
Abstract
Single nucleotide polymorphisms (SNPs) and genes associated with susceptibility to hepatitis B virus (HBV) infection that have been identified by genome-wide association studies explain only a limited portion of the known heritability, indicating more genetic variants remain to be discovered. In this study, we adopted a new research strategy to identify more susceptibility genes and variants for HBV infection. We first performed genetic association analysis of 300 sib-pairs and 3,087 case-control samples, which revealed that 36 SNPs located in 31 genes showed nominal associations with HBV infection in both samples. Of these genes, we selected SEC24D for further molecular analysis according to the following two main lines of evidence. First, a time course analysis of the expression profiles from HBV-infected primary human hepatocytes (PHH) demonstrated that SEC24D expression increased markedly as time passed after HBV infection (P = 4.0 × 10−4). Second, SNP rs76459466 in SEC24D was adversely associated with HBV risk (ORmeta = 0.82; Pmeta = 0.002), which again indicated that SEC24D represents a novel susceptibility gene for HBV infection. Moreover, SEC24D appeared to be protective against HBV infection in vitro. Consistently, we found that SEC24D expression was significantly enhanced in non-infected liver tissues (P = 0.002). We conclude that SEC24D is a novel candidate gene linked to susceptibility to HBV infection.
Similar content being viewed by others
Introduction
Hepatitis B virus (HBV) infection is a serious global health issue and shows marked geographic diversity. Thus, in the United States and northern European countries, the prevalence of chronic HBV infection (CHBVI) is estimated to be <0.5%, but this figure is as high as 10–12% in China and South Korea1,2. Despite the availability of a potent HBV vaccine and effective antiviral drugs for two decades, hepatitis B maintains at high prevalence worldwide, with more than 240 million people infected3. Chronic hepatitis B can cause liver cirrhosis and hepatocellular carcinoma and is responsible for more than 0.5–1.0 million deaths per year4.
Chronic HBV infection (CHBVI) and viral clearance are influenced by multiple genetic and environmental factors, including viral and host factors5,6,7. Twin and segregation studies indicate that host genetic components strongly influence the outcome of HBV infection8. Recently, genome-wide association study (GWAS) has been used to identify genetic variants for numerous complex human diseases such as HBV infection and clearance. Several loci, located in human leukocyte antigen-C (HLA-C)9, HLA-DP10, HLA-DQ11, HLA-DOA, complement factor B (CFB), NOTCH412, euchromatic histone lysine methyltransferase 2 (EHMT2), transcription factor 19 (TCF19)13, and two non-HLA loci, CD4012 and ubiquitin conjugating enzyme E2 L3 (UBE2L3)9, have been reported to be significantly associated with HBV-related diseases. However, as observed in many human disorders, these single nucleotide polymorphisms (SNPs) account for only a small proportion of the apparent genetic variance, implying many susceptibility loci remain to be identified for HBV-related diseases14.
To further elucidate disease-predisposing genes for HBV infection, we employed an integrative functional genomics strategy, which can be summarized briefly as follows. We first conducted a variant discovery in 300 sib-pairs, followed by replication of top candidate SNPs in 3,087 case-control samples. The SEC24D gene was then selected for analysis based on the expression analysis and association study. Through a series of in vitro experiments, we found SEC24D to be an antiviral gene for HBV infection.
Methods
Subjects
The three hundred sib-pairs used for the discovery stage were recruited between 2010 and 2012 at the First Affiliated Hospital of Zhejiang University School of Medicine and other neighbor medical hospitals or centers. Among these siblings, 300 CHBVI subjects were defined as seropositive for either hepatitis B surface antigen (HBsAg) or HBV-DNA, and the corresponding sib-controls were negative for both. More detailed descriptions of the demographic and phenotypic characteristics of these subjects are shown in Table 1 and Supplementary Fig. 1A.
