
Abstract
Despite extensive studies suggesting increased susceptibility to HIV during the secretory phase of the menstrual cycle, the molecular mechanisms involved remain unclear. Our goal was to analyze transcriptomes of the endocervix and ectocervix during the proliferative and secretory phases using RNA sequencing to explore potential molecular signatures of susceptibility to HIV. We identified 202 differentially expressed genes (DEGs) between the proliferative and secretory phases of the cycle in the endocervix (adjusted p < 0.05). The biofunctions and pathways analysis of DEGs revealed that cellular assembly and epithelial barrier function in the proliferative phase and inflammatory response/cellular movement in the secretory phase were among the top biofunctions and pathways. The gene set enrichment analysis of ranked DEGs (score = log fold change/p value) in the endocervix and ectocervix revealed that (i) unstimulated/not activated immune cells gene sets positively correlated with the proliferative phase and negatively correlated with the secretory phase in both tissues, (ii) IFNγ and IFNα response gene sets positively correlated with the proliferative phase in the ectocervix, (iii) HIV restrictive Wnt/β-catenin signaling pathway negatively correlated with the secretory phase in the endocervix. Our data show menstrual cycle phase-associated changes in both endocervix and ectocervix, which may modulate susceptibility to HIV.
Similar content being viewed by others

Introduction
Sex hormones control female reproductive tract (FRT) mucosal immune system and cyclical changes in hormone levels are implicated in the “window of vulnerability” theory, suggesting that HIV transmission is more likely to occur within 7–10 days after ovulation during the progesterone (P4) dominating secretory phase of the menstrual cycle rather than during the rest of the cycle1,2,3,4. This theory was supported by SHIV and SIV transmission studies in rhesus and pigtail macaques5,6,7,8,9,10. However, in vitro HIV challenge of human cervical tissues from subjects in the proliferative and secretory phases of the cycle demonstrated contrasting outcomes11,12,13. Our studies utilizing tissues collected over broad periods during the proliferative and secretory phases of the cycle showed similar HIV-1BaL infection level in both phases of the cycle13. Tissue challenge close to ovulation at the peak of estradiol (E2) during the proliferative phase and at the peak of P4 concentrations during the secretory phase could have potentially revealed differences in tissue infection. Our results demonstrating an inverse association between serum E2 concentrations and endocervical and ectocervical tissue infection support this possibility and suggest E2-mediated protection against HIV acquisition13. Overall, these data underlie the need for studies to understand better how susceptibility to HIV is affected by the menstrual cycle in different mucosal areas of the FRT.
Ectocervical, endocervical and vaginal mucosa are distinct anatomical sites, all susceptible to HIV infection, as suggested by SIV transmission studies in rhesus macaques14, but with significant differences in HIV target T cell, macrophage and dendritic cell distributions15,16. Potential biomarkers influencing HIV transmission in the lower FRT have been explored by several groups. CD45+, CD3+, CD4+, CD8+, CCR5+, CXCR4+, HLA-DR+, CD1a+, CD68+ and GalCer+ populations in the ectocervix and/or vaginal tissue were found not to be affected by the menstrual cycle12,15,17,18,19. However, an increase in frequencies of CCR5+ CD4+ T cells in endocervical cytobrush specimens was reported in the luteal phase20.
Analysis of the transcriptome using microarrays demonstrated significant differences in endocervical21, but not in vaginal12, tissue gene expression between the phases of the cycle. The endocervical microarray data revealed genes associated with cell-matrix interactions, amino acid and lipid metabolism, and immune regulation in the follicular phase tissues. The luteal phase was primarily associated with genes involved in chromatin remodeling, inflammation, angiogenesis, oxidative stress and immune cell regulation21. To our knowledge, there is no published data on ectocervical gene expression changes during the menstrual cycle.
We aimed to analyze the ectocervical and endocervical tissue transcriptomes during the proliferative and secretory phases of the cycle using RNA sequencing (RNAseq) to explore potential signatures of susceptibility to HIV. Hysterectomy tissue specimens from subjects not using hormonal contraception/treatment for gynecological conditions were utilized. Several paired ecto- and endocervical specimens were included, minimizing intersubject variability. We chose RNAseq as a high-throughput means with low background signal and large dynamic range22,23.
Differentially expressed genes (DEGs) between proliferative and secretory phases of the cycle were identified in the endocervix and were subjected to the analysis of pathways, molecular networks and biofunctions24. To inquire into changes in gene expression irrespective of p value cut off, full ranked gene lists without any cutoff based on proliferative vs. secretory phase expression were subjected to gene set enrichment analysis (GSEA) against the Hallmark gene sets (H)25 and Immunologic Signatures gene sets (collection C7)26 from the Molecular Signatures Database (MSigDB). These analyses identified menstrual cycle phase-specific changes in both ectocervix and endocervix.
Results
Gene expression in the ecto- and endocervix and DEGs in the proliferative vs. secretory phases of the cycle
The study subjects were categorized into proliferative or secretory phase groups based on the histological assessment of the endometrium (Supplementary Table 1). Serum E2 and P4 concentrations were measured (Supplementary Table 1) and confirmed subjects’ categorization. To present data in unbiased way, an unsupervised analysis using multidimensional scaling (MDS) reduction of gene expression profiles in ectocervical (n = 10) and endocervical (n = 15) tissues was performed and is shown in Fig. 1a.

Ectocervical and endocervical tissue gene expression in the proliferative and secretory phases of the cycle. (a) MDS plot of endocervical and ectocervical gene expression. Top 500 genes were plotted based on expression differences (logFC) between the samples. 1084Q* ectocervical tissue sample clustered close to endocervical tissues and was excluded from subsequent analysis. (b,c) MDS plots of endocervical (b) and ectocervical (c) gene expression in the proliferative and secretory phases. (d,e) Volcano plots of endocervical (d) and ectocervical (e) gene expression in the proliferative vs. secretory phase of the cycle. In red are upregulated DEGs and in green are downregulated DEGs.
Ectocervix and endocervix samples tended to group separately on the MDS plot (Fig. 1a). Also, an intermediate cluster of six samples (1040Y ectocervix, 1068A endocervix, 1069B endocervix, 1092Y ectocervix, 1072E endocervix, 1073F endocervix) was noted. The paired ectocervical/endocervical samples from subjects in this cluster (1040Y, 1068A, 1092Y, 1072E) are located at a distance, pointing to the differences in gene expression between the two mucosal sites within an individual subject. Five of the six samples in the intermediate cluster were from subjects in the proliferative phase.
