
Abstract
Two new harziane diterpene lactones, possessing a 6/5/7/5-fused carbocyclic core containing a lactone ring system, harzianelactones A and B (1 and 2), and five new harziane diterpenes, harzianones A–D (3–6) and harziane (7), were isolated from the soft coral-derived fungus Trichoderma harzianum XS-20090075. Their structures were determined by extensive NMR spectroscopic data, ECD and OR calculations, as well as X-ray diffraction. The isolated compounds exhibited potent phytotoxicity against seedling growth of amaranth and lettuce. Harziane diterpenes were rarely reported for their remarkably bioactivities, and it was the first report to study the phytotoxicity of harziane diterpenes, which provide a new application of such compounds in agriculture for future research.
Similar content being viewed by others
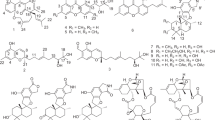

Introduction
Increasing concerns for the management of weeds have been caused by scientists, as they can bring out greater reduction in crop yields than plant diseases and pests1. Nowadays more than half of the pesticides used are herbicides1,2. With the increasing attention to food safety and environmental protection, it is desiderate to develop new types of bio-herbicides with high efficiency and low toxicity.
Trichoderma spp. are one of the most commonly disseminated fungi in nature, and are distributed around the world ranging from the tundra to the tropics. They have been widely used as biocontrol agents (T. harzianum, T. atroviride, and T. asperellum), and commercially marketed as biopesticides, due to their capacity to parasitize in other fungi and to compete with deleterious plant microorganisms3. However, there are few Trichoderma spp. products sold in the commercial market, and limited studies are focused on the phtotoxicity of compounds from Trichoderma spp4.
Marine fungi have gained more and more attention over the past decades, as the recognition that they are a quite diverse group and an excellent source of natural products, possessing prominent bioactivities, including antibacterial, antifungal, antiviral, anti-inflammatory, antitumor, and insecticidal5. Marine-derived Trichoderma spp. have been reported to represent a potential source for producing compounds with novel structures and remarkable bioactivities, such as trichodermamides A and B6, dithioaspergillazine A7, tandyukisins E and F8, as well as harzianone9. Therefore, it has huge potential to find new phytotoxic compounds from marine-derived Trichoderma spp.
During our efforts to find novel bioactive compounds from coral-derived fungi in the South China Sea10,11,12,13, a T. harzianum XS-20090075 strain attracted our attention because the finger-print for the extract of the fungal culture on HPLC showed abundant peaks with interesting UV absorption spectra at around 250 nm, and the fungal extracts showed obvious phytotoxicity. Further chemical examination on the EtOAc extract resulted in the discovery of two new harziane diterpene lactones, harzianelactones A and B (1 and 2), and five new harziane diterpenes, harzianones A–D (3–6), and harziane (7) (Fig. 1). Herein, we describe the isolation, structure elucidation, and phytotoxicity of these harziane diterpenes.

Chemical structures of harzianelactones A and B (1 and 2), harzianones A–D (3–6), and harziane (7).
