
Abstract
Recent studies have proposed that nucleic acids act as potential cofactors for protein aggregation and prionogenesis. By means of sedimentation, transmission electron microscopy, circular dichroism, static and dynamic light scattering, we have studied how RNA can influence the aggregation of the murine recombinant prion protein (rPrP). We find that RNA, independent of its sequence, source and size, modulates rPrP aggregation in a bimodal fashion, affecting both the extent and the rate of rPrP aggregation in a concentration dependent manner. Analogous to RNA-induced liquid-liquid phase transitions observed for other proteins implicated in neurodegenerative diseases, high protein to RNA ratios stimulate rPrP aggregation, while low ratios suppress it. However, the latter scenario also promotes formation of soluble oligomeric aggregates capable of seeding de novo rPrP aggregation. Furthermore, RNA co-aggregates with rPrP and thereby gains partial protection from RNase digestion. Our results also indicate that rPrP interacts with the RNAs with its N-terminus. In summary, this study elucidates the proposed adjuvant role of RNA in prion protein aggregation and propagation, and thus advocates an auxiliary role of the nucleic acids in protein aggregation in general.
Similar content being viewed by others


Introduction
Prion diseases such as Transmissible Spongiform Encephalopathies (TSE) are a group of infectious disorders, which irreversibly cripple the nervous system of humans and other mammals1,2. The agents responsible for those fatal conditions are commonly referred to as prions or prion proteins (PrP). Mammalian prions are ubiquitous proteins, which share the chemical properties of all polypeptides, but surprisingly can maintain several, marginally stable, conformational states at once. In the case of TSEs, the infectious form of the prion is called scrapie (PrPSc). This disease-associated conformation is amyloidogenic and self-sustaining over time and across cell divisions3. In recent years the number of proteins suspected of prion-like behavior has grown significantly. Some of those clearly serve a physiological purpose and are considered as functional prions4,5. Others are toxic in their prion-like state and have been linked to other non-infectious, degenerative disorders, such as Alzheimer’s, Parkinson’s disease and cancer6,7.
While the prevalence of proteinopathies becomes more evident, the necessity to understand the prion nature and evolutionary significance grows8,9. The outstanding characteristic of every prion is its ability to exist in two distinct physical states at the least10. In the case of TSEs, conversion of the cellular prion protein (PrPC) to its infectious form (PrPSc) is the hallmark of disease onset. The two states of the protein remain chemically identical but differ significantly in their biophysical properties. Whereas PrPC is mostly α-helical, soluble and susceptive to protease digestion and chaotropic agents, PrPSc favors a β-sheet rich conformation11,12,13,14, which renders it amyloidogenic, partially protease-resistant and insensitive to chaotropes1.
The most decisive of the criteria, commonly employed in PrPC/PrPSc discrimination, is the ability of the oligomeric scrapie protein to recruit native PrPC and induce further aggregation. This ability is at the center stage of prion self-replication and maintenance of heritable phenotypic change15. It is also the inspiration behind the “protein-only” hypothesis, which states that the misfolded PrPSc alone is sufficient for PrPC to PrPSc conversion16,17,18. This theory has been widely accepted in the field and even a study demonstrating propagation of an infectious prion in the absence of any cofactors has been recently published19. Despite that, a number of reports have emerged suggesting nucleic acids as the putative cofactors for prion formation and propagation20,21,22,23. A recent study has characterized the interaction between PrP and nucleic acids24. Furthermore, studies on RNA dependent phase separation of other neurodegenerative disease-related proteins have also been reported25,26. Apart from demonstrating that nucleic acids could facilitate prion conversion27,28,29,30,31,32,33,34,35, we recently reported that DNA and RNA modulate the aggregation of the tumor suppressor protein p5335,36,37,38. It is, therefore, becoming evident that nucleic acids, particularly RNA, have pronounced effect on protein folding, misfolding and aggregation, in some cases inhibiting and in other cases stimulating the processes39,40.