One ectocervical sample from a subject in the proliferative phase (1084Q) clustered with endocervical samples (Fig. 1a) and was excluded from the subsequent analysis. Based on routine pre-operative clinical cancer screening, this subject had positive HPV testing (Supplementary Table 1) and HPV infection was previously reported to be associated with altered gene expression in cervical epithelium27,28.
Phase-based gene expression profiles in the endocervix and ectocervix were mixed, with higher degree of separation in the endocervix (Fig. 1b,c), likely due to larger sample size in this group.
202 DEGs (adjusted p values < 0.05; 102 upregulated and 100 downregulated) were identified in the endocervix based on comparison of n = 8 vs. n = 7 samples from subjects in the proliferative and secretory phases, respectively (Supplementary Tables 2, 3). In the ectocervix, no DEGs were identified comparing n = 5 proliferative and n = 4 secretory phase samples. The volcano plots of gene expression in the proliferative vs. secretory phase according to the fold change and p values depict DEGs in the endocervix and lack of DEGs in the ectocervix (Fig. 1d,e).
DEGs in the proliferative vs. secretory phases of the cycle in the endocervix
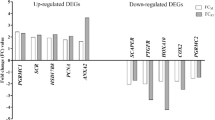
The upregulated DEGs in the proliferative vs. secretory endocervix ranked by fold change are listed the Supplementary Table 2. The complete data can be found in the NCBI’s Gene Expression Omnibus database (Accession Number:GSE122248)29. FOSL1, a regulator of cell proliferation and differentiation, which is induced by estrogen30, was the most upregulated gene in the proliferative endocervix. Consistent with regulatory role of estrogen in lipid metabolism31,32, several genes involved in lipid metabolism (FADS1, SREBF2, PI4K2A, LDLR)33,34,35,36 were upregulated. Expression of Cathepsin G was increased in accord with data demonstrating induction of serine proteases by estrogen37. Cyclin Y family member CCNYL1 enhancing Wnt/β-catenin signaling in mitosis38, was also induced. Several genes involved in epithelial cell barrier function, microtubule cytoskeleton organization, cell division, microtubule structure/DNA stability, actin dynamics and cell motility, structure and intracellular signaling (TUBB2A, TUBA1B, TUBB, TUBA1C, CFL1, CDC42, Actin β (ACTB), MYL6)39,40,41,42,43 were upregulated.
The downregulated DEGs in the proliferative vs. secretory endocervix (accordingly representing upregulated genes in the secretory phase) ranked by fold change are listed in the Supplementary Table 3. The most downregulated gene in the proliferative phase was a serine proteinase inhibitor SERPINA5. Mucosal serpins were previously reported to be differentially regulated during the menstrual cycle, with some increasing in the proliferative phase (Serpin A1) and some (Serpin A3) remaining similar between the phases of the cycle44. The downregulated DEGs in the proliferative phase included genes associated with inflammatory pathways such as PLA2 group 6 gene (PLA2G6). PLA2 promotes inflammation by catalyzing arachidonic acid pathway and regulates speed and directionality of monocytes’ chemotaxis45. ENPP3, a ectonucleotide pyrophosphatase/phosphodiesterase 3, which is expressed on basophils and mast cells46 and a marker of cell activation, was downregulated. ENPP3 expression in uterus is suggested to be regulated by P4 and is increased during midsecretory phase when P4 is high47. ADHFE1, an enzyme mediating oxidative stress48, was downregulated. Also, a decrease in expression of broad range of nucleic acid binding zinc finger proteins was detected.
Biological processes, pathways and molecular networks in the proliferative and secretory phase of the cycle in the endocervix
Biofunctions and pathways analysis using all DEGs
The analysis revealed that the top two molecular and cellular functions associated with the DEGs in the proliferative phase in the endocervix were (i) DNA replication, recombination and repair (p = 1.86E-03-5.83E-05) and (ii) cell death and survival (p = 7.66E-03-8.35E-05). The canonical pathway analysis revealed 32 pathways that were significantly changed between the phases of the cycle (Fig. 2a and Supplementary Table 4). Many of the pathways belonged to the categories representing cytoskeleton remodeling, cell and organ development.

Canonical pathways associated with the proliferative or secretory phases of the cycle in endocervix. (a) Pathways significantly changed in the proliferative vs. secretory phases. The y-axis displays the −log (p value) which was calculated by right-tailed Fisher’s exact test. The default cut off -log (p value) of 1.3 was applied. (−) z scores indicate down-regulation in the proliferative endocervix and (+) z scores indicate upregulation of the pathways in the proliferative endocervix. Some z scores were unavailable or unpredicted as the eligibility criteria of z score algorithm were not met. Ratio denotes the number of significantly changed genes compared with the total number of genes within the pathway. (b,c) Pathways identified by ORA using upregulated sets of DEGs in the proliferative (b) and secretory (c) phases.
In the proliferative endocervix, the analysis predicted activation state of pathways involving selected Rho family GTPases linked with multiple aspects of cellular regulation including the actin system, cellular morphology, migration, gene transcription, the cell cycle and phagocytosis49. Rho A and Cdc42 signaling was induced and the negative regulator of Rho family (RhoGDI) signaling was inhibited (Fig. 2a). Expectedly, regulation of actin-based motility by Rho and actin cytoskeleton signaling pathways, Fcγ receptor-mediated phagocytosis in macrophages and monocytes and ILK signaling were also activated. Several pathways were significantly different between the phases of the cycle, however, the z scores indicating activation state for these pathways were either unavailable or unpredicted as the eligibility criteria of z score algorithm were not met (Fig. 2a). These pathways were linked to epithelial barrier function (remodeling of epithelial adherens junctions, epithelial adherens junction signaling, gap junction and tight junction signaling), metabolism (guanine and guanosine salvage I, PRPP biosynthesis I, pyrimidine ribonucleotides interconversion, spermidine and putrescin synthesis, tRNA charging, Rapoport-Luebering glycolytic shunt, ethanol degradation II, non-adrenaline and adrenalin degradation), immune regulation (phagosome maturation, clathrin-mediated endocytosis, TWEAK signaling, role of PKR in IFN induction and antiviral response), cell cycle/DNA repair/cell growth (14-3-3-mediated signaling, TWEAK signaling, PAK signaling).