The replication sample was recruited from the same medical facilities and included 1,648 CHBVI participates and 1,439 unrelated controls (Table 1 and Supplementary Fig. 1B). A detailed description of this independent replication sample has been provided in previous publications from our group15,16.
All the participants were of Chinese Han ethnicity. Informed written consent was obtained from every participant, and the demographic and clinical data were collected by structured questionnaires. This project was approved by the Ethical Committee of the First Affiliated Hospital of Zhejiang University School of Medicine.
Whole exome-sequencing analysis in the discovery sample
Genomic DNA was extracted from 3 ml of peripheral blood from each subject using the Qiagen DNA purification kit. Libraries were prepared according to the operational manual provided by the manufacturer, and the enriched coding exons were captured using a TruSeq Exome Enrichment Kit (Illumina, San Diego, USA) and sequenced by the Illumina HiSeq2000 system. Paired-end sequencing was carried out for 100 bases from each end of about 200-bp insert fragment libraries using standard Illumina protocols, and sequencing reads were aligned to hg19 from UCSC Genome Browser (http://genome.ucsc.edu/) using the Burrows-Wheeler Aligner (BWA) with default parameters17. After removing PCR duplicates by Picard tools (http://broadinstitute.github.io/picard/), the median sequencing depth of all samples was 56 × (see Supplementary Fig. 2). Of the targeted exon regions, 90.73% were covered at an average of ≥10× with genotype quality scores of ≥30. Single nucleotide variations (SNVs) were identified by the Genome Analysis Toolkit (GATK)18,19. The statistics of each variant, including allele balance, depth of coverage, strand balance, and multiple quality metrics, were annotated using the GATK Variant Annotator18,19. These statistics were then used in an adaptive error model to estimate the probability that each SNV is a true one using the GATK Variant Quality Score Realibrator (VQSR)18,19. Functional annotation of variants was performed using the ANNOVAR20, and the annotation database was downloaded from the UCSC Genome Browser.
Stringent quality control steps were performed to ensure robust association analysis. Single nucleotide polymorphisms were excluded from further analysis if they had a minor allele frequency (MAF) of <0.01 and a P value of <1 × 10−6 for Hardy-Weinberg equilibrium (HWE). After this appropriate quality control, a total of 98,357 SNPs remained for further analysis.
Genotyping in the replication stage
All of the 4,000 SNPs selected at the discovery stage were genotyped using the Illumina iSelect custom genotyping array according to the Illumina Infinium HD Assay Ultra Manual. Among them, 121 SNPs failed to be designed in the custom array. In addition, 291 ancestry informative markers (AIMs) from different chromosomes were included in the iSelect array and used to assess population admixture for the replication samples. An SNP was excluded if it had: (1) a call rate of <0.95; (2) an MAF of <0.05; and (3) a P value of <1 × 10−6 with the HWE test in the replication samples. Those SNPs located on sex chromosomes also were excluded. Any sample was removed if it had a call rate of <0.95. After these quality control steps, 2,925 SNPs on autosomal chromosomes from 3,064 samples remained for further analysis.
Gene expression analysis
Gene expression profiles of three independent datasets were downloaded from Gene Expression Omnibus (GEO). Dataset 1 (Accession Number GSE72068) was used to perform time course analysis, which consists of determining 20 mRNA expression profiles in primary human hepatocytes (PHH) obtained at different times after HBV infection using an Illumina HumanHT-12 V4.0 expression beadchip. Dataset 2 (Accession Number GSE36250) was used to measure the differential expression of candidate genes of interest, which consists of examining 123 mRNA expression profiles of liver samples by the NimbleGen Custom Gene Expression HX3 Microarray. Dataset 3 (Accession Number GSE22058) was used for pathway enrichment analysis, which consists of study of expression data from 96 liver specimens from patients with HBV-related HCC employing the Rosetta/Merck Human RSTA Custom Affymetrix 1.0 microarray. The specimens were divided into two groups on the basis of mean SEC24D expression. The up quartile was defined as the SEC24D high-expression group and the down quartile as the low-expression group. The genes with a false-discovery rate Q value < 0.001 and |fold change| >1.5 were considered highly differentially expressed. Pathway enrichment analysis was carried out with the DAVID tool (v. 6.8)21.
