, and antibiotic use, that could affect the severity of RSV infection but were not considered in our study. Third, this study was only able to provide associations between genes and microbiota without exploring the potential causal relationship.
In this study, we performed dimension reduction and differential-association analyses to quantify patterns in the nasal microbiome and host transcriptome that are associated with RSV infection severity. We also used a network-based approach to integrate these two data types and identify higher-order associations among genes and clades. This integrative analysis allowed us to systematically quantify relationships among the nasal microbiome, host transcriptome, and disease severity for RSV infection. Our results suggest that certain associations between the microbiome and transcriptome are modulated based on RSV infection severity, and that particular microbial taxa impact host immune response, with a key role for mitochondrial genes. Overall, these findings on microbiome-transcriptome associations provides novel insights into how the immune system mounts a response against RSV. Based on our findings, a key future direction for our group is to study the potential mechanisms by which the nasal microbiome impacts host immune response, such as how gene regulation is altered or otherwise affected by microbial composition, or how nasal bacteria influence the severity of RSV from a pathophysiologic perspective.
Methods
Study participants
RSV-infected infants were enrolled from three cohorts in order to capture the full spectrum of disease severity. A birth cohort was enrolled at the University of Rochester Medical Center’s (URMC), Strong Memorial Hospital and Highland Hospital, and Rochester General Hospital (RGH) for two winter seasons, extending from August 15 to February 1 for 2012–2013 and 2013–2014, and followed by passive and active surveillance for development of RSV infection during the winter months (November 1–April 1). A second cohort was enrolled in pediatric offices or the emergency room at URMC’s Golisano Children’s Hospital or RGH when respiratory symptoms were present. The third cohort was enrolled on admission to the hospital with documented RSV infection. Eligible infants were full-term (>36 weeks gestation), healthy infants born after May 1 and less than 10 months of age at infection.
RSV-infected infants underwent evaluation by two members of the study team (a physician and a nurse). Demographic data, illness symptoms, findings on physical examination, results of laboratory and radiograph results were recorded. A nasal swab was obtained using a medium sized flocked swab (Copan Diagnostics Inc. Murrieta, CA, cat. no. 501CS01) and placed in 2 ml of sterile UV-inactivated water for quantitative RSV reverse-transcriptase polymerase chain reaction (RT-PCR). A 2–3 ml sample of heparinized blood was collected for CD4 gene expression studies as previously described12.
RSV severity score
RSV disease severity was measured using the Global Respiratory Severity Score (GRSS) on a scale from 0–10, with higher scores representing more severe infection67. The GRSS was developed using an unbiased data-driven approach with nine clinical variables (including the infant’s general appearance, the presence of wheezing, rales, retractions, cyanosis, lethargy, and poor air movement). The maximal age-adjusted respiratory rate, as well as the worst room air oxygen saturation, are also included in the score. The GRSS is highly predictive of other potential parameters of severity, such as need for hospitalization and duration of hospitalization67. The GRSS has an excellent ability to discriminate between mild and severe disease with AUC (area under the receiver operating characteristic curve) ~0.961. An infant with a GRSS ≤ 3.5 (or > 3.5) is classified as having mild (or severe) infection, respectively. For more information see67. We note that using continuous or discrete severity scores in our analyses resulted in similar results, so for simplicity we treated RSV infection severity as a binary variable.
CD4+ T cell extraction
CD3+, CD4+, CD8− T cells were isolated from freshly collected peripheral blood samples as previously described12,68. Briefly, within 24 hours of collection, Ficoll-purified peripheral blood mononuclear cells (PBMCs) were stained and sorted into major lymphocyte populations. Sorted cells were immediately lysed and homogenized in RNA extraction buffer and stored for later purification.
Transcriptomic data collection and analysis
RNA extraction, processing and normalization
RNA-Seq data for CD4+ T cells were collected at two distinct time points, with 46 samples collected during the acute visit (Visit 1) and 34 samples collected on follow-up day 12–16 (Visit 2). RNA purification, library preparation, sequencing and data processing were essentially the same as previously described12,68,69. Briefly, library preparation was performed using the NexteraXT library kit (Illumina, San Diego, CA) following the SMARter Ultra Low amplification kit (Clontech, Mountain View, CA). Libraries were sequenced using the Illumina HiSeq 2500 at a target depth of ~20 million 100-bp single end reads per sample. Raw reads were mapped to Human Genome GRCh38 (annotation of GENCODE 23) and normalized by FPKM (fragments per kilobase of transcript per million reads). The transcriptome data contained reads for 11,576 genes with unique Gene Symbol annotations across 80 total samples.