Pathway overrepresentation analysis (ORA) using overabundant gene sets
Next, we explored ORA50 using overabundant gene sets as an alternative strategy to identify pathways associated with the proliferative or secretory phases. In agreement with analysis of all DEGs, ORA identified pathways involved in epithelial barrier function (remodeling of epithelial adherens junctions (p = 9.3E-10), epithelial adherens junction signaling (p = 3.51E-07) associated with the proliferative phase (Fig. 2b). In the secretory phase, metabolic pathways were overrepresented (Fig. 2c).
Network and biofunctions analysis using overabundant gene sets
To explore significant biological functions and to determine non-directional relationships among the upregulated DEGs in the proliferative and secretory phases, the network and biofunctions analysis tool on overabundant gene sets was employed. Consistent with the analyses above, lipid, mineral and vitamin metabolism, cellular assembly and organization biofunctions were enhanced in the proliferative endocervix (Table 1). Cell cycle, cell signaling, inflammatory response and cellular movement were enhanced in the secretory endocervix (Table 1).
GSEA
To incorporate information on all genes expression without introducing p value cut off, endocervix and ectocervix limma outputs genes were ranked (score = log FC/p value) and then used as input to run against the Hallmark gene sets (H) (n = 50)25 and Immunologic Signatures gene sets (collection C7)(n = 4872)26 of the GSEA MSigDB. The top gene sets, which positively or negatively correlated with a particular phase of the cycle in the endo- and ectocervix are shown in Tables 2, 3 and Supplementary Figures 1, 2.
The Hallmark database analysis detected positive correlation of growth and proliferation related gene sets (mTOR1 signaling, oxidative phosphorylation, MYC targets V1 and epithelial-mesenchymal transition)51,52,53,54 and TNFα signaling with the proliferative phase in the endocervix. In the secretory endocervix, negative correlation with Wnt/β-catenin signaling, was revealed. Similar to the results in the endocervix, cell growth related gene sets (MYC Targets V1, G2M Checkpoint and E2F target)55,56,57,58 positively correlated with proliferative phase in the ectocervix. Also, a positive correlation between gene sets induced in response to IFNγ and IFNα and the proliferative phase in ectocervix was noted. In contrast, negative correlation between several cell proliferation-related gene sets (KRAS Signaling, angiogenesis and epithelial-mesenchymal transition)59,60,61 and the secretory phase in the ectocervix was detected.
The Immunologic Signatures database analysis revealed positive correlations between (i) unstimulated NK cells, uninfected/resting neutrophils gene sets and the proliferative phase in the endocervix, (ii) resting/untreated CD4 T cells gene sets and the proliferative phase in ectocervix. Negative correlations were observed between (i) unstimulated immune cells (DCs, NK cells) gene sets and the secretory phase in endocervix, and (ii) unstimulated/untreated DCs and CD4 T cells, CD56+brightCD62L+ NK cells and the secretory phase in the ectocervix.
Discussion
To our knowledge this is the first report to explore ectocervical and endocervical tissue transcriptomes relative to the menstrual cycle phases using RNAseq. The conducted analyses of DEGs in the endocervix and full ranked data analysis in the endocervix and ectocervix revealed complementary hypothesis-generating information on how susceptibility to HIV may be regulated during the menstrual cycle.
The unbiased analysis of gene expression pattern between ectocervix and endocervix and within each tissue during the proliferative and secretory phases of the cycle showed variable separation between the groups. RNAseq capability to detect weakly expressed genes more efficiently than other platforms62,63 and small sample size could have contributed to the observed gene expression pattern on the MDS plots. Studies involving larger sample size will help characterize better tissue-associated and phase-associated gene expression patterns in the ectocervix and endocervix.
The functional annotation of DEGs identified between the proliferative and secretory phase of the cycle in the endocervix is largely in agreement with findings by microarray21. However, RNAseq identified more DEGs than reported by the microarray (n = 110). Based on the manual comparison, only six DEGs (ECHDC2, PNPLA7, DRAP1, FKBP1A, IGFBP4, ACTB) overlapped between the studies. Similar with the microarray data, we did not see any of the mucin genes among the DEGs.
Among identified DEGs, literature review revealed several genes influencing HIV pathogenesis through protein-protein interactions. This includes genes which regulate HIV infectivity (EHD4, AP2M1)64,65,66, entry/assembly/budding (INSIG1, PI4K2A)67,68, proteasomal degradation (LSP1)69, HIV transcription/latency (TFAP4, MED27, FOSL1, PSMC3, PSMD11, L3MBTL1)70,71,72,73,74,75, viral transport (ALYREF, MAPRE1 (EB1))76,77, and actin dynamic involved in multiple steps during HIV life cycle (CFL1)78,79. Majority of these genes were upregulated and TFAP4, L3MBTL1 were downregulated in the proliferative phase. No phase-associated HIV enhancing or inhibitory pattern of these genes was evident.
In the proliferative endocervix, pathways involved in barrier function maintenance (remodeling of epithelial adherens junctions, epithelial adherence junction signaling) were overrepresented possibly indicating effective epithelial barrier, that can mediate protection against HIV acquisition80. Several pathways cascaded to activation of cytoskeleton development (Rho A and actin cytoskeleton signaling) and immune responses (Fcγ receptor-mediated phagocytosis in macrophages and monocytes, ILK signaling). Growth and proliferation-related gene sets, TNFα signaling, unstimulated immune cells gene sets (neutrophils, NK cells) positively correlated with the proliferative phase. Unstimulated immune cells gene signatures point to HIV non-permissive environment. Resting neutrophils (vs. activated) bind HIV less effectively and are less efficient at HIV transfer to lymphocytes81. Presence of resting NK cells is an indicator of non-inflammatory environment in the mucosa during the proliferative phase, as NK cells are activated by proinflammatory cytokines82. Downregulation of SERPINA5 in the proliferative phase deserves further exploration as a balance between serine proteases and their inhibitors was suggested to influence susceptibility to HIV83.
In the secretory endocervix, enhanced expression of genes related to inflammation, oxidative stress, and immune cell migration, potentially contributing to increased susceptibility to HIV84,85, was evident. Unstimulated/resting DCs and NK cells gene sets negatively correlated with the secretory phase. Furthermore, the E2-induced restrictive for HIV replication Wnt/β-catenin signaling pathway86 gene set also negatively correlated with the secretory phase.