Results and Discussion
Harzianelactone A (1) was obtained as a colorless oil with the molecular formula of C20H30O3 by HRESIMS, requiring six degrees of unsaturation. The 1H NMR spectrum (Table 1) showed three protons on oxygenated carbons at δH 3.89 (d, J = 9.0 Hz), 3.80 (d, J = 8.0 Hz), and 3.73 (d, J = 8.0 Hz), four methyl singlets at δH 2.33 (s), 1.43 (s), 0.85(s), and 0.84 (s), as well as one methyl doublet at δH 1.15 (d, J = 8.0 Hz). The 13C NMR (Table 2) and DEPT spectra in combination with HMQC data revealed one lactone carbonyl (δC 171.3), one oxymethylene carbon (δC 78.4), one oxymethine carbon (δC 73.7), four methylenes (δC 39.8, 32.1, 28.2, and 28.0), three methines (δC 50.5, 41.3, and 40.0), five methyl groups (δC 24.9, 22.9, 22.6, 21.2, 20.7), and five nonprotonated carbons (δC 155.9, 131.0, 49.7, 46.3, 44.9) including two quaternary olefinic ones. The aforementioned data corresponded to two degrees of unsaturation, and the remaining four degrees of unsaturation suggested the existence of four rings. The planar structure of 1 was elucidated on the basis of COSY and HMBC experiments (Fig. 2). The spin systems of H-14/H-15/H-2/H-3/H-4/H-5/H-18 in the COSY cross peaks and the correlations from H-3 to C-1, C-5, and C-15, from H-4 to C-2, C-6 and C-18, from H-5 to C-1 and C-14, from H-16 and H-17 to C-2 and C-6, and from H-18 to C-6 in the HMBC spectrum, led to the construction of a five-membered ring B and a six-membered ring A with a hydroxy and a methyl group anchored to C-4 and C-5, respectively. The seven-membered ring C with two methyl groups connected to C-9 and C-13 was further constructed according to the HMBC correlations from H-7 to C-9 and C-14, from H-8 to C-6 and C-10, from H-19 to C-10 and C-14, and from H-20 to C-8 and C-10. The lactone carbonyl (δC 171.3), in addition to the HMBC correlations from H-12 to C-10, C-11, C-14, and C-19, and from H-15 to C-13 indicated the existence of a five-membered lactone ring D connected to ring C. Finally, the connection of ring B and ring C was confirmed by the HMBC correlations from H-7 and H-14 to C-1, from H-7 to C-5, and from H-15 to C-13. Therefore, the planar structure of 1 was determined.

COSY and key HMBC correlations of 1 and 2.
The NOESY correlations of H3-16 with H-2 and H-14 indicated a cis-relationship of them (Fig. 3). Hence, ring D was oriented on the opposite face of ring C relative to these protons. The NOESY correlation of H-4/H3-18 indicated an anti-relationship of 4-OH and H3-18. The vicinity of H-5 and C-19 was deduced by the correlation of H-5/H3-19 in the NOESY spectrum.

Selected NOESY correlations of 1 and 2.
The absolute configuration of 1 was established by comparison of its calculated and observed ECD and optical rotations (OR) data (see Supplementary Information). The predicted ECD for (2R, 4S, 5S, 6S, 13S, 14R)-1 was in agreement with the experimental result of 1 (λmax (Δε) 200 (−1.55), 239 (+7.03) nm) (Fig. 4). The computed ORs in the gas phase were –38.8 for (2S, 4R, 5R, 6R, 13R, 14S)-1, and +38.8 for (2R, 4S, 5S, 6S, 13S, 14R)-1, respectively, and the experimental value was +34.0. Based on both of ECD and OR calculations, the absolute configuration of 1 was assigned as 2R, 4S, 5S, 6S, 13S, 14R.

Experimental ECD spectra of 1 and calculated ECD spectra for (2S, 4R, 5R, 6R, 13R, 14S)-1 and (2R, 4S, 5S, 6S, 13S, 14R)-1.
Harziane diterpenes are a unique class of terpenes, and only 16 such skeletons have been reported9,14,15,16,17,18,19,20. The cyclization mechanism of these unique diterpenes was illuminated by studies of selectively 13C- and 2H-labeled synthetic mevalonolactone isotopologues21. Distinguishing 1 from classic harziane diterpenes such as harzianone and harziandione was the D ring, which is a product of a Baeyer-Villiger monooxygenase catalyzed oxidation of a 6/5/7/4 fused tetra-cyclic skeleton; only two such harziane diterpene lactones have been discovered17,22. Moreover, only one report has appeared on the absolute configuration of such a harziane diterpene lactone (harzianelactone) by comparison its optical rotation data with that of the classic harziane diterpene, harzianone17. This report is the first to determine the absolute configuration of a harziane diterpene lactone by comparison of calculated and observed ECD spectra.