When PrP is overexpressed in the cytoplasm of the cells treated with proteasome inhibitors, it converts to an alternative isoform and accumulates in the perinuclear organelles (aggresomes), together with RNA41,42. Another type of membraneless structure – an organelle-like ribonucleoprotein particle, is formed when cytosolic PrP co-aggregates with RNA43. RNA is also detected in highly infectious brain isolates from TSE-positive Syrian hamsters44. Together with the demonstrations that RNA can induce scrapie phenotype in vitro45,46 this suggests a strong possibility for macromolecular interplay between RNA and PrP, in vivo. Transcriptome extracts of murine neuroblastoma (N2a) cells are shown to co-aggregate with rPrP to form large, partially protease resistant complexes, in vitro30,47. Finally, Maharana et al.25 show that high protein to RNA ratios promote phase separation and aggregation of the prion-like RNA binding proteins such as TDP43 and FUS, whereas low ratios prevent it. In accordance with these findings, the emerging concept is that the amyloid fibrils arise from more dynamic liquid droplets and glassy-solid states48,49, where RNA acts as phase modulator26,35,39,50. These observations hint at an auxiliary, previously unassumed function of RNA, in addition to its traditional role in the central dogma of molecular biology.
In the present study, we investigate the role of RNA in the aggregation of the full-length, recombinant, murine PrP (rPrP23–231 variant, referred hereafter as rPrP) by using static light scattering (LS), dynamic light scattering (DLS), sedimentation, transmission electron microscopy (TEM) and circular dichroism (CD). Furthermore, we follow the aggregation kinetics of rPrP, alone and in the presence of various RNAs, by monitoring the time-dependent increase in LS in a stopped-flow instrument. Our results suggest a modulatory role of RNA in rPrP aggregation, where high protein to RNA ratios lead to the formation of large amorphous aggregates of rPrP, whereas low ratios prevent it, and in turn, generate soluble, oligomeric species that can seed de novo rPrP aggregation. Furthermore, we observe that RNA co-aggregates with rPrP and gets partial resistance to RNase digestion. We therefore propose that RNA is an active player in rPrP aggregation, capable of modulating the outcome by direct interaction.
Results
Bimodal action of RNA on the aggregation of rPrP studied by LS and DLS
Equilibrium aggregation of rPrP (5 μM) alone or in the presence of various RNAs and ribonucleoside tri-phosphates (rNTPs) was studied by recording LS at 400 nm after incubating the samples for at least 10 minutes at room temperature. The RNAs include in vitro transcribed mRNAs for dihydrofolate reductase (DHFR) from Escherichia coli and Green Fluorescent Protein + (GFP+), domains V, IV and II from 23S ribosomal RNA (rRNA) of E. coli and bulk-tRNAs isolated from E. coli MRE600.
All rRNA domains and mRNAs tested here stimulated aggregation of rPrP in a concentration dependent manner (Fig. 1A). At low RNA concentration the LS signal increased over ten-fold compared to the rPrP only sample. With increase in RNA concentration, however, the aggregation decreased gradually (Fig. 1A). This phenomenon is analogous to the recently reported effect of RNA on the aggregation of the central core domain of the tumor suppressor protein p5336. However, unlike p53C, at the highest RNA concentration tested here, LS values still remained ~two-fold higher than the protein only reaction. Similar results obtained with different RNAs also suggest that RNA interaction for rPrP aggregation is not dependent on any particular sequence or secondary structure.

Equilibrium aggregation of rPrP (A,B) and rPrP90–231 (C) in the presence of various RNAs (as indicated) followed by LS at 400 nm in a steady state fluorimeter. Aggregation was achieved by incubating the protein (5 μM) without or with RNAs for 10 minutes at the room temperature without agitation. AU – arbitrary units.
Interestingly, tRNAs showed the strongest impact on the aggregation of rPrP among all RNAs tested here. Even though a relatively higher concentration of tRNAs was needed to stimulate rPrP aggregation, the addition of tRNAs led to a 20-fold increase in rPrP aggregation (Fig. 1B). Despite that, the overall pattern of tRNA induced rPrP aggregation remained similar to that of other RNAs. After the initial spike in aggregation with 2.5 μM tRNA, LS gradually decreased with increase in tRNA concentration. Similar to what we observed for p53C36, free rNTPs up to a rPrP to rNTP ratio = 1:100 exerted no effect on the aggregation of rPrP (data not shown). Thus, we conclude that the modulation of rPrP aggregation is a function of the structured RNAs.