Differential expression analysis
Differential gene expression analysis was performed on FPKM normalized RNA-Seq data using the Bioconductor70 limma package (version 3.30.13)71. We note that principal component (PC) analysis indicated an association of the leading PC with enrollment season. Several methods have been developed to directly regress out signals related to batch in expression data72,73,74. We used ComBat function from sva (version 3.26.0) to correct for differences from enrollment season75. In our differential-expression analysis we compared groups based on RSV infection severity (mild vs. severe) while correcting for sex and race. RSV viral load from nasal swab and nasal wash did not show any association with severity of infection26 and were not included as covariates. Genes were considered significantly differentially expressed (DE) if their Benjamini-Hochberg corrected p-value was less than 0.05.
Differential variability analysis
We used var.test() in R (3.4.3 (2017–11–30)) to perform an F-test on log2-transformed gene expression data in order to statistically quantify differences in variance in gene expression levels between the mild and severe RSV-infected groups. Genes were considered significantly differentially variable if their Benjamini-Hochberg corrected p-value was less than 0.05.
Gene Ontology pathway analysis
We used the topGO package (version 2.30.1)41 in R to assess the enrichment of Gene Ontology (GO) pathways in a given gene list. Gene lists analyzed included (1) genes that were identified as significantly differentially-variable (see above), as well as (2) the top 100 genes associated with a gene principal component (PC) from principal component analysis on transcriptome data, based on the associated PC-loadings (see below). In the topGO (version 2.30.1) analysis, we used the 11,576 genes with measured reads in our RNA-Seq data as a background.
Microbiome data collection and analysis
16S rRNA sequencing
Nasal swab specimens were collected during acute RSV-related illness (Visit 1) and one month later (Visit 3). As described in69, bacterial 16 S rRNA from these samples was extracted, amplified, and sequenced, and the resulting data were used to determine the taxonomic compositions, in terms of the relative abundances of those present operational taxonomic units (OTUs). Briefly, the V3-V4 hypervariable regions were targeted for amplification and sequenced using an Illumina MiSeq platform according to a paired end 2 × 300 bp read protocol. Preliminary read processing and quality control were performed using the Quantitative Insights into Microbial Ecology (QIIME) software package76, and a closed-reference OTU picking was done with USEARCH and the GreenGenes reference database76. The final microbiome data contained information for 1,022 distinct OTUs across 91 samples.
Multivariate association Analysis with Linear modeling (MaAsLin)
We used MaAsLin (version 0.0.5) to test for significant relationships between microbial clusters and clinical outcome (severe or mild infection). MaAsLin is a multivariate statistical framework that finds associations between clinical metadata and potentially high-dimensional experimental data76. In contrast to transcriptomic data, the application of batch correction methods to microbiome data is still nascent77. Therefore, when we applied MaAsLin we adjusted for potential confounding factors, including sex and race, as well as enrollment season, which was removed by batch-correction in our transcriptomic data analysis. All microbiome samples from both Visit 1 and Visit 3 were used when running MaAsLin.
Integrative analysis of transcriptomic and microbiomic data
Principal Component Analysis (PCA) on transcriptomic and microbiomic data
To maximize statistical power in the integrative analysis, we first performed dimension reduction47. In particular, we applied principal component analysis (PCA) to host transcriptomics data to create a handful of gene principal components (gPCs), and to the nasal microbiomics data to create a few clade principal components (cPCs)76. To achieve that, we used the prcomp() function in the stats (version 3.4.3) package in R to perform PCA on the OTU relative abundance profiles and the FPKM-normalized RNA-Seq gene expression profiles (after applying batch-effect correction for enrollment season). In this integrative analysis, we restricted ourselves to samples from 40 subjects with both microbiome and transcriptome data collected during Visit 1. The top 13 gPCs and top 10 cPCs, which explained 95% of the variance in the transcriptomic and microbiomic data, respectively, were selected for further analysis.
Correlation between gPCs and cPCs
We constructed a Spearman correlation matrix comparing the top 13 gPCs and the top 10 cPCs, using cor() function from the stats (version 3.4.3) package in R. We treated this global 13×10 correlation matrix as the weighted adjacency matrix of a complete bipartite graph (Gα, where the subscript α denotes the fact that Gα is derived using all the 40 input samples) that contains two types of nodes, gPCs and cPCs.