Harzianelactone B (2) was also isolated as a colorless oil and was assigned the same molecular formula C20H30O3 as 1 by HRESIMS results. Extensive analysis of the 1D and 2D NMR data indicated that 2 was also a harziane diterpene lactone, possessing a 6/5/7/5-fused tetra-cyclic ring scaffold like 1. Compared to 1, the disappearance of the oxymethylene signals in the 1H (δH 3.80, 3.73) and 13C NMR (δC 78.4) spectra, and its replacement with ketomethylene signals at δH 2.46, 2.33, and δC 47.3, and the significant downfield shift of C-10, as well as the different UV absorption (2: λmax = 204 nm; 1: λmax = 237 nm), in combination with biogenetic considerations, suggested that C-10 is connected to the O-atom of the ester carbonyl. Detailed analysis of the HMBC correlations from H-8 and H3-20 to C-10, from H-12 to C-11, from H-14 to C-12, and from H-15 to C-13 (Fig. 2) also confirmed the structure. The relative configuration of 2 was deduced to be identical to that of 1 from the assignments of the cross-peaks in its NOESY spectrum (Fig. 3). The computed OR was +21.7 for (2R, 4S, 5S, 6S, 13S, 14R)-2, and the experimental value was +18.9 (see Supplementary Information). Therefore, the absolute configuration of 2 was determined as 2R, 4S, 5S, 6S, 13S, 14R. This is the first report of the absolute configuration of harziane diterpene lactones with the acyloxy group connected to C-10, such as 2.
Harzianone A (3) was obtained as a colorless oil. Its molecular formula of C20H30O2 (six degrees of unsaturation) was determined by HRESIMS and NMR data (Tables 1, 2). The UV absorption and the IR spectrum as well as the NMR data showed that 3 was also a harziane diterpene. Inspection of its NMR data revealed that 3 was similar to harzianone, which was isolated from an alga-endophytic isolate of T. longibrachiatum9. The difference between these two compounds was on the A ring, especially the chemical shifts of C-2 to C-4. In the 1H and 13C NMR spectra, the replacement of a methylene in harzianone with an oxymethine (δH 3.82; δC 73.5), and the downfield shift of C-2 to C-4 indicated that there was a hydroxy group anchored at one of these three carbons. The COSY correlations of H-2/H-3/H-4/H-5/H-18 and the HMBC correlations from H-4 to C-2, C-3, C-5, C-6 and C-18 suggested the hydroxy group was attached to C-4. Therefore, 3 has a 6/5/7/4-fused tetra-cyclic ring scaffold different from 1 and 2. Analysis of the NOESY spectrum allowed the relative configuration of 3 to be the same as those of 1 and 2. The positive first Cotton effect at 340 nm (Δε + 4.09) and the negative second one at 251 nm (Δε −3.11) (Fig. 5) in the ECD spectrum was consistent with that of harzianone9, thus indicating a 2 R,4 S,5 S,6 S,13 S,14 S absolute configuration of 3.

Experimental ECD spectra of 3–6.
As illustrated above, 1 and 2 might formed from 3 through a Baeyer-Villiger monooxygenase catalyzed oxidation. The different oxygenation position would from 1 and 2, respectively. On the basis of biogenetic considerations, 1−3 should have the same configurations, which were in accordance with the description we discussed above.
Harzianone B (4) was afforded as a colorless oil, and had a molecular formula of C20H30O3 evidenced from its HRESIMS spectrum. Analysis of the NMR data demonstrated that the structure of 4 resemble that of 3. The 1H and 13C NMR spectra of 4 displayed signals of four methyl groups, while those of 3 revealed five ones. Compared to 3, the C-20 methyl group was replaced by an oxymethylene in 4, which was defined by the HMBC correlations from H-20 to C-8, C-9, and C-10. As expected, subsequent analyses of the coupling constants, NOESY correlations, and experimental ECD data (Fig. 5) indicated that 4 has the same absolute configuration (2R, 4S, 5S, 6S, 13S, 14S) as that of 3.
Harzianone C (5) was isolated as colorless crystal needles. The molecular formula, C20H30O2, was assigned to be the same as that of 3 by its HRESIMS. The 1H and 13C NMR spectra of 5 showed similar characteristic signals to 3 (Tables 1, 2), except for the chemical shifts around the oxymethine group. In the 1H NMR spectrum, the oxymethine proton appeared as a multiplet, which was different from the doublets for 1−4, and indicated the position of the hydroxy group was changed in 5. In the HMBC spectrum, the correlations of H-4 with C-6, of H-15 with C-3, and of H3-18 with C-4 indicated the hydroxyl group was attached to C-3. The NOESY correlations from H-3 to H3-18 suggested that 3-OH and H3-18 are on the opposite sites of ring A. The relative configurations of the other chiral centers were confirmed to be the same as those of 3. The ECD spectrum of 5 showed the same pattern as those of 3 and 4 (Fig. 5), suggesting that their chirality centers have the same absolute configurations. In addition, an X-ray crystallographic study (Fig. 6) was performed to confirm unambiguously the structure and determined the absolute configuration of 5 as 2S, 4S, 5S, 6S, 11R, 13S, 14S.