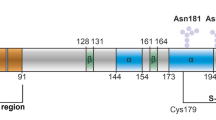
To investigate the role of the unstructured N-terminal region of the prion protein, we subjected a truncated variant of rPrP (rPrP90–231), which lacks the octapeptide repeats in the N-terminal domain, to the aggregation assay in the presence of DHFR mRNA and domain V rRNA. In comparison to rPrP, rPrP90–231 aggregated to a much lesser extent on its own, as reflected by the LS values (Fig. 1C). More interestingly, neither of the two RNAs tested, showed any influence on the aggregation of rPrP90–231. This result is indicative of the importance of the N-terminus of rPrP in nucleating its aggregation and also confirms that RNA interactions involve the N-terminus of rPrP, as proposed earlier30.
We also evaluated the hydrodynamic radius (Rh) and size distribution of the rPrP aggregates that are formed in the presence of low concentration of RNA (rPrP:RNA = 1:0.05) by employing DLS. Regardless of the type of RNA, all aggregated species displayed low polydispersity and yielded good correlation functions, demonstrating high sample homogeneity. The Rh values of different samples were on average ~100 nm, which indicated the presence of high molecular weight aggregates (Table 1). Interestingly tRNA containing samples formed significantly larger aggregates, with Rh value ~700 nm, approximately seven times larger than those with other RNAs. This result corroborates with our LS data where highest level of aggregation of rPrP was seen with tRNA (Fig. 1B). At high RNA concentration (rPrP:RNA = 1:2), the samples were polydisperse with no indication of large aggregates.
Fast kinetics analysis of the RNA induced and seeded aggregation of rPrP
We have followed the kinetics of rPrP aggregation in the presence and absence of RNA at varying rPrP to RNA ratios. For that, we monitored the time-dependent change in LS at 400 nm after rapid mixing of rPrP and DHFR mRNA in a stopped-flow instrument. rPrP aggregated alone at a very slow rate with kobs = 0.007 s−1 (Fig. 2A,B). Addition of very small amount of RNA, at rPrP to RNA ratio = 1:0.005, accelerated the rate of aggregation about 200 times, resulting in kobs = 1.3 ± 0.2 s−1. The rate of rPrP aggregation peaked (kobs = 4.5 ± 0.005 s−1) at a rPrP to RNA ratio of 1:0.01. In line with the steady-state LS measurements (Fig. 1), not only the extent but also the rate of rPrP aggregation decreased with further increase in RNA concentration. At 1:0.25 rPrP to RNA ratio the rate of rPrP aggregation decreased to 1.1 s−1 and finally dropped to 0.14 s−1 at 1:0.5 rPrP to RNA ratio. These results clearly suggest bimodal effect of RNA on rPrP aggregation, similar to the results obtained with p53C36.

Fast kinetics of rPrP aggregation at room temperature followed by LS in a stopped-flow instrument. (A) Time course and (B) rates of aggregation of rPrP (2 μM) alone or with DHFR mRNA at various rPrP to RNA ratios. (C) Time course and (D) rates of rPrP (2 μM) aggregation after seeding the reaction with 10% (v/v) SN from rPrP aggregation reactions with various rPrP to DHFR mRNA ratios. The inset in (C) shows LS curves beyond 150 sec. AU – arbitrary units. The error bars in (B,D) indicate standard deviation from three independent measurements.
To probe the nature of rPrP aggregates produced at different protein to RNA ratios, we designed a seeding experiment, where supernatant (SN) from rPrP aggregation reactions, prepared without or with DHFR mRNA, was treated with RNAse A and rapidly mixed with native rPrP in a stopped-flow instrument at a 10-fold dilution. The time-dependent change in LS was recorded to obtain the kinetics of seeded rPrP aggregation. SN from the protein only sample did not enhance the rate of de novo rPrP aggregation (Fig. 2C) (kobs = 0.01 ± 0.001 s−1). Also, SN from low RNA reaction (rPrP to RNA = 1:0.2) did not show any stimulation of rPrP aggregation (kobs = 0.02 ± 0.005 s−1). In contrast, SN from a high RNA reaction (rPrP to RNA = 1:0.5) increased the rate of rPrP aggregation more than two-fold (kobs = 0.026 ± 0.005 s−1). However, a much more dramatic increase of the rate of de novo rPrP aggregation occurred when SN from a 1:1 rPrP to DHFR mRNA reaction was used for seeding (Fig. 2C). In this case, the rate of rPrP aggregation became 20-fold higher than the protein only reaction, with kobs = 0.232 ± 0.005 s−1 (Fig. 2D). This result suggests that in abundance of RNA (low protein to RNA ratio) soluble, oligomeric rPrP seeds are formed, which could nucleate de novo rPrP aggregation.