Linear Interpolation to Obtain Network Estimates for Single Samples (LIONESS)
In order to relate gPC/cPC associations to clinical variables, we applied the LIONESS method to Gα to construct sample-specific (infant-specific) correlation networks49. LIONESS works under the assumption that the global correlation network represents a linear combination of N different networks, one from each of the N input samples. Therefore, to construct the network for sample q, we first exclude sample q and calculate the Spearman correlation matrix using the remaining samples (G(α−q)). We then use the LIONESS equation, \({G}_{q}=N({G}_{\alpha }-{G}_{(\alpha -q)})+{G}_{(\alpha -q),}\) to find the network estimate for that sample (Gq). We applied LIONESS to the nasal microbiome and host transcriptome samples collected from the same set of 40 infants during Visit 1. The end result of this analysis was 40 sample-specific networks, i.e., bipartite graphs relating gPCs and cPCs.
Analysis of sample-specific networks
We separated the sample-specific LIONESS networks based on their severity class and compared the distribution of each edge’s weight between mild and severe groups using limma (version 3.34.9) correcting for sex and race. All edges with p-value < 0.05 were considered nominally significant.
Data Availability
The Institutional Review Boards of the University of Rochester and Rochester General Hospital approved the study. For each infant, one parent provided written informed consent at enrollment. All methods were performed in accordance with the relevant guidelines and regulations. The data analyzed in the manuscript are available on dbGap (phs001201.v1.p1).
References
Shi, T. et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390, 946–958, https://doi.org/10.1016/S0140-6736(17)30938-8 (2017).
Karron, R. A. & Black, R. E. Determining the burden of respiratory syncytial virus disease: the known and the unknown. Lancet 390, 917–918, https://doi.org/10.1016/S0140-6736(17)31476-9 (2017).
Svensson, C., Berg, K., Sigurs, N. & Trollfors, B. Incidence, risk factors and hospital burden in children under five years of age hospitalised with respiratory syncytial virus infections. Acta paediatrica 104, 922–926, https://doi.org/10.1111/apa.13061 (2015).
Sonego, M., Pellegrin, M. C., Becker, G. & Lazzerini, M. Risk factors for mortality from acute lower respiratory infections (ALRI) in children under five years of age in low and middle-income countries: a systematic review and meta-analysis of observational studies. PloS one 10, e0116380, https://doi.org/10.1371/journal.pone.0116380 (2015).
Hasegawa, K., Tsugawa, Y., Brown, D. F., Mansbach, J. M. & Camargo, C. A. Jr. Trends in bronchiolitis hospitalizations in the United States, 2000–2009. Pediatrics 132, 28–36, https://doi.org/10.1542/peds.2012-3877 (2013).
Shi, T. et al. Risk factors for respiratory syncytial virus associated with acute lower respiratory infection in children under five years: Systematic review and meta-analysis. Journal of global health 5, 020416, https://doi.org/10.7189/jogh.05.020416 (2015).
Welliver, R. C. Sr. et al. Fatality rates in published reports of RSV hospitalizations among high-risk and otherwise healthy children. Current medical research and opinion 26, 2175–2181, https://doi.org/10.1185/03007995.2010.505126 (2010).
Meissner, H. C. Viral Bronchiolitis in Children. The New England journal of medicine 374, 62–72, https://doi.org/10.1056/NEJMra1413456 (2016).
Blanken, M. O. et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. The New England journal of medicine 368, 1791–1799, https://doi.org/10.1056/NEJMoa1211917 (2013).
Sigurs, N. et al. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 65, 1045–1052, https://doi.org/10.1136/thx.2009.121582 (2010).
Backman, K., Piippo-Savolainen, E., Ollikainen, H., Koskela, H. & Korppi, M. Adults face increased asthma risk after infant RSV bronchiolitis and reduced respiratory health-related quality of life after RSV pneumonia. Acta paediatrica 103, 850–855, https://doi.org/10.1111/apa.12662 (2014).
Mariani, T. J. et al. Association of Dynamic Changes in the CD4 T-Cell Transcriptome With Disease Severity During Primary Respiratory Syncytial Virus Infection in Young Infants. The Journal of infectious diseases 216, 1027–1037, https://doi.org/10.1093/infdis/jix400 (2017).
Mejias, A. et al. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS medicine 10, e1001549, https://doi.org/10.1371/journal.pmed.1001549 (2013).
Fjaerli, H. O. et al. Whole blood gene expression in infants with respiratory syncytial virus bronchiolitis. BMC infectious diseases 6, 175, https://doi.org/10.1186/1471-2334-6-175 (2006).