Cells culture and transfection
The human hepatoma cell lines HepG2 and HepG2.2.15 were purchased from the China Center for Type Culture Collection (CCTCC). All cells were cultured in Dulbecco Modified Eagle Medium (HyClone, Logan, UT USA) containing 10% fetal bovine serum (GIBCO, Waltham, MA USA), and penicillin G 100 U/ml and streptomycin 100 μg/ml (GIBCO) at 37 °C in a humidified incubator with 5% CO2. The HepG2.2.15 cells were supplemented with G418 400 μg/ml (GIBCO) to maintain the stably transfected dimeric HBV-DNA. The HBV-producing plasmid pGEM-4Z-HBV1.3, which contains 1.3 U of the HBV genome (subtype ayw)22, was a gift from Dr. Shick Ryu Wang (Addgene plasmid # 65459). The SEC24D cDNA was cloned into the SacII and EcoRI sites of the expression vector pEGFP-C3 (Clontech, Palo Alto, CA USA). The recombinant plasmid was sequenced to confirm the accuracy by Sangon Biotech (Shanghai, China). For inhibition of gene expression, the cells were transfected with siRNA duplexes, which were synthesized by RiboBio Inc. (Guangzhou, China). All the transfection reactions were established using the Lipofectamine 3000 Transfection Reagent (Invitrogen, Carlsbad, CA USA) according to the manufacturer’s instruction.
Western blotting analysis
After 48 h of transfection, cell lysates were collected using a RIPA lysis buffer with protease inhibitors (Tiangen, Beijing, China). The lysates, containing 12 μg of protein, were separated on SDS-PAGE and transferred to PVDF membranes (Millipore, Bedford, USA). The membrane was probed with a designated primary antibody (anti-SEC24D and anti-beta actin; Abcam, Cambridge, UK) overnight at 4 °C and further incubated with the corresponding horseradish peroxidase-conjugated secondary antibody (Bioker, Hangzhou, China) for 1 h at room temperature. The immunoreactive bands were labeled with Clarity Western ECL Substrate (Bio-Rad, Richmond, USA). Beta-actin was used as a protein loading control. The signal intensity was quantified by ImageJ software (National Institutes of Health, Bethesda, MD USA).
Detection of HBV-DNA, HBsAg, and HBeAg
After 48 h of transfection, cell supernatant liquid was collected by centrifugation at 3,000 rpm for 10 min at 4 °C. The HBV DNA load was measured by quantitative real-time PCR using the Fluorescence Quantitative PCR Detection Kit for HBV-DNA (Acon, Hangzhou, China). The HBsAg and HBeAg concentrations were quantified by the chemiluminescent microparticle immunoassay using an ARCHITECT Reagent Kit (Abbott, Chicago, IL USA). All the assays were performed at least three times following the manufacturer’s instructions.
Data analysis
We carried out a liberalization of the sibling transmission/disequilibrium test (sTDT)23 for 300 sib-pairs under an additive genetic model adjusted for age, sex, and the first five principal components (PCs). In the replication stage, association of SNPs with HBV infection was performed under an additive genetic model using PLINK (v. 1.07)24 with age, sex, and the first five PCs as covariates. The population admixture of samples was assessed by PC analysis (PCA) as implemented in EIGENSTRAT25. Meta-analysis of the data generated from family and case-control samples was carried out to assess the pooled genetic effects using the Mantel-Haenszel method26. Heterogeneity was examined with Cochran’s Q test27. When the P value of the Q test was < 0.1, we considered there to be strong evidence for heterogeneity between samples. Time course analysis was performed using BRB-ArrayTools software28. For signal intensity of WB analysis, SEC24D expression, HBV-DNA load, and HBsAg and HBeAg concentrations, significant difference was determined by the two-tailed Student’s t-test. A P value < 0.05 was considered statistically significant.