The analysis of the ectocervix did not reveal DEGs between the follicular and secretory phases of the cycle. These data concur with the microarray results obtained in the vaginal mucosa, showing no difference in vaginal gene expression (greater than two fold) between the follicular and luteal phases12 with an exception of one gene (HAL), which was significantly upregulated in the luteal phase. Similar to data in the proliferative endocervix, the full ranked data analysis revealed positive correlation between cell growth-associated gene sets, not-activated CD4 T cells gene sets and proliferative ectocervix. IFNα and IFNγ induced gene sets also positively correlated with the proliferative phase in ectocervix. Vaginal administration of Type I IFN (IFNβ) was shown to prevent SHIV infection in macaques87 and blockade of IFN-I receptor led to reduced anti-viral gene expression, increased SIV reservoir size and accelerated CD4 depletion with progression to AIDS88. However, it needs to be noted that the role of IFNs in HIV transmission and pathogenesis is complex89,90.
The secretory phase of the cycle in ectocervix negatively correlated with unstimulated DCs; resting CD4 T cells, which are less susceptible to HIV than activated CD4 T cells91; and CD56+brightCD62L+ NK cells, which polyfunctionality potentially relates to resistance to HIV-1 infection92. These data are indicative of higher susceptibility to HIV during the secretory phase in the ectocervix.
It needs to be acknowledged that although next generation sequencing technologies have advanced sequence-based research with the advantages of high-throughput, high-sensitivity, and high-speed, RNAseq analysis harbors limitations22,23. Messenger RNA concentrations do not necessarily reflect protein concentrations, which are influenced by transcription and translation rates and protein half-life. However, RNAseq transcriptome data can be used as a template for generation of proteomics database93.
Our study corroborates and expands the microarray data in the endocervix21 and is concurrent with cervico-vaginal lavage fluids proteome data, demonstrating dominance of pathways associated with inflammation, including chemotaxis and recruitment of leucocytes in the secretory phase94. The study offers insights into how susceptibility to HIV may be regulated during the menstrual cycle in the endocervix and ectocervix. Overall, competent epithelial barrier function, cytoskeletal organization, phagocytosis in monocytes/macrophages, immune responses, lack of inflammation and cellular activation (NK cells, neutrophils) may render endocervix resistant to infection with HIV during the proliferative phase. In contrast, in the secretory endocervix, compromised epithelial barrier function, inflammation, influx of leucocytes, cellular activation (NK cells, DCs), oxidative stress and decreased HIV-restrictive Wnt/β-catenin signaling may lead to enhanced susceptibility to HIV. Similarly, in the ectocervix, lack of cellular activation (T cells) and IFN γ and IFNα-induced responses potentially contribute to protection against HIV during the proliferative phase and cellular activation (T cells, DC), loss of CD56+bright CD62L+ NK cells may lead to HIV permissive environment in the secretory phase.
Our study revealed gene expression signatures characteristic of menstrual cycle phase in the endocervical and ectocervical mucosa. This data may contribute to better understanding of susceptibility to mucosal HIV infection and other sexually transmitted infections as well as to development of prevention strategies.
Materials and Methods
Subjects
The project was approved by the Icahn School of Medicine at Mount Sinai Program for the Protection of Human Subjects (protocol #11-01380) and The Population Council IRB. The research was performed in accordance with relevant guidelines and regulations. Cervical tissues, blood and urine were obtained from women undergoing hysterectomies for non-malignant conditions (menometrorrhagia, leiomyomas, chronic pelvic pain, and pelvic organ prolapse) at Mount Sinai Hospital, the primary teaching hospital of the medical school. Subjects were enrolled after providing written informed consent. This is a sub-analysis from the 16 subjects who did not use either (i) hormonal contraception and/or (ii) any hormonal treatments for gynecological conditions within the 3 months prior to surgery. Age, race, phase of the cycle, histopathology report [cervical inflammation, metaplasia (for endocervical mucosa), parakeratosis (for ectocervical mucosa)], HSV-2 status and Pap cytology and HPV co-testing results of subjects included in individual data sets are summarized in Supplementary Table 1. The cycle phase was determined by assessing the histopathology of hematoxylin-and-eosin stained sections of the endometrial mucosa by board-certified diagnostic gynecologic pathologists who perform this assessment routinely. Cervical inflammation was defined as the presence of white blood cells within the cervical tissue (epithelium and stroma of the mucosa).
Surgical tissue collection
Cervical tissues not used for clinical diagnosis which would otherwise be discarded were collected and released by pathologists within 1-2 hours (hrs) of surgery. Endocervix and ectocervix, excluding transformation zone, were utilized. Tissue samples were transported to the laboratory in RPMI on ice and a portion of mucosa (~3–5 mm) was placed in RNAlater (Ambion) and then stored at −80 °C before RNA isolation.
Radioimmunoassay (RIA)
Serum was kept at −80 °C before RIA. Hormone concentrations were determined using ImmuChem Double antibody 125I RIA kits for E2 (LLOQ 10 pg/ml) and for P4 (LLOQ 200 pg/ml) (MP Biomedicals, LLC, NY, USA).
HSV-2 IgG AB Herpeselect test
Serum was kept at −80 °C before being assayed by Quest Diagnostics.
Total RNA Extraction, Library Preparation & RNA Sequencing
Total RNA was isolated from tissues frozen in RNAlater (Ambion) following manufacturer’s instructions (Qiagen RNeasy Fibrous Tissue Mini Kit). After extraction, the quality and the purity of the RNA were measured by the Agilent Bioanalyzer (Agilent, Santa Clara, CA). RNA was labeled and sequenced at the Rockefeller University Genomics center by using Illumina TruSeq technology (75 bp, > 30 M coverage). The FASTQ files were first quality controlled through FastQC v0.11.1595 with default parameters. Cutadapt v1.9.1 was used to locate and remove the adapter sequences from each high-throughput sequencing reads before mapping96 as applied to trim the low quality bases and TrueSeq adapters (–times = 2–quality-base = 33 —quality-cutoff = 5–format = FASQT–minimum-length = 25 -a AAAAAAAAAAAAA TTTTTTTTTTTTT -a AGATCGGAAGAG -a CTCTTCCGATCT). Trimmed FASQT files were aligned to the human genome (GRCH37) using STAR v2.4.297 aligner with default parameters. The alignment results were then evaluated through Qualimap v.2.2 (https://doi.org/10.1093/bioinformatics/bts503)98 to ensure that all the samples had a consistent coverage, alignment rate, and no obvious 5′ or 3′ bias. Aligned reads were then summarized through feature Counts v1.599. The gene model used for this purpose was that from Ensembl at gene level. The uniquely mapped reads (NH ‘tag’ in bam file) that overlapped with an exon (feature) by at least 1 bp were counted and then the counts of all exons annotated to an Ensembl gene (meta features) and were summed into a single number. edgeR* v 3.16.5100 was used to normalize the samples and Voom from limma# v 3.30.11 was applied to estimate the differential log fold change in the expression of genes. An adjusted p value of less than 0.05 (p adj. < 0.05) was used to shortlist the genes that have a significant expression change.