Quantitative analysis of rPrP aggregation in the presence of RNA by sedimentation assay
Complementary to LS, we quantified the fractions of soluble and insoluble rPrP in the presence and absence of three different RNAs by sedimentation assay with low-speed centrifugation followed by SDS-PAGE (Fig. 3A). Analysis of the relative amounts of rPrP in the SN and the pellet fractions in a rPrP only reaction (Fig. 3B) showed that only ~20% of total rPrP protein aggregates in the absence of RNA and the rest of the protein (~ 80%) remains in the soluble fraction. In stark contrast, the distribution of rPrP between the soluble and insoluble fractions reversed completely upon addition of a small amount of RNA. At the rPrP to RNA ratio at 1:0.01, ~20% rPrP remained in the soluble fraction and ~80% partitioned in the insoluble pellet. At rPrP to RNA ratio of 1:0.2, ~90% of rPrP aggregated, thereby partitioning almost exclusively to the insoluble fraction. However, with further increase in RNA, the amount of insoluble aggregated rPrP gradually decreased and the fraction of soluble rPrP increased. At the highest RNA concentration tested here (rPrP:RNA = 1:2) the distribution of rPrP between the soluble and insoluble fractions became almost equal (Fig. 3A,B). This result corroborates strongly with our LS data and strengthens the conclusion that RNA modulates rPrP aggregation in a bimodal fashion.

Distribution of rPrP in the supernatant (SN) and the pellet (P) fractions in the presence of RNA, assessed by SDS-PAGE. rPrP samples (10 μM) with and without RNAs at various ratios were incubated 10 min at room temperature and then centrifuged at 20,000 g. Equal volumes of supernatant and buffer equilibrated pellet were ran on a 15% SDS acrylamide gel (A). The amount of protein in each sample was estimated as a percentage of total protein available in each supernatant/pellet pair (B).
Characterization of the rPrP aggregates with TEM
To probe the morphology of rPrP aggregates formed in the presence and absence of RNA we subjected the rPrP samples to TEM analysis. Samples of rPrP alone or with low RNA (rPrP:RNA = 1:0.02) and high RNA (rPrP:RNA = 1:0.5) concentrations were briefly incubated at room temperature. The experiments were conducted with both GFP+ mRNA and domain IV rRNA. As shown in Fig. 4, the rPrP-only samples contained only few aggregated particles of a size range far below 100 nm. The addition of RNA in low concentration resulted in the formation of densely packed, amorphous aggregates, reaching a couple of micrometers in size (Fig. 4). In contrast, when RNA concentration was increased to obtain 1:0.5 rPrP to RNA ratio, the average size of the aggregates visibly decreased and they appeared to be distributed more evenly. In accord with previous data31,47, we did not detect any fibrillar structures in rPrP aggregates under our experimental conditions. Our TEM results demonstrate that in the presence of low concentration of RNA, rPrP undergoes massive aggregation - typical for liquid-liquid phase separation behavior. Alternatively, with high concentration of RNA there is only limited exclusion of rPrP from solution, which corroborates well with our LS and sedimentation data.

TEM images of rPrP (5 μM) obtained after 10 min incubation at room temperature in the presence of GFP + mRNA and domain IV of the 23S rRNA, at protein to RNA ratios of 1:0.02 and 1:0.5. Scale bars correspond to 100 nm.
Demonstration of direct RNA-rPrP interaction by co-sedimentation analysis
To test whether RNA interacts with rPrP during aggregation, we incubated rPrP with DHFR mRNA at 1:0.2, 1:0.1 and 1:0.05 rPrP to RNA ratios, which according to our sedimentation assay induce high levels of rPrP aggregation (Fig. 3A). In this experiment, we kept the RNA concentration fixed and varied rPrP concentration to achieve the desired ratios so that the sedimentation of RNA can be quantified. The SN and the pellet fractions were separated after low speed centrifugation and the RNA content was analyzed by agarose gel electrophoresis.