X-ray ORTEP diagrams of compounds 5 and 7.
Harzianone D (6) was obtained as a colorless oil. The NMR spectral features suggested that 6 was closely related to 3. The additional carbonyl group (δC 216.9) and the disappearance of the oxymethine group (δC 73.5; δH 3.82 in 3) in 6 indicated that the hydroxy group at C-4 in 3 was replaced by a carbonyl group in 6, which was confirmed by the HMBC correlations from H-2, H-3, H-5, and H-18 to C-4. The relative configuration was determined as the same as 1−5 through NOESY spectrum. Similar cotton effects observed for 6 (Δε 223 + 4.16, Δε 280 −1.36) to 3 and 4 in their ECD spectra (Fig. 5) indicated that they shared the same absolute configurations of (2R, 5S, 6S, 13S, 14S).
Harziane (7) was obtained as colorless crystals. Its molecular formula, C20H32O2, was deduced from its HRESIMS data with five indices of hydrogen deficiency, one fewer than those of 1−5. The 1H and 13C NMR data (Tables 1, 2) of 7 and 3 were very similar with each other except for those in the vicinity of C-11. In the 13C NMR spectra, the signals for α,β-unsaturated ketone (δC 200.0) in 3 was disappeared and one more oxymethine (δC 67.8) was emerged in 7. These evidences as well as the unsaturation degrees of these two compounds indicated that the ketone carbonyl group at C-11 in 3 was replaced by an oxymethine group in 7, which was confirmed by the HMBC correlations from H-11 to C-9, C-10, and C-13. The relative configuration of all chiral centers but C-11 was determined by the NOESY spectrum of 7 like 1−6, and the correlation of H-11 with H3-18 indicated they were cis-oriented. To clarify its absolute stereochemistry, 7 was recrystallized in a dichloromethane/methanol (20:1) mixture to yield crystals. The low-temperature X-ray diffraction (CuKα) of the single crystals (Fig. 6) revealed that 7 had a (2R, 4S, 5S, 6S, 11R, 13S, 14S)-configuration.
Harziane diterpenes have rarely been reported to have significant bioactivities. In the present study, compounds 1−5 and 7 were evaluated for their phytotoxic and antibacterial activities. All the tested compounds showed obvious phytotoxicity against the seedling growth of amaranth and lettuce at a concentration of 200 ppm (Table 3). Compounds 1, 3, 4, and 5 were more effective as they could completely inhibit seed germination against amaranth at 200 μg/mL, and this strong phytotoxicity was still evident at lower concentrations (50 μg/mL), compared to the positive control glyphosate. No compound was found to inhibit the root growth of lettuce at 200 ppm. It seemed that the isolated compounds caused weaker inhibition to lettuce than to amaranth, and have stronger toxicity on the growth of root growth than hypocotyl. Although there are three reports on the phytotoxicity of crude extracts of Trichoderma spp.4,23,24, no one had studied the phytotoxicity of compounds from Trichoderma spp. Thus, this is the first report of the phytotoxic compounds from Trichoderma spp., and the phytotoxicity of harziane diterpenes is also reported for the first time. None of the isolated compounds exhibited antibacterial activities.
Conclusions
In summary, the present chemical investigation on the soft coral-derived T. harzianum XS-20090075 resulted in the discovery of a series of harziane diterpenes (1–7). Compounds 1 and 2 represent a unique type of harziane diterpene lactone derived from harziane diterpenes though Baeyer-Villiger monooxygenase catalyzed oxidations. Harziane diterpenes have rarely been studied, and only 18 such compounds have been reported, including two harziane diterpene lactones. In this study, the structures of harziane diterpenes were determined by NMR spectroscopic data, ECD and OR calculations, together with X-ray diffraction. The phytotoxicity of compounds from Trichoderma sp. was evaluated for the first time, and the isolated compounds exhibited potent phytotoxicity towards amaranth and lettuce.