RNAseq data accession number
Raw sequence reads were uploaded to the Gene Expression Omnibus (GEO) Database (GEO Series Accession Number: GSE122248).
Analysis of DEGs
MDS plots
MDS plots were prepared through plot MDS function within edgeR (https://doi.org/10.1093/bioinformatics/btp616), which is a widely accepted RNAseq analytical package. It plots samples on a two-dimensional scatterplot so that distances on the plot approximate the expression differences (logFC) between the samples. Top 500 genes were plotted, which is the default value from the package.
Ingenuity pathway analysis (IPA)
To identify relevant cellular pathways differentially expressed between the proliferative and secretory phases of the cycle, list of DEGs containing gene identifiers, corresponding p values and log2 FC were subjected to the the IPA24 core analysis (Ingenuity Systems®) using default settings and uploading the DEGs, corresponding fold change expression values and FDR adjusted p values (aka q values)(<0.05). The analysis derives its results from the Ingenuity Knowledge Database, which is manually curated from published data. The canonical pathway component of the core analysis converted gene expression data into pathway illustrations and identified the top canonical pathways associated with the genes differentially expressed between two phases. −(log) p -value were calculated using the right-tailed Fisher’s Exact Test. A cut-off of 1.3 was applied to display only significantly changed pathways.
For ORA, we performed separate IPA core analysis with upregulated DEG datasets in each phase. Top gene networks associated with each phase were generated by default algorithm, which randomly selects focus genes from the uploaded list and draws connections on the basis of biological function from its knowledge database. The analysis also indicates the relevance of molecules to its assigned network.
GSEA
Rank list file was created from limma output after proliferative vs. secretory phase gene expression comparison in the endocervix and ectocervix with each gene ranked by logFC/p value. Then the rank lists were used as an input of GSEA and analyzed through “Run GSEA on a Pre-Ranked” gene list option against gene sets databases (Hallmark gene sets25 and Immunologic Signatures gene sets26).
References
Rodriguez-Garcia, M., Patel, M. V. & Wira, C. R. Innate and adaptive anti-HIV immune responses in the female reproductive tract. Journal of reproductive immunology 97, 74–84, https://doi.org/10.1016/j.jri.2012.10.010 (2013).
Wira, C. R. & Fahey, J. V. A new strategy to understand how HIV infects women: identification of a window of vulnerability during the menstrual cycle. Aids 22, 1909–1917, https://doi.org/10.1097/QAD.0b013e3283060ea4 (2008).
Wira, C. R. et al. Sex hormone regulation of innate immunity in the female reproductive tract: the role of epithelial cells in balancing reproductive potential with protection against sexually transmitted pathogens. American journal of reproductive immunology 63, 544–565, https://doi.org/10.1111/j.1600-0897.2010.00842.x (2010).
Wira, C. R., Rodriguez-Garcia, M. & Patel, M. V. The role of sex hormones in immune protection of the female reproductive tract. Nature reviews. Immunology 15, 217–230, https://doi.org/10.1038/nri3819 (2015).
Sodora, D. L., Gettie, A., Miller, C. J. & Marx, P. A. Vaginal transmission of SIV: assessing infectivity and hormonal influences in macaques inoculated with cell-free and cell-associated viral stocks. AIDS research and human retroviruses 14(Suppl 1), S119–123 (1998).
Morris, M. R. et al. Relationship of menstrual cycle and vaginal infection in female rhesus macaques challenged with repeated, low doses of SIVmac251. Journal of medical primatology 44, 301–305, https://doi.org/10.1111/jmp.12177 (2015).
Kenney, J. et al. Short communication: a repeated simian human immunodeficiency virus reverse transcriptase/herpes simplex virus type 2 cochallenge macaque model for the evaluation of microbicides. AIDS research and human retroviruses 30, 1117–1124, https://doi.org/10.1089/aid.2014.0207 (2014).
Vishwanathan, S. A. et al. High susceptibility to repeated, low-dose, vaginal SHIV exposure late in the luteal phase of the menstrual cycle of pigtail macaques. Journal of acquired immune deficiency syndromes 57, 261–264, https://doi.org/10.1097/QAI.0b013e318220ebd3 (2011).
Kersh, E. N. et al. SHIV susceptibility changes during the menstrual cycle of pigtail macaques. Journal of medical primatology 43, 310–316, https://doi.org/10.1111/jmp.12124 (2014).
Derby, N. et al. An intravaginal ring that releases three antiviral agents and a contraceptive blocks SHIV-RT infection, reduces HSV-2 shedding, and suppresses hormonal cycling in rhesus macaques. Drug Deliv Transl Res 7, 840–858, https://doi.org/10.1007/s13346-017-0389-0 (2017).
Saba, E. et al. Productive HIV-1 infection of human cervical tissue ex vivo is associated with the secretory phase of the menstrual cycle. Mucosal immunology 6, 1081–1090, https://doi.org/10.1038/mi.2013.2 (2013).
Thurman, A. R. et al. Comparison of Follicular and Luteal Phase Mucosal Markers of HIV Susceptibility in Healthy Women. AIDS research and human retroviruses 32, 547–560, https://doi.org/10.1089/AID.2015.0264 (2016).
Calenda, G. et al. Mucosal Susceptibility to Human Immunodeficiency Virus Infection in the Proliferative and Secretory Phases of the Menstrual Cycle. AIDS research and human retroviruses 35, 335–347, https://doi.org/10.1089/AID.2018.0154 (2019).
Stieh, D. J. et al. Vaginal challenge with an SIV-based dual reporter system reveals that infection can occur throughout the upper and lower female reproductive tract. PLoS pathogens 10, e1004440, https://doi.org/10.1371/journal.ppat.1004440 (2014).