Christiaansen, A. F., Knudson, C. J., Weiss, K. A. & Varga, S. M. The CD4 T cell response to respiratory syncytial virus infection. Immunologic research 59, 109–117, https://doi.org/10.1007/s12026-014-8540-1 (2014).
Hacking, D. & Hull, J. Respiratory syncytial virus–viral biology and the host response. The Journal of infection 45, 18–24 (2002).
Openshaw, P. J. & Chiu, C. Protective and dysregulated T cell immunity in RSV infection. Current opinion in virology 3, 468–474, https://doi.org/10.1016/j.coviro.2013.05.005 (2013).
Raiden, S. et al. Respiratory Syncytial Virus (RSV) Infects CD4+ T Cells: Frequency of Circulating CD4+ RSV+ T Cells as a Marker of Disease Severity in Young Children. The Journal of infectious diseases 215, 1049–1058, https://doi.org/10.1093/infdis/jix070 (2017).
Bennett, J. E., Dolin, R. & Blaser, M. J. Infectious Disease Essentials. (2017).
Stewart, C. J. et al. Associations of Nasopharyngeal Metabolome and Microbiome with Severity among Infants with Bronchiolitis. A Multiomic Analysis. American journal of respiratory and critical care medicine 196, 882–891, https://doi.org/10.1164/rccm.201701-0071OC (2017).
Teo, S. M. et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell host & microbe 17, 704–715, https://doi.org/10.1016/j.chom.2015.03.008 (2015).
de Steenhuijsen Piters, W. A. et al. Nasopharyngeal Microbiota, Host Transcriptome, and Disease Severity in Children with Respiratory Syncytial Virus Infection. American journal of respiratory and critical care medicine 194, 1104–1115, https://doi.org/10.1164/rccm.201602-0220OC (2016).
Silverman, E. K. & Loscalzo, J. Network medicine approaches to the genetics of complex diseases. Discovery medicine 14, 143–152 (2012).
Fodha, I. et al. Respiratory syncytial virus infections in hospitalized infants: association between viral load, virus subgroup, and disease severity. J Med Virol 79, 1951–1958, https://doi.org/10.1002/jmv.21026 (2007).
Garcia-Maurino, C. et al. Viral Load Dynamics and Clinical Disease Severity in Infants With Respiratory Syncytial Virus Infection. J Infect Dis 219, 1207–1215, https://doi.org/10.1093/infdis/jiy655 (2019).
Walsh, E. E. et al. Virus-Specific Antibody, Viral Load, and Disease Severity in Respiratory Syncytial Virus Infection. J Infect Dis 218, 208–217, https://doi.org/10.1093/infdis/jiy106 (2018).
Zhang, W. et al. Human respiratory syncytial virus infection is inhibited by IFN-induced transmembrane proteins. J Gen Virol 96, 170–182, https://doi.org/10.1099/vir.0.066555-0 (2015).
Smith, S. E. et al. Interferon-induced Transmembrane Protein 1 restricts replication of virus that enter cells via the plasma membrane. J Virol, https://doi.org/10.1128/JVI.02003-18 (2018).
Ioannidis, I. et al. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J Virol 86, 5422–5436, https://doi.org/10.1128/JVI.06757-11 (2012).
Goswami, R. et al. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res 23, 1025–1042, https://doi.org/10.1038/cr.2013.98 (2013).
Bakre, A. et al. Respiratory syncytial virus modifies microRNAs regulating host genes that affect virus replication. J Gen Virol 93, 2346–2356, https://doi.org/10.1099/vir.0.044255-0 (2012).
Ampuero, S. et al. Time-course of transcriptome response to respiratory syncytial virus infection in lung epithelium cells. Acta Virol 62, 310–325, https://doi.org/10.4149/av_2018_225 (2018).
Gonzalez-Sanz, R. et al. ISG15 Is Upregulated in Respiratory Syncytial Virus Infection and Reduces Virus Growth through Protein ISGylation. J Virol 90, 3428–3438, https://doi.org/10.1128/JVI.02695-15 (2016).
Reed, J. L. et al. Innate immune signals modulate antiviral and polyreactive antibody responses during severe respiratory syncytial virus infection. J Infect Dis 199, 1128–1138, https://doi.org/10.1086/597386 (2009).
Russell, C. D., Unger, S. A., Walton, M. & Schwarze, J. The Human Immune Response to Respiratory Syncytial Virus Infection. Clin Microbiol Rev 30, 481–502, https://doi.org/10.1128/CMR.00090-16 (2017).