Methods
General experimental procedures
Optical rotations were measured using a P-1020 polarimeter (JASCO). UV spectrua were obtained with a DU 640 spectrophotometer (Beckman). ECD spectra were acquired on a JASCO J-815-150S CD spectrometer. IR spectra were obtained via a Nicolet-Nexus-470 spectrometer. NMR spectra were recorded on an Agilent DD2 NMR spectrometer (500 MHz). ESIMS and HRESIMS spectra were obtained by a Q-TOF (Micromass) and a LTQ Orbitrap XL (Thermo Scientific) spectrometer, respectively. Single-crystal analysis were performed on a Gemini A Ultra system using Cu Kα radiation (Aglient Technologies). A 1525 separation module (Waters) equipped with a C18 (Kromasil, 5 μm, 10 × 250 mm) column was used for semi-preparative HPLC. ODS (Unicorn; 45–60 μm), Sephadex LH-20 (Amersham Biosciences), and silica gel (200–300 mesh; Qing Dao Hai Yang Chemical Group Co.) were applied for column chromatography. TLC (G60, F-254; Yan Tai Zi Fu Chemical Group Co.) was used in the compounds detection.
Fungal materia
The fungal strain (XS-20090075) was isolated from the inner part of an unidentified soft coral, and was identified as T. harzianum by morphological characteristics and ITS sequence. A voucher specimen was deposited at School of Medicine and Pharmacy, Ocean University of China, PR China (KU866299).
Extraction and isolation
The fungus XS-20090075 was fermented at room temperature for four weeks in 100 conical flask (1 L) containing 80 g rice and 120 mL H2O with 3% salinity. The culture medium was extracted by EtOAc and CH2Cl2−MeOH (v/v, 1:1) for three times, and the solution was concentrated under reduced pressure to afford a residue. The residue was mixed with 1000 mL of H2O, and extracted with ethyl acetate to yield the crude extract (18.5 g). The extract was fractioned by silica gel column chromatography (CC) eluted with gradient EtOAc in petroleum ether (0%–100%), and then with MeOH/EtOAc (10%–50%) to yield six fractions (Fr. 1−Fr. 6). Fr. 1 was first repeatedly chromatographed on silica gel column by EtOAc/petroleum ether (10%), and then separated by ODS eluted with MeOH−H2O (30−80%) to afford Fr. 1-1−Fr. 1–5. Fr. 1–3 was further purified over semipreparative RP-HPLC (MeOH/H2O, 80/100) to yiled 2 (24.0 mg), 3 (94.3 mg), and 6 (4.2 mg). Fr. 2 was first separated by silica gel CC (EtOAc/petroleum ether = 20/80), and the eluent were combined, concentrated, and submitted to Sephadex LH-20 CC (CH2Cl2/MeOH, v/v, 1/1), followed by purification on HPLC with 55% MeOH−H2O to afford 1 (8.9 mg) and 5 (22.8 mg). Fr. 3 was chromatographed on silica gel CC (EtOAc/petroleum ether = 20%) and separated by ODS CC using 50% MeOH−H2O to obtain Fr. 3-1–Fr. 3-3. Fr.3-1 was futher purified on HPLC (65% MeOH−H2O) to give 4 (38.2 mg). Fr. 3-2 was purified by semipreparative RP-HPLC (MeOH/H2O, 80/20) to yield 7 (6.4 mg).
Harzianelactone A (1)
colorless oil; [α]20D + 33.8 (c 0.42, MeOH); UV (MeOH) λmax (log ε) 237 (3.81) nm; ECD (1.57 mM, MeOH) λmax (Δε) 200 (−1.55), 239 (+7.03) nm; IR (KBr) νmax 3425, 2933, 2362, 2340, 1738, 1653, 1029 cm–1; 1H and 13C NMR data, Tables 1, 2; HRESIMS m/z 319.2263 [M + H]+ (calcd for C20H31O3, 319.2268).