Results
Association study and expression analysis identified SEC24D as a candidate gene for susceptibility to HBV infection
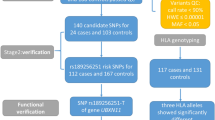
As shown in Fig. 1 and Table 2, we carried out association analyses for both the family and the case-control samples. First, 442,078 SNVs were identified by WES analysis. Second, we performed sTDT for the 98,357 SNPs remaining after quality controls. Third, the top 4,000 SNPs were selected for replication in 1648 CHBVIs and 1439 unrelated controls, which revealed that 36 SNPs across 31 genes were nominally associated with HBV infection in both samples (all P values < 0.05).

Workflow of the integrative functional genomics methodology to identify the susceptibility gene for HBV infection. Abbreviations: SNV = single nucleotide variation; SNP = single nucleotide polymorphism; MAF = minor allele frequency; HWE = Hardy-Weinberg equilibrium; sTDT = sibling transmission/disequilibrium test.
To identify which genes are more likely to affect HBV infection, we performed time course analysis based on different time points of the expression data from HBV-infected PHHs. We found that only three genes showed significant time-dependent changes in expression in response to HBV (Fig. 2 and Supplementary Fig. 3). The expression of SEC24D was cumulative and generally elevated, dependent on the time after HBV infection (P = 4 × 10−4). At day 12, it had the largest change (>1.5-fold), indicating a potential correlation between SEC24D and HBV infection. The extent of expression of MICAL1 (microtubule associated monooxygenase, calponin and LIM domain containing 1) and SDAD1 also displayed significant dynamic changes (P = 4 × 10−4 and P = 4 × 10−2, respectively). However, MICAL1 expression was increased within 24 h and decreased after that, indicating different roles in early (24 h and before) and late (post-24 h) responses to HBV infection. For SDAD1, the expression changes also displayed a trend of ascending at first and descending latter, again suggesting different responses to HBV infection at different time points and some type of adaptation at day 6. Considering the potentially complex roles of MICAL1 and SDAD1 in HBV infection and the main objective of this report, we confined our attention to SEC24D.

Time course analysis after HBV infection. Gene expression data (log2 transformed) were extracted from GEO dataset (Accession Number GSE72068). The mean extent of gene expression for each time point was plotted separately for the HBV and mock-infection groups. Red line represents the HBV-infected group, and blue one represents the mock group. Error bar represents standard deviation (SD).
We found a significant association of SEC24D polymorphism with HBV infection (Table 2). In the family sample, when comparing CHBVI with the corresponding sib-control group, SNP rs76459466 (G > T) was negatively associated with HBV infection risk (odds ratio [OR] = 0.64; 95% confidence interval [CI] 0.46, 0.88; P = 5.9 × 10−3). This association was replicated in an independent case-control sample, which showed that rs76459466 was associated with a significantly lower HBV infection risk (OR = 0.86; 95% CI 0.75, 0.99; P = 3.4 × 10−2). As there was no significant heterogeneity between the two samples (P value of Q test > 0.1), meta-analysis was performed on the results from both samples together. We found that the rs76459466 T carriers had a lower risk of HBV infection than the non-carriers (ORmeta = 0.82; 95% CI = 0.72, 0.93; Pmeta = 2.0 × 10−3). Thus, both genetic association studies and gene expression analyses robustly indicated a potential role of SEC24D in HBV infection.
SEC24D inhibits HBV replication
SEC24D is a member of the SEC24 subfamily and correlates with vesicle trafficking. According to the RNA-Seq Atlas database29, SEC24D is expressed in various tissues, including the liver. However, the specific role of SEC24D in HBV infection has not been illuminated. Therefore, we investigated its impact on HBV infection using in vitro functional experiments.