Panapasa, J. A., Cox, R. J., Mohn, K. G., Aqrawi, L. A. & Brokstad, K. A. The expression of B & T cell activation markers in children’s tonsils following live attenuated influenza vaccine. Hum Vaccin Immunother 11, 1663–1672, https://doi.org/10.1080/21645515.2015.1032486 (2015).
Li, X. et al. Mucolipin 1 positively regulates TLR7 responses in dendritic cells by facilitating RNA transportation to lysosomes. Int Immunol 27, 83–94, https://doi.org/10.1093/intimm/dxu086 (2015).
Rinkenberger, N. & Schoggins, J. W. Mucolipin-2 Cation Channel Increases Trafficking Efficiency of Endocytosed Viruses. MBio 9, https://doi.org/10.1128/mBio.02314-17 (2018).
Zhang, X. et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7, 12109, https://doi.org/10.1038/ncomms12109 (2016).
Gardinassi, L. G. A Cross-Study Biomarker Signature of Human Bronchial Epithelial Cells Infected with Respiratory Syncytial Virus. Advances in virology 2016, 3605302, https://doi.org/10.1155/2016/3605302 (2016).
Alexa, A., Rahnenfuhrer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607, https://doi.org/10.1093/bioinformatics/btl140 (2006).
Österreicher, F. & Vajda, I. {A new class of metric divergences on probability spaces and its applicability in statistics}. Annals of the Institute of Statistical Mathematics 55, 639–653, https://doi.org/10.1007/BF02517812 (2003). doi:citeulike-article-id:9377855.
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and environmental microbiology 71, 8228–8235, https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Koren, O. et al. A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS computational biology 9, e1002863, https://doi.org/10.1371/journal.pcbi.1002863 (2013).
Morgan, X. C. et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome biology 13, R79, https://doi.org/10.1186/gb-2012-13-9-r79 (2012).
Morgan, X. C. & Huttenhower, C. Chapter 12: Human microbiome analysis. PLoS computational biology 8, e1002808, https://doi.org/10.1371/journal.pcbi.1002808 (2012).
Morgan, X. C. et al. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome biology 16, 67, https://doi.org/10.1186/s13059-015-0637-x (2015).
Chen, C.-Y. et al. Sexual dimorphism in gene expression and regulatory networks across human tissues. bioRxiv. https://doi.org/10.1101/082289 (2016).
Kuijjer, M. L., Tung, M. G., Yuan, G., Quackenbush, J. & Glass, K. Estimating Sample-Specific Regulatory Networks. iScience 14, 226–240, https://doi.org/10.1016/j.isci.2019.03.021 (2019).
Inchley, C. S., Osterholt, H. C., Sonerud, T., Fjaerli, H. O. & Nakstad, B. Downregulation of IL7R, CCR7, and TLR4 in the cord blood of children with respiratory syncytial virus disease. The Journal of infectious diseases 208, 1431–1435, https://doi.org/10.1093/infdis/jit336 (2013).
McNamara, P. S. & Smyth, R. L. The pathogenesis of respiratory syncytial virus disease in childhood. Br Med Bull 61, 13–28 (2002).
Wang, S. Z. et al. Shedding of L-selectin and PECAM-1 and upregulation of Mac-1 and ICAM-1 on neutrophils in RSV bronchiolitis. Am J Physiol 275, L983–989 (1998).
Hishiki, H. et al. Incidence of bacterial coinfection with respiratory syncytial virus bronchopulmonary infection in pediatric inpatients. Journal of infection and chemotherapy: official journal of the Japan Society of Chemotherapy 17, 87–90, https://doi.org/10.1007/s10156-010-0097-x (2011).
Thorburn, K., Harigopal, S., Reddy, V., Taylor, N. & van Saene, H. K. High incidence of pulmonary bacterial co-infection in children with severe respiratory syncytial virus (RSV) bronchiolitis. Thorax 61, 611–615, https://doi.org/10.1136/thx.2005.048397 (2006).
Resch, B., Gusenleitner, W. & Mueller, W. D. Risk of concurrent bacterial infection in preterm infants hospitalized due to respiratory syncytial virus infection. Acta paediatrica 96, 495–498, https://doi.org/10.1111/j.1651-2227.2007.00226.x (2007).
Durbin, J. E. et al. The role of IFN in respiratory syncytial virus pathogenesis. Journal of immunology 168, 2944–2952 (2002).