Harzianelactone B (2)
colorless oil; [α]20D + 18.9 (c 0.42, MeOH); UV (MeOH) λmax (log ε) 204 (3.62) nm; IR (KBr) νmax 3398, 2931, 2362, 2340, 1779, 1703, 1029 cm−1; 1H and 13C NMR data, Tables 1, 2; ESIMS m/z 319.3 [M + H]+, 341.3 [M + Na]+, 637.4 [2 M + H]+, 659.5 [2 M + Na]+; HRESIMS m/z 319.2271 [M + H]+ (calcd for C20H31O3, 319.2268).
Harzianone A (3)
colorless oil; [α]20D + 72.1 (c 0.42, MeOH); UV (MeOH) λmax (log ε) 259 (3.98) nm; ECD (1.65 mM, MeOH) λmax (Δε) 251 (−3.11), 340 (+4.09) nm; IR (KBr) νmax 3400, 2932, 2361, 1735, 1669, 1441, 1260, 1150, 1027 cm−1; 1H and 13C NMR data, Tables 1, 2; HRESIMS m/z 303.2316 [M + H]+ (calcd for C20H31O2, 303.2319).
Harzianone B (4)
colorless oil; [α]20D + 32.2 (c 0.41, MeOH); UV (MeOH) λmax (log ε) 203 (3.49), 256 (3.64) nm; ECD (1.57 mM, MeOH) λmax (Δε) 248 (−6.98), 345 (+7.02) nm; IR (KBr) νmax 3425, 2935, 2363, 1722, 1689, 1029 cm−1; 1H and 13C NMR data, Tables 1, 2; HRESIMS m/z 319.2262 [M + H]+ (calcd for C20H31O3, 319.2268).
Harzianone C (5)
colorless crystals; mp 168−169 °C; [α]20D + 14.7 (c 0.46, MeOH); UV (MeOH) λmax (log ε) 256 (3.58) nm; ECD (1.65 mM, MeOH) λmax (Δε) 254 (−6.21), 337 (+4.66) nm; IR (KBr) νmax 3398, 2932, 2363, 1737, 1659, 1444, 1382, 1020 cm−1; 1H and 13C NMR data, Tables 1, 2; HRESIMS m/z 303.2318 [M + H]+ (calcd for C20H31O2, 303.2319).
Harzianone D (6)
colorless oil; [α]20D + 52.6 (c 0.28, MeOH); UV (MeOH) λmax (log ε) 255 (2.72) nm; ECD (1.67 mM, MeOH) λmax (Δε) 255 (−2.83), 338 (+3.02) nm; IR (KBr) νmax 2948, 2356, 1728, 1655, 1438, 1260, 1022 cm−1; 1H and 13C NMR data, Tables 1, 2; HRESIMS m/z 301.2161 [M + H]+ (calcd for C20H29O2, 301.2162).
Harziane (7)
colorless crystals; mp 214−215 °C; [α]20D + 5.1 (c 0.48, MeOH); UV (MeOH) λmax (log ε) 206 (3.58) nm; IR (KBr) νmax 3404, 2929, 2362, 1653, 1382, 1033 cm−1; 1H and 13C NMR data, Tables 1, 2; HRESIMS m/z 287.2364 [M + H − H2O]+ (calcd for C20H31O, 287.2369).
X-ray Crystallographic Analysis of 5 and 7
The Single-crystal X-ray diffraction data were recorded on a Xcalibur, Atlas, Gemini ultra diffractometer at 120 K. Crystallographic data for 5 (deposition NO. CCDC 1573734) and 7 (deposition NO. CCDC 1573693) have been deposited in the Cambridge Crystallographic Data Centre. Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB21EZ, UK [fax: + 44(0)-1233-336033 or e-mail: deposit@ccdc.cam.ac.uk].
Crystal data for 5
C20H30O2, Mr = 302.44, monoclinic, a = 7.13670 (10) Å, b = 13.7978 (3) Å, c = 8.4352(2) Å, α = 90.00°, β = 94.516 (2)°, γ = 90.00°, V = 828.04 (3) Å3, space group P21, Z = 2, Dx = 1.213 mg/m3, μ = 0.586 mm−1, and F (000) = 332. Crystal size: 0.42 × 0.25 × 0.13 mm3. Reflections collected/unique: 8011/2950 [R(int) = 0.0230]. The final indices were R1 = 0.0295, wR2 = 0.0733 (I > 2σ(I)). Flack parameter = 0.13 (19).