To explore the potential effect of SEC24D on HBV infection, we investigated the amounts of HBV-DNA, HBsAg, and HBeAg in the cell medium after SEC24D overexpression or inhibition. The HepG2 cells were transfected by pGEM-4Z-HBV1.3, together with either pEGFP-C3-SEC24D or pEGFP-C3 control plasmids (Figs 3A and 4A–C). Compared with the cells treated with control vectors, the cells with overexpressed SEC24D plasmid showed a significant drop in HBV-DNA load to 70.3 ± 5.8%, HBsAg to 59.0 ± 5.1%, and HBeAg to 85.1 ± 3.7%. For the SEC24D inhibition (Figs 3B and 5A–C), we used two independent siRNAs, which led to markedly enhanced amounts of HBV-DNA to 152.8 ± 12.9% and 129.8 ± 7.0%, HBsAg to 134.6 ± 11.2% and 115.7 ± 7.4%, and HBeAg to 129.1 ± 7.9% and 112.2 ± 3.3%. Further, we replicated the antiviral effect of SEC24D efficiently resisting HBV in HepG2.2.15 cells (Supplementary Figs 4–6). The HBV markers were significantly reduced by SEC24D overexpression but increased by SEC24D inhibition.

Western blotting analysis for amount of SEC24D protein in HepG2 cells. Cells (~2 × 105) were transfected by pGEM-4Z-HBV1.3, together with pEGFP-C3-SEC24D (SEC24D) or pEGFP-C3 control vectors (Control) (A) or with SEC24D-specific siRNAs (siRNA1 and siRNA2) or negative control siRNAs (NCRNAs) (B). Cell lysates were collected after 48 h transfection. Error bar represents SD; *P < 0.05, **P < 0.01, and ***P < 0.001.

Overexpressed SEC24D inhibited HBV replication in HepG2 cells. Amount of HBV-DNA was detected by quantitative real-time PCR (A), and the quantities of HBsAg (B) and HBeAg (C) were tested by chemiluminescent microparticle immunoassay. All the supernatant liquids were collected after 48 h of transfection. Error bar represents SD; *P < 0.05, **P < 0.01, and ***P < 0.001.

Inhibition of SEC24D-enhanced HBV replication in HepG2 cells. SEC24D expression was inhibited by two independent siRNAs. The amount of HBV-DNA was detected by quantitative real-time PCR (A), and the amounts of HBsAg (B) and HBeAg (C) were measured by chemiluminescent microparticle immunoassay. All the supernatant liquids were collected after 48 h of transfection. Error bar represents SD; *P < 0.05, **P < 0.01, and ***P < 0.001.
Decreased SEC24D expression in infected liver tissues
To further confirm the role of SEC24D in HBV infection, we examined whether a differential degree of SEC24D expression existed in liver tissues from woodchucks by searching public database. The woodchuck can be naturally infected with woodchuck hepatitis virus (WHV), a hepadnavirus that is genetically close to human HBV. It is often used as an animal model for studying the pathogenesis of CHBVI and HBV-related HCC development in human30. We investigated SEC24D expression in the infected (n = 60) and non-infected (n = 63) liver tissues of WHV models (Fig. 6). When compared with the control group (mean log2 normalized expression value of 11.90), we found obviously lower SEC24D expression (mean log2 normalized expression value of 12.15) in the infected liver (fold change 1.2; P = 0.002). Consistent with our previous findings, these data support the protective role of SEC24D in HBV infection.

Expression of SEC24D in liver tissues of infected and control groups (Accession Number GSE36533). Top bar is maximum expression, lower bar is minimum observation, top of box is third quartile, bottom of box is first quartile, middle bar is median value.
Discussion
During the past several years, GWAS has been commonly used to investigate the genetic predisposition to common diseases, but the identified susceptibility variants can explain only a small proportion of the known heritability. Using new research strategies would be helpful to identify the real causal variants. In this study, we combined WES data, iSelect-based array data, and GEO expression profiles followed by in vitro experiments to identify novel susceptibility genes.