Pudney, J., Quayle, A. J. & Anderson, D. J. Immunological microenvironments in the human vagina and cervix: mediators of cellular immunity are concentrated in the cervical transformation zone. Biology of reproduction 73, 1253–1263, https://doi.org/10.1095/biolreprod.105.043133 (2005).
Lee, S. K., Kim, C. J., Kim, D. J. & Kang, J. H. Immune cells in the female reproductive tract. Immune Netw 15, 16–26, https://doi.org/10.4110/in.2015.15.1.16 (2015).
Chandra, N. et al. Depot medroxyprogesterone acetate increases immune cell numbers and activation markers in human vaginal mucosal tissues. AIDS research and human retroviruses 29, 592–601, https://doi.org/10.1089/aid.2012.0271 (2013).
Yeaman, G. R. et al. Chemokine receptor expression in the human ectocervix: implications for infection by the human immunodeficiency virus-type I. Immunology 113, 524–533, https://doi.org/10.1111/j.1365-2567.2004.01990.x (2004).
Poppe, W. A., Drijkoningen, M., Ide, P. S., Lauweryns, J. M. & Van Assche, F. A. Lymphocytes and dendritic cells in the normal uterine cervix. An immunohistochemical study. European journal of obstetrics, gynecology, and reproductive biology 81, 277–282 (1998).
Byrne, E. H. et al. Association between injectable progestin-only contraceptives and HIV acquisition and HIV target cell frequency in the female genital tract in South African women: a prospective cohort study. The Lancet. Infectious diseases 16, 441–448, https://doi.org/10.1016/S1473-3099(15)00429-6 (2016).
Yildiz-Arslan, S., Coon, J. S., Hope, T. J. & Kim, J. J. Transcriptional Profiling of Human Endocervical Tissues Reveals Distinct Gene Expression in the Follicular and Luteal Phases of the Menstrual Cycle. Biol Reprod 94, 138, https://doi.org/10.1095/biolreprod.116.140327 (2016).
Ozsolak, F. & Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nature reviews. Genetics 12, 87–98, https://doi.org/10.1038/nrg2934 (2011).
Han, Y., Gao, S., Muegge, K., Zhang, W. & Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinformatics and biology insights 9, 29–46, https://doi.org/10.4137/BBI.S28991 (2015).
Kramer, A., Green, J., Pollard, J. Jr. & Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530, https://doi.org/10.1093/bioinformatics/btt703 (2014).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems 1, 417–425, https://doi.org/10.1016/j.cels.2015.12.004 (2015).
Godec, J. et al. Compendium of Immune Signatures Identifies Conserved and Species-Specific Biology in Response to Inflammation. Immunity 44, 194–206, https://doi.org/10.1016/j.immuni.2015.12.006 (2016).
Kang, S. D. et al. Effect of Productive Human Papillomavirus 16 Infection on Global Gene Expression in Cervical Epithelium. Journal of virology 92, https://doi.org/10.1128/JVI.01261-18 (2018).
Karim, R. et al. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PloS one 6, e17848, https://doi.org/10.1371/journal.pone.0017848 (2011).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210 (2002).
O’Donnell, A. J., Macleod, K. G., Burns, D. J., Smyth, J. F. & Langdon, S. P. Estrogen receptor-alpha mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocrine-related cancer 12, 851–866, https://doi.org/10.1677/erc.1.01039 (2005).
Gonzalez-Bengtsson, A., Asadi, A., Gao, H., Dahlman-Wright, K. & Jacobsson, A. Estrogen Enhances the Expression of the Polyunsaturated Fatty Acid Elongase Elovl2 via ERalpha in Breast Cancer Cells. PloS one 11, e0164241, https://doi.org/10.1371/journal.pone.0164241 (2016).
Knopp, R. H. & Zhu, X. Multiple beneficial effects of estrogen on lipoprotein metabolism. The Journal of clinical endocrinology and metabolism 82, 3952–3954, https://doi.org/10.1210/jcem.82.12.4472 (1997).
Bommer, G. T. & MacDougald, O. A. Regulation of lipid homeostasis by the bifunctional SREBF2-miR33a locus. Cell metabolism 13, 241–247, https://doi.org/10.1016/j.cmet.2011.02.004 (2011).
Wang, L. et al. Fatty acid desaturase 1 gene polymorphisms control human hepatic lipid composition. Hepatology 61, 119–128, https://doi.org/10.1002/hep.27373 (2015).
Balla, T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiological reviews 93, 1019–1137, https://doi.org/10.1152/physrev.00028.2012 (2013).
Go, G. W. & Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. The Yale journal of biology and medicine 85, 19–28 (2012).
Dai, R. et al. Neutrophils and neutrophil serine proteases are increased in the spleens of estrogen-treated C57BL/6 mice and several strains of spontaneous lupus-prone mice. PloS one 12, e0172105, https://doi.org/10.1371/journal.pone.0172105 (2017).
Zeng, L. et al. Essential Roles of Cyclin Y-Like 1 and Cyclin Y in Dividing Wnt-Responsive Mammary Stem/Progenitor Cells. PLoS genetics 12, e1006055, https://doi.org/10.1371/journal.pgen.1006055 (2016).
Watson, J. R., Owen, D. & Mott, H. R. Cdc42 in actin dynamics: An ordered pathway governed by complex equilibria and directional effector handover. Small GTPases 8, 237–244, https://doi.org/10.1080/21541248.2016.1215657 (2017).
Janke, C. The tubulin code: molecular components, readout mechanisms, and functions. The Journal of cell biology 206, 461–472, https://doi.org/10.1083/jcb.201406055 (2014).
Perrin, B. J. & Ervasti, J. M. The actin gene family: function follows isoform. Cytoskeleton 67, 630–634, https://doi.org/10.1002/cm.20475 (2010).
Heissler, S. M. & Sellers, J. R. Myosin light chains: Teaching old dogs new tricks. Bioarchitecture 4, 169–188, https://doi.org/10.1080/19490992.2015.1054092 (2014).
Bravo-Cordero, J. J., Magalhaes, M. A., Eddy, R. J., Hodgson, L. & Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nature reviews. Molecular cell biology 14, 405–415, https://doi.org/10.1038/nrm3609 (2013).
Rahman, S. et al. Mucosal serpin A1 and A3 levels in HIV highly exposed sero-negative women are affected by the menstrual cycle and hormonal contraceptives but are independent of epidemiological confounders. American journal of reproductive immunology 69, 64–72, https://doi.org/10.1111/aji.12014 (2013).