Crystal data for 7
C20H32O2, Mr = 304.46, monoclinic, a = 18.9468 (16) Å, b = 8.3433 (2) Å, c = 13.245 (4) Å, α = 90.00°, β = 124.739 (8)°, γ = 90.00°, V = 1720.6 (5) Å3, space group C2, Z = 4, Dx = 1.175 mg/m3, μ = 0.564 mm−1, and F (000) = 672. Crystal size: 0.21 × 0.20 × 0.19 mm3. Reflections collected/unique: 9303/3064 [R(int) = 0.0261]. The final indices were R1 = 0.0307, wR2 = 0.0748 (I > 2σ(I)). Flack parameter = 0.13 (19).
Phytotoxicity bioassays
Phytotoxicity against seeding growth of amaranth (Amaranthus retroflexus L.) and lettuce (Lactuca sativa) was assayed by the method reported previously25. Glyphosate was used as the positive control.
Antibacterial assays
The antibacterial activity was evaluated by the conventional broth dilution assay26. Five pathogenic bacterial strains, including Gram-positive Kocuria rhizophila (ATCC 9341), Staphyloccocus aureus (ATCC 27154), and Gram-negative Escherichia coli (ATCC 25922), Ralstonia solanacearum, Vibrio anguillarum (ATCC 19019), and V. Parahemolyticus (ATCC 17802) were used, and ciprofloxacin and streptomycin sulfate were used as positive controls.
References
Dayan, F. E., Cantrell, C. L. & Duke, S. O. Natural products in crop protection. Bioorg. Med. Chem. 17, 4022–4034 (2009).
Cantrell, C. L., Dayan, F. E. & Duke, S. O. Natural products as sources for new pesticides. J. Nat. Prod. 75, 1231–1242 (2012).
Silva, R. N., Steindorff, A. S. & Monteiro V. N. Biotechnology and biology of Trichoderma (ed. Gupta, V. K., Schmoll, M., Herrera-Estrella, A., Upadhyay, R. S., Druzhinina, I. & Tuohy, M. G.) 363–376 (Elsevier, 2014).
Javaid, A. & Ali, S. Herbicidal activity of culture filtrates of Trichoderma spp. against two problematic weeds of wheat. Nat. Prod. Res. 25, 730–740 (2011).
Blunt, J. W., Copp, B. R., Keyzers, R. A., Munro, M. H. G. & Prinsep, M. R. Marine natural products. Nat. Prod. Rep. 35, 8–53 (2018).
Jans, P. E. et al. Cytotoxicity and mechanism of action of the marine-derived fungal metabolite trichodermamide B and synthetic analogues. J. Nat. Prod. 80, 676–683 (2017).
Yamazaki, H., Rotinsulu, H., Takahashi, O., Kirikoshi, R. & Namikoshi, M. Induced production of a new dipeptide with a disulfide bridge by longterm fermentation of marine-derived Trichoderma cf. brevicompactum. Tetrahedron Lett. 57, 5764–5767 (2016).
Suzue, M., Kikuchi, T., Tanaka, R. & Yamada, T. Tandyukisins E and F, novel cytotoxic decalin derivatives isolated from a marine sponge-derived fungus. Tetrahedron Lett. 57, 5070–5073 (2016).
Miao, F. P., Liang, X. R., Yin, X. L., Wang, G. & Ji, N. Y. Absolute configurations of unique harziane diterpenes from Trichoderma species. Org. Lett. 14, 3815–3817 (2012).
Liu, Q. A. et al. Antifouling and fungicidal resorcylic acid lactones from the sea anemone-derived fungus Cochliobolus lunatus. J. Agric. Food Chem. 62, 3183–3191 (2014).
Zhao, D. L. et al. Azaphilone and diphenyl ether derivatives from a gorgonian-derived strain of the fungus Penicillium pinophilum. J. Nat. Prod. 78, 2310–2314 (2015).
Jia, Y. L. et al. (+)- and (−)-Pestaloxazine A, a pair of antiviral enantiomeric alkaloid dimers with a symmetric spiro[oxazinane-piperazinedione] skeleton from Pestalotiopsis sp. Org. Lett. 17, 4216–4219 (2015).