In the discovery stage, we used sTDT analysis of WES data to identify SNPs that were significant in 300 CHBVIs compared with 300 unaffected siblings. The primary reason for choosing sib-pairs as the discovery sample for exome-sequencing analysis was the fact that the family-based design has an advantage over the case-control design for its robustness to population stratification31. Then the top 4000 SNPs with the smallest P values were selected for subsequent replication. In the replication stage, these SNPs were genotyped in an independent sample consisting of 1648 CHBVIs and 1439 unrelated controls. We found that 36 nominal SNPs located in 31 genes were validated (P < 0.05) with the same association directions observed in the discovery stage. To narrow down the candidates for the causative genes, we performed time course analysis to investigate the expression of genes that displayed significant time-related changes induced by HBV. We found that, as time went on, only SEC24D expression was markedly increased after HBV infection. Moreover, based on the aforementioned genetic association study, SNP rs76459466 in SEC24D was significantly associated with HBV infection in both family and case-control samples, which also supports SEC24D as a susceptibility gene for HBV infection. Taken together, the association study and the time course analysis indicated that SEC24D is a potential candidate gene for encouraging HBV infection.
SEC24D is located on chromosome 4q26 and encodes the protein involved in vesicle trafficking that is supposed to affect the HBV infection process32. SEC24D inhibition is involved in enteropathogenic Escherichia coli- and enterohemorrhagic E. coli-induced diseases33. However, there is no report regarding the association between SEC24D and HBV infection. By overexpressing or inhibiting SEC24D expression, we examined whether the amounts of HBV markers changed. By enhancing SEC24D expression, we found that the protein significantly reduced the amounts of HBV-DNA, HBsAg, and HBeAg. Consistently, inhibition of SEC24D by two independent siRNAs produced significantly greater amounts of HBV-DNA, HBsAg, and HBeAg. These data indicate that SEC24D plays a protective role against HBV infection. Furthermore, the expression profiles of WHV models in liver tissue showed decreased expression in infected tissues compared with non-infected tissues, which further supports the antiviral role of SEC24D in HBV-exposed persons. Considering the important role of SEC24D in HBV infection, we then explored the possible biological pathway involved by analyzing expression data of liver tissues from 96 patients with HBV-related HCC (Supplementary Tables 1 and 2). When comparing samples with high SEC24D expression with those with low expression, we found 262 differential genes (false discovery rate [FDR] Q value < 0.005, |fold change| > 1.5) that were significantly changed after SEC24D dysregulation. Interestingly, the following pathway enrichment analysis showed the most significant pathway to be fatty acid degradation. It has been reported that fatty acid biosynthesis is involved in replication of the hepatitis C virus genome, as well as HBV proliferation34. Moreover, saturated fatty acids could inhibit HBV replication mediated by the innate immune response via Toll-like receptor 435. Thus, we speculate that SEC24D inhibits HBV replication through increasing the amount of saturated fatty acid. However, such an antiviral signaling pathway needs to be verified.
Although we identified SEC24D as a novel gene for HBV infection, some of limitations in this study need to be considered. First, we restricted our search to the candidate genes with constantly enhanced expression after HBV infection. However, some genes may be involved only in the early response to HBV, and their expression would not keep increasing at all time points. Thus, more comprehensive studies are needed to uncover new genes. Second, although we selected the top 4000 SNPs for replication from WES analysis of family samples, only 36 SNPs showed nominal association with HBV infection. Such a relatively low replication might be attributable to differences in both techniques (sequencing and array) and samples (family vs. case-control). Third, although we found that SNP rs76459466 in SEC24D played a protective role in HBV infection, we did not investigate the potential biological functions of this SNP. To provide clearer pathogenic insights into HBV infection, the biological functions of this and other SNPs in this gene merit further investigation. In spite of these potential limitations, we integrated genetic association studies, expression data, and in vitro functional assays to minimize false-positive association36.