Diamond, M. S. & Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nature reviews. Immunology 13, 46–57, https://doi.org/10.1038/nri3344 (2013).
Huang, I. C. et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS pathogens 7, e1001258, https://doi.org/10.1371/journal.ppat.1001258 (2011).
Brass, A. L. et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254, https://doi.org/10.1016/j.cell.2009.12.017 (2009).
Anand, S. K. & Tikoo, S. K. Viruses as modulators of mitochondrial functions. Adv Virol 2013, 738794, https://doi.org/10.1155/2013/738794 (2013).
Ashton-Rickardt, P. G. Mitochondria Apply the Brake to Viral Immunity. Cell Metab 23, 967–968, https://doi.org/10.1016/j.cmet.2016.05.018 (2016).
Garaude, J. et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol 17, 1037–1045, https://doi.org/10.1038/ni.3509 (2016).
Weinberg, S. E., Sena, L. A. & Chandel, N. S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417, https://doi.org/10.1016/j.immuni.2015.02.002 (2015).
Franco-Obregon, A. & Gilbert, J. A. The Microbiome-Mitochondrion Connection: Common Ancestries, Common Mechanisms, Common Goals. mSystems 2, https://doi.org/10.1128/mSystems.00018-17 (2017).
Gruber, J. & Kennedy, B. K. Microbiome and Longevity: Gut Microbes Send Signals to Host Mitochondria. Cell 169, 1168–1169, https://doi.org/10.1016/j.cell.2017.05.048 (2017).
Ichinohe, T. et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci USA 108, 5354–5359, https://doi.org/10.1073/pnas.1019378108 (2011).
Caserta, M. T. et al. Development of a Global Respiratory Severity Score for Respiratory Syncytial Virus Infection in Infants. J Infect Dis 215, 750–756, https://doi.org/10.1093/infdis/jiw624 (2017).
Misra, R. S. et al. Flow-based sorting of neonatal lymphocyte populations for transcriptomics analysis. Journal of immunological methods 437, 13–20, https://doi.org/10.1016/j.jim.2016.07.001 (2016).
Chu, C. Y. et al. The Healthy Infant Nasal Transcriptome: A Benchmark Study. Scientific reports 6, 33994, https://doi.org/10.1038/srep33994 (2016).
Huber, W. et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nature methods 12, 115–121, https://doi.org/10.1038/nmeth.3252 (2015).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43, e47, https://doi.org/10.1093/nar/gkv007 (2015).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127, https://doi.org/10.1093/biostatistics/kxj037 (2007).
Leek, J. T. & Storey, J. D. A general framework for multiple testing dependence. Proc Natl Acad Sci USA 105, 18718–18723, https://doi.org/10.1073/pnas.0808709105 (2008).
Leek, J. T. & Storey, J. D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet 3, 1724–1735, https://doi.org/10.1371/journal.pgen.0030161 (2007).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883, https://doi.org/10.1093/bioinformatics/bts034 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336, https://doi.org/10.1038/nmeth.f.303 (2010).
Gibbons, S. M., Duvallet, C. & Alm, E. J. Correcting for batch effects in case-control microbiome studies. PLoS Comput Biol 14, e1006102, https://doi.org/10.1371/journal.pcbi.1006102 (2018).
Acknowledgements
This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272201200005C. The authors declare no competing interests.
Author information
Authors and Affiliations
Contributions
A.R.S., L.T., T.J.M., S.T.W., E.K.S., K.G. and Y.Y.L. designed the analysis. A.R.S. performed the transcriptome and integrative analysis of transcriptome and microbiome, L.T. performed the microbiome analysis. C.C., X.Q., L.W., J.H.W., A.G., S.R.G., M.T.C., E.W., T.M. were responsible for collection and post-processing of clinical, microbial and transcriptional data. A.R.F. and D.J.T. provided input on the study design. A.R.S., L.T., K.G. and Y.Y.L. prepared the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sonawane, A.R., Tian, L., Chu, CY. et al. Microbiome-Transcriptome Interactions Related to Severity of Respiratory Syncytial Virus Infection. Sci Rep 9, 13824 (2019). https://doi.org/10.1038/s41598-019-50217-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50217-w
This article is cited by
-
Low pathogenic avian influenza virus infection retards colon microbiota diversification in two different chicken lines
Animal Microbiome (2021)
-
Genes and regulatory mechanisms associated with experimentally-induced bovine respiratory disease identified using supervised machine learning methodology
Scientific Reports (2021)
-
Das neue Verständnis pulmonaler Infektionen
Der Pneumologe (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