Chen, M., Zhang, W., Shao, C. L., Chi, Z. M. & Wang, C. Y. DNA methyltransferase inhibitor induced fungal biosynthetic products: diethylene glycol phthalate ester oligomers from the marine-derived fungus Cochliobolus lunatus. Mar. Biotechnol. 18, 409–417 (2016).
Ghisalberti, E. L., Hockless, D. C. R., Rowland, C. & White, A. H. Harziandione, a new class of diterpene from Trichoderma harzianum. J. Nat. Prod. 55, 1690–1694 (1992).
Mannina, L. et al. A new fungal growth inhibitor from Trichoderma viride. Tetrahedron 53, 3135–3144 (1997).
Adelin, E. et al. Bicyclic and tetracyclic diterpenes from a Trichoderma symbiont of Taxus baccata. Phytochemistry 97, 55–61 (2014).
Zhang, M. et al. Two new diterpenoids from the endophytic fungus Trichoderma sp. Xy24 isolated from mangrove plant Xylocarpus granatum. Chin. Chem. Lett. 27, 957–960 (2016).
Zhang, M. et al. Two furanharzianones with 4/7/5/6/5 ring system from microbial transformation of harzianone. Org. Lett. 19, 1168–1171 (2017).
Zhang, M. et al. Microbial oxidation of harzianone by Bacillus sp. IMM-006. Tetrahedron 73, 7195–7199 (2017).
Song, Y. P., Fang, S. T., Miao, F. P., Yin, X. L. & Ji, N. Y. Diterpenes and sesquiterpenes from the marine algicolous fungus Trichoderma harzianum X-5. J. Nat. Prod. 81, 2553–2559 (2018).
Barra, L. & Dickschat, J. S. Harzianone biosynthesis by the biocontrol fungus Trichoderma. Chembiochem 18, 2358–2365 (2017).
Xie, Z. L. et al. Trichodermaerin, a new diterpenoid lactone from the marine fungus Trichoderma erinaceum associated with the sea star Acanthaster planci. Nat. Prod. Commun. 8, 67–68 (2013).
Kuang, W. F., Wang, C. F. & Mao, W. L. Screening and evaluation of herbicidal metabolites produced by Trichoderma spp. Afr. J. Microbiol. Res. 10, 866–872 (2016).
Javaid, A., Shafique, G., Ali, S. & Shoaib, A. Effect of culture medium on herbicidal potential of metabolites of Trichoderma species against Parthenium hysterophorus. Int. J. Agric. Biol. 15, 119–124 (2013).
Zhang, Q. et al. Potential allelopathic indole diketopiperazines produced by the plant endophytic Aspergillus fumigatus using the one strain-many compounds method. J. Agric. Food Chem. 61, 11447–11452 (2013).
Zhu, A. et al. new anti-vibrio prenylxanthones from the marine-derived fungus Aspergillus sp. ZA-01. Mar. Drugs 16, 312 (2018).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81673350; 41806194; U1706210; U1606403), the Scientific and Technological Innovation Project Financially Supported by Qingdao National Laboratory for Marine Science and Technology (No. 2016ASKJ08), the Fundamental Research Funds for the Central Universities of China (No. 201762017), and the Taishan Scholars Program, China.
Author information
Authors and Affiliations
Contributions
D.L.Z. contributed to identification of the compounds and manuscript preparation. T.S. and C.Y.W. (Chao-Yi Wang) contributed to fungi identification, fermentation, extraction and isolation of the compounds. L.J.Y. contributed to bioassays of the compounds. C.L.S. and C.Y.W. (Chang-Yun Wang) conceived and designed research. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, DL., Yang, LJ., Shi, T. et al. Potent Phytotoxic Harziane Diterpenes from a Soft Coral-Derived Strain of the Fungus Trichoderma harzianum XS-20090075. Sci Rep 9, 13345 (2019). https://doi.org/10.1038/s41598-019-49778-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49778-7
This article is cited by
-
Chemistry and biology of marine-derived Trichoderma metabolites
Natural Products and Bioprospecting (2024)
-
Contouring Multifaceted Biological Activities and Applications of Trichoderma spp. for Managing Plant Health
Journal of Crop Health (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