In conclusion, we first revealed SEC24D as a novel gene crucial for HBV infection by employing an integrated functional genomics strategy. Future studies, such as pathway analysis and SNP functional study, are needed to better define the mechanisms of this gene’s actions in HBV infection.
Declarations
Ethics approval and consent to participate: This project was approved by the Ethical Committee of the First Affiliated Hospital of Zhejiang University School of Medicine. Informed written consent was obtained from every participant.
Data Availability
We downloaded bioinformatics tools and data from public domains with the details given in the Methods section. All materials generated from this project are available upon the request. The WES data has been submitted to NCBI databank with accession code PRJNA553618. The genotyping data is provided in Supplementary Dataset.
References
Schweitzer, A., Horn, J., Mikolajczyk, R. T., Krause, G. & Ott, J. J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386, 1546–1555, https://doi.org/10.1016/S0140-6736(15)61412-X (2015).
Custer, B. et al. Global epidemiology of hepatitis B virus. Journal of clinical gastroenterology 38, S158–168 (2004).
Ott, J. J., Stevens, G. A., Groeger, J. & Wiersma, S. T. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30, 2212–2219, https://doi.org/10.1016/j.vaccine.2011.12.116 (2012).
European Association For The Study Of The, L. EASL clinical practice guidelines. Management of chronic hepatitis B virus infection. Journal of hepatology 57, 167–185, https://doi.org/10.1016/j.jhep.2012.02.010 (2012).
Frodsham, A. J. Host genetics and the outcome of hepatitis B viral infection. Transplant immunology 14, 183–186, https://doi.org/10.1016/j.trim.2005.03.006 (2005).
Ganem, D. & Prince, A. M. Hepatitis B virus infection–natural history and clinical consequences. The New England journal of medicine 350, 1118–1129, https://doi.org/10.1056/NEJMra031087 (2004).
Thursz, M. Genetic susceptibility in chronic viral hepatitis. Antiviral research 52, 113–116 (2001).
Lin, T. M. et al. Hepatitis B virus markers in Chinese twins. Anticancer research 9, 737–741 (1989).
Hu, Z. et al. New loci associated with chronic hepatitis B virus infection in Han Chinese. Nature genetics 45, 1499–1503, https://doi.org/10.1038/ng.2809 (2013).
Kamatani, Y. et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nature genetics 41, 591–595, https://doi.org/10.1038/ng.348 (2009).
Mbarek, H. et al. A genome-wide association study of chronic hepatitis B identified novel risk locus in a Japanese population. Human molecular genetics 20, 3884–3892, https://doi.org/10.1093/hmg/ddr301 (2011).
Jiang, D. K. et al. Genetic variants in five novel loci including CFB and CD40 predispose to chronic hepatitis B. Hepatology 62, 118–128, https://doi.org/10.1002/hep.27794 (2015).
Kim, Y. J. et al. A genome-wide association study identified new variants associated with the risk of chronic hepatitis B. Human molecular genetics 22, 4233–4238, https://doi.org/10.1093/hmg/ddt266 (2013).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753, https://doi.org/10.1038/nature08494 (2009).
Jiang, X. et al. Association of STAT4 polymorphisms with hepatitis B virus infection and clearance in Chinese Han population. Amino acids 48, 2589–2598, https://doi.org/10.1007/s00726-016-2283-3 (2016).
Tao, J. et al. Fine mapping analysis of HLA-DP/DQ gene clusters on chromosome 6 reveals multiple susceptibility loci for HBV infection. Amino acids 47, 2623–2634, https://doi.org/10.1007/s00726-015-2054-6 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43, 491–498, https://doi.org/10.1038/ng.806 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164, https://doi.org/10.1093/nar/gkq603 (2010).
Dennis, G. Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3 (2003).