Carnevale, K. A. & Cathcart, M. K. Calcium-independent phospholipase A(2) is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. Journal of immunology 167, 3414–3421 (2001).
Buhring, H. J., Streble, A. & Valent, P. The basophil-specific ectoenzyme E-NPP3 (CD203c) as a marker for cell activation and allergy diagnosis. International archives of allergy and immunology 133, 317–329, https://doi.org/10.1159/000077351 (2004).
Boggavarapu, N. R. et al. Compartmentalized gene expression profiling of receptive endometrium reveals progesterone regulated ENPP3 is differentially expressed and secreted in glycosylated form. Scientific reports 6, 33811, https://doi.org/10.1038/srep33811 (2016).
Mishra, P. et al. ADHFE1 is a breast cancer oncogene and induces metabolic reprogramming. The Journal of clinical investigation 128, 323–340, https://doi.org/10.1172/JCI93815 (2018).
Hall, A. Rho GTPases and the actin cytoskeleton. Science 279, 509–514 (1998).
Hong, G., Zhang, W., Li, H., Shen, X. & Guo, Z. Separate enrichment analysis of pathways for up- and downregulated genes. Journal of the Royal Society, Interface 11, 20130950, https://doi.org/10.1098/rsif.2013.0950 (2014).
Saxton, R. A. & Sabatini, D. M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371, https://doi.org/10.1016/j.cell.2017.03.035 (2017).
Wilson, D. F. Oxidative phosphorylation: regulation and role in cellular and tissue metabolism. The Journal of physiology 595, 7023–7038, https://doi.org/10.1113/JP273839 (2017).
Zajac-Kaye, M. Myc oncogene: a key component in cell cycle regulation and its implication for lung cancer. Lung cancer 34(Suppl 2), S43–46 (2001).
Micalizzi, D. S. & Ford, H. L. Epithelial-mesenchymal transition in development and cancer. Future oncology 5, 1129–1143, https://doi.org/10.2217/fon.09.94 (2009).
Barnum, K. J. & O’Connell, M. J. Cell cycle regulation by checkpoints. Methods in molecular biology 1170, 29–40, https://doi.org/10.1007/978-1-4939-0888-2_2 (2014).
Ren, B. et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes &. development 16, 245–256, https://doi.org/10.1101/gad.949802 (2002).
Thurlings, I. & de Bruin, A. E2F Transcription Factors Control the Roller Coaster Ride of Cell Cycle Gene Expression. Methods in molecular biology 1342, 71–88, https://doi.org/10.1007/978-1-4939-2957-3_4 (2016).
Dang, C. V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Molecular and cellular biology 19, 1–11, https://doi.org/10.1128/mcb.19.1.1 (1999).
Jancik, S., Drabek, J., Radzioch, D. & Hajduch, M. Clinical relevance of KRAS in human cancers. Journal of biomedicine & biotechnology 2010, 150960, https://doi.org/10.1155/2010/150960 (2010).
Tahergorabi, Z. & Khazaei, M. A review on angiogenesis and its assays. Iranian journal of basic medical sciences 15, 1110–1126 (2012).
Micalizzi, D. S., Farabaugh, S. M. & Ford, H. L. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. Journal of mammary gland biology and neoplasia 15, 117–134, https://doi.org/10.1007/s10911-010-9178-9 (2010).
Wang, C. et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nature biotechnology 32, 926–932, https://doi.org/10.1038/nbt.3001 (2014).
Zhao, S., Fung-Leung, W. P., Bittner, A., Ngo, K. & Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PloS one 9, e78644, https://doi.org/10.1371/journal.pone.0078644 (2014).
Bregnard, C. et al. Comparative proteomic analysis of HIV-1 particles reveals a role for Ezrin and EHD4 in the Nef-dependent increase of virus infectivity. Journal of virology 87, 3729–3740, https://doi.org/10.1128/JVI.02477-12 (2013).
Lubben, N. B. et al. HIV-1 Nef-induced down-regulation of MHC class I requires AP-1 and clathrin but not PACS-1 and is impeded by AP-2. Molecular biology of the cell 18, 3351–3365, https://doi.org/10.1091/mbc.e07-03-0218 (2007).
Craig, H. M., Reddy, T. R., Riggs, N. L., Dao, P. P. & Guatelli, J. C. Interactions of HIV-1 nef with the mu subunits of adaptor protein complexes 1, 2, and 3: role of the dileucine-based sorting motif. Virology 271, 9–17, https://doi.org/10.1006/viro.2000.0277 (2000).
van ‘t Wout, A. B. et al. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1-infected T cells. Journal of virology 79, 10053–10058, https://doi.org/10.1128/JVI.79.15.10053-10058.2005 (2005).
Gerber, P. P. et al. Rab27a controls HIV-1 assembly by regulating plasma membrane levels of phosphatidylinositol 4,5-bisphosphate. The Journal of cell biology 209, 435–452, https://doi.org/10.1083/jcb.201409082 (2015).
Smith, A. L. et al. Leukocyte-specific protein 1 interacts with DC-SIGN and mediates transport of HIV to the proteasome in dendritic cells. The Journal of experimental medicine 204, 421–430, https://doi.org/10.1084/jem.20061604 (2007).
Apcher, G. S. et al. Human immunodeficiency virus-1 Tat protein interacts with distinct proteasomal alpha and beta subunits. FEBS letters 553, 200–204 (2003).
Imai, K. & Okamoto, T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. The Journal of biological chemistry 281, 12495–12505, https://doi.org/10.1074/jbc.M511773200 (2006).
Imbeault, M., Giguere, K., Ouellet, M. & Tremblay, M. J. Exon level transcriptomic profiling of HIV-1-infected CD4(+) T cells reveals virus-induced genes and host environment favorable for viral replication. PLoS pathogens 8, e1002861, https://doi.org/10.1371/journal.ppat.1002861 (2012).
Ruiz, A. et al. Characterization of the influence of mediator complex in HIV-1 transcription. The Journal of biological chemistry 289, 27665–27676, https://doi.org/10.1074/jbc.M114.570341 (2014).
Li, J. et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nature communications 7, 11730, https://doi.org/10.1038/ncomms11730 (2016).
Boehm, D. et al. SMYD2-Mediated Histone Methylation Contributes to HIV-1 Latency. Cell host & microbe 21, 569–579 e566, https://doi.org/10.1016/j.chom.2017.04.011 (2017).