Wang, H., Kim, S. & Ryu, W. S. DDX3 DEAD-Box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. Journal of virology 83, 5815–5824, https://doi.org/10.1128/JVI.00011-09 (2009).
Schaid, D. J. & Rowland, C. Use of parents, sibs, and unrelated controls for detection of associations between genetic markers and disease. American journal of human genetics 63, 1492–1506, https://doi.org/10.1086/302094 (1998).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575, https://doi.org/10.1086/519795 (2007).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics 38, 904–909, https://doi.org/10.1038/ng1847 (2006).
DerSimonian, R. & Laird, N. Meta-analysis in clinical trials. Control Clin Trials 7, 177–188 (1986).
Higgins, J. P., Thompson, S. G., Deeks, J. J. & Altman, D. G. Measuring inconsistency in meta-analyses. Bmj 327, 557–560, https://doi.org/10.1136/bmj.327.7414.557 (2003).
Simon, R. et al. Analysis of gene expression data using BRB-ArrayTools. Cancer informatics 3, 11–17 (2007).
Krupp, M. et al. RNA-Seq Atlas–a reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics 28, 1184–1185, https://doi.org/10.1093/bioinformatics/bts084 (2012).
Allweiss, L. & Dandri, M. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. Journal of hepatology 64, S17–31, https://doi.org/10.1016/j.jhep.2016.02.012 (2016).
Ott, J., Kamatani, Y. & Lathrop, M. Family-based designs for genome-wide association studies. Nature reviews. Genetics 12, 465–474, https://doi.org/10.1038/nrg2989 (2011).
Xie, N. et al. Comprehensive proteomic analysis of host cell lipid rafts modified by HBV infection. Journal of proteomics 75, 725–739, https://doi.org/10.1016/j.jprot.2011.09.011 (2012).
Thanabalasuriar, A. et al. Sec. 24 interaction is essential for localization and virulence-associated function of the bacterial effector protein NleA. Cellular microbiology 14, 1206–1218, https://doi.org/10.1111/j.1462-5822.2012.01789.x (2012).
Kapadia, S. B. & Chisari, F. V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proceedings of the National Academy of Sciences of the United States of America 102, 2561–2566, https://doi.org/10.1073/pnas.0409834102 (2005).
Zhang, R. N. et al. Saturated Fatty Acid inhibits viral replication in chronic hepatitis B virus infection with nonalcoholic Fatty liver disease by toll-like receptor 4-mediated innate immune response. Hepatitis monthly 15, e27909, https://doi.org/10.5812/hepatmon.15(5)2015.27909 (2015).
Mancuso, N. et al. Integrating Gene Expression with Summary Association Statistics to Identify Genes Associated with 30 Complex Traits. American journal of human genetics 100, 473–487, https://doi.org/10.1016/j.ajhg.2017.01.031 (2017).
Acknowledgements
We acknowledge the invaluable contributions of personal information and blood samples by all participants in the study. We thank Dr. David L. Bronson for excellent editing of this manuscript. This study was in part supported by the China Precision Medicine Initiative (2016YFC0906300), National Science and Technology Major Project (No. 2012ZX10002004) and the Chinese High Technology Research & Development program (No. 2012AA020204), Research Center for Air Pollution and Health of Zhejiang University, and Ministry of Science and Technology of China (2012AA020405).
Author information
Authors and Affiliations
Contributions
X.J., K.S., J.T. and L.L. participated clinical data collection; X.J., B.Z., J.Z., H.H., K.S. and R.F. performed laboratory experiments; X.J., Y.X., K.S. and X.Z. participated data analysis; X.J. and K.S. participated paper writing; M.D.L. and L.L. conceived the study and was involved every step of this study; and all authors approved the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, X., Zhang, B., Zhao, J. et al. Identification and characterization of SEC24D as a susceptibility gene for hepatitis B virus infection. Sci Rep 9, 13425 (2019). https://doi.org/10.1038/s41598-019-49777-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49777-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