Taniguchi, I., Mabuchi, N. & Ohno, M. HIV-1 Rev protein specifies the viral RNA export pathway by suppressing TAP/NXF1 recruitment. Nucleic acids research 42, 6645–6658, https://doi.org/10.1093/nar/gku304 (2014).
Dumas, A. et al. The HIV-1 protein Vpr impairs phagosome maturation by controlling microtubule-dependent trafficking. The Journal of cell biology 211, 359–372, https://doi.org/10.1083/jcb.201503124 (2015).
Menager, M. M. & Littman, D. R. Actin Dynamics Regulates Dendritic Cell-Mediated Transfer of HIV-1 to T Cells. Cell 164, 695–709, https://doi.org/10.1016/j.cell.2015.12.036 (2016).
Ospina Stella, A. & Turville, S. All-Round Manipulation of the Actin Cytoskeleton by HIV. Viruses 10, https://doi.org/10.3390/v10020063 (2018).
Burgener, A., McGowan, I. & Klatt, N. R. HIV and mucosal barrier interactions: consequences for transmission and pathogenesis. Current opinion in immunology 36, 22–30, https://doi.org/10.1016/j.coi.2015.06.004 (2015).
Gabali, A. M., Anzinger, J. J., Spear, G. T. & Thomas, L. L. Activation by inflammatory stimuli increases neutrophil binding of human immunodeficiency virus type 1 and subsequent infection of lymphocytes. Journal of virology 78, 10833–10836, https://doi.org/10.1128/JVI.78.19.10833-10836.2004 (2004).
Zwirner, N. W. & Domaica, C. I. Cytokine regulation of natural killer cell effector functions. BioFactors 36, 274–288, https://doi.org/10.1002/biof.107 (2010).
Van Raemdonck, G. et al. Increased Serpin A5 levels in the cervicovaginal fluid of HIV-1 exposed seronegatives suggest that a subtle balance between serine proteases and their inhibitors may determine susceptibility to HIV-1 infection. Virology 458-459, 11–21, https://doi.org/10.1016/j.virol.2014.04.015 (2014).
Selhorst, P. et al. Cervicovaginal Inflammation Facilitates Acquisition of Less Infectious HIV Variants. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 64, 79–82, https://doi.org/10.1093/cid/ciw663 (2017).
Masson, L. et al. Genital inflammation and the risk of HIV acquisition in women. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 61, 260–269, https://doi.org/10.1093/cid/civ298 (2015).
Al-Harthi, L. Interplay between Wnt/beta-catenin signaling and HIV: virologic and biologic consequences in the CNS. J Neuroimmune Pharmacol 7, 731–739, https://doi.org/10.1007/s11481-012-9411-y (2012).
Veazey, R. S. et al. Prevention of SHIV transmission by topical IFN-beta treatment. Mucosal immunology 9, 1528–1536, https://doi.org/10.1038/mi.2015.146 (2016).
Sandler, N. G. et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511, 601–605, https://doi.org/10.1038/nature13554 (2014).
Utay, N. S. & Douek, D. C. Interferons and HIV. Infection: The Good, the Bad, and the Ugly. Pathog Immun 1, 107–116, https://doi.org/10.20411/pai.v1i1.125 (2016).
Roff, S. R., Noon-Song, E. N. & Yamamoto, J. K. The Significance of Interferon-gamma in HIV-1 Pathogenesis, Therapy, and Prophylaxis. Frontiers in immunology 4, 498, https://doi.org/10.3389/fimmu.2013.00498 (2014).
Pan, X., Baldauf, H. M., Keppler, O. T. & Fackler, O. T. Restrictions to HIV-1 replication in resting CD4+ T lymphocytes. Cell research 23, 876–885, https://doi.org/10.1038/cr.2013.74 (2013).
Lima, J. F., Oliveira, L. M. S., Pereira, N. Z., Duarte, A. J. S. & Sato, M. N. Polyfunctional natural killer cells with a low activation profile in response to Toll-like receptor 3 activation in HIV-1-exposed seronegative subjects. Scientific reports 7, 524, https://doi.org/10.1038/s41598-017-00637-3 (2017).
Luge, T. & Sauer, S. Generating Sample-Specific Databases for Mass Spectrometry-Based Proteomic Analysis by Using RNA Sequencing. Methods Mol Biol 1394, 219–232, https://doi.org/10.1007/978-1-4939-3341-9_16 (2016).
Birse, K. et al. Molecular Signatures of Immune Activation and Epithelial Barrier Remodeling Are Enhanced during the Luteal Phase of the Menstrual Cycle: Implications for HIV Susceptibility. Journal of virology 89, 8793–8805, https://doi.org/10.1128/JVI.00756-15 (2015).
Andrews, S. FastQC: a quality control tool for high throughput sequence data, http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads, https://journal.embnet.org/index.php/embnetjournal/article/view/200 (2011).
Dobin, A. & Gingeras, T. R. Mapping RNA-seq Reads with STAR. Current protocols in bioinformatics 51(11), 14 11–19, https://doi.org/10.1002/0471250953.bi1114s51 (2015).
Okonechnikov, K., Conesa, A. & Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294, https://doi.org/10.1093/bioinformatics/btv566 (2016).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, https://doi.org/10.1093/bioinformatics/btt656 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, https://doi.org/10.1093/bioinformatics/btp616 (2010).
Acknowledgements
We thank Florian Hladik and Sean Hughes for critical reading of the manuscript. This project was funded by the National Institutes of Health grant RO1AI110370.
Author information
Authors and Affiliations
Contributions
S.M., IPA analysis, graphs, writing. Y.L. raw RNAseq data analysis, GSEA analysis, statistical analysis. H.H. raw RNAseq data analysis, graphs. G.V. clinical samples processing, RNA extraction. G.C. clinical samples processing, contributed to IPA analysis. A.R., L.M., P.B. clinical samples processing. L.M., N.K., RIA .T.K. histopathological examination. R.S. conceptualization, project administration, resources, supervision, funding acquisition. N.T. conceptualization, writing, funding acquisition, project administration, supervision, formal data analysis, visualization, investigation.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mukhopadhyay, S., Liang, Y., Hur, H. et al. Comparative transcriptome analysis of the human endocervix and ectocervix during the proliferative and secretory phases of the menstrual cycle. Sci Rep 9, 13494 (2019). https://doi.org/10.1038/s41598-019-49647-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49647-3
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