DHFR mRNA alone (without rPrP) was perfectly soluble (Fig. 5A). However, when rPrP aggregation reactions were conducted in the low RNA conditions, RNA co-precipitated with rPrP and populated mostly in the insoluble pellet fraction. The amount of RNA in the pellet increased with decrease in RNA concentration (rPrP to RNA ratio 1:0.2 → 1:0.1 → 1:0.05). At a rPrP to RNA ratio of 1:0.05, which leads to a very high rPrP aggregation, almost all available RNA accumulated in the pellet (Fig. 5A). The experiment with domain IV of the 23S rRNA produced similar results (data not shown). These experiments suggest direct interaction between RNA and rPrP during aggregation.

Sedimentation and RNase A digestion assays demonstrating RNA-rPrP interaction during rPrP aggregation. (A) Distribution of DHFR mRNA in the supernatant (SN) and pellet (P) at different rPrP to RNA ratios as assessed by agarose gel electrophoresis. For quantitative comparison, the RNA concentration was kept constant (1 μM) and rPrP concentration was varied to achieve different rPrP to RNA ratios. (see materials and methods for details). (B) RNase A resistance assay with DHFR mRNA (1 μM) and domain IV rRNA (1 μM) after incubation without and with rPrP (20 μM) at 1:0.05 rPrP to RNA ratio. Equal volumes of the sample were run in a 1% agarose gel.
To further investigate the nature of the RNA-rPrP complexes we performed RNase protection assays at rPrP to RNA ratio of 1:0.05, with both DHFR mRNA and domain IV rRNA at 1 μM concentration. The reactions were treated with 25 μg/mL RNase A for 30 minutes at 37 °C. While RNase treatment completely digested the RNAs in the absence of rPrP, partial resistance to RNase was observed for both the RNAs coaggregated with rPrP (Fig. 5B). These results indicate that the RNAs are partially protected from RNases in the rPrP aggregates.
Characterization of RNA-rPrP interaction with CD
To further elucidate the nature of interaction between RNA and rPrP during co-aggregation, we conducted CD experiments at a 1:0.05 protein to RNA ratio using domain V rRNA (Fig. 6A), DHFR mRNA (Fig. 6B) and GFP+ mRNA (Fig. 6C). The results show that in all cases the rPrP-RNA complex spectra (blue traces) and the difference spectra (complex spectra minus RNA spectra – dotted lines), diverge significantly from the spectrum of rPrP alone (red traces) (Fig. 6). They also differ from the arithmetically derived cumulative spectra for rPrP and RNA (yellow). Compared to the protein-only spectra and cumulative spectra, the rPrP-RNA complex spectra show some loss of ellipticity. Most likely the RNAs contributed to a significant decrease in the helicity of the otherwise predominantly α-helical native CD spectrum of the protein. In conclusion, it appears that RNA not only co-aggregates, but directly interacts with rPrP, causing significant conformational changes in the protein.

CD spectra of rPrP (5 μM) aggregated in the presence of different RNAs (A) domain V rRNA, (B) DHFR mRNA, (C) GFP+ mRNA, at 1:0.05 protein to RNA ratio. For each sample, three spectra were collected; protein alone (red trace); RNA alone (green); rPrP-RNA complex (blue trace). The sum of the protein alone and RNA alone spectra produced the cumulative spectra (yellow trace); RNA spectra subtracted from the complex spectra (blue trace) generated the difference spectra (dotted line).
Discussion
Since the description of PrP as the infectious agent behind scrapie, its biological function and implication in a variety of human proteinopathies, ranging from amyloidosis to cancer, have been reported51. Also, it has gradually become evident that nucleic acids are involved in the propagation of prions40,52. Of the nucleic acid pool available in an eukaryotic cell, RNA was categorically proven to stimulate prion protein conversion and formation of protease resistant prion fibrils45,47,53. RNA isolated from scrapie associated fibrils was shown to aggregate recombinant PrP to a prion-like conformation44,46. It was also demonstrated that certain anti-prion compounds specifically target the RNA-mediated protein folding activity of the ribosome52,54,55,56,57. These arguments support the proposal that one of the putative factors involved in mediating prion protein conversion is RNA.
In the present study, we used in vitro transcribed RNAs to clarify the role of RNA in the aggregation of rPrP. In line with previous reports30,31 we observed that rPrP hardly aggregates at physiological pH and salt concentration. However, RNAs of various source and length, significantly influence rPrP aggregation in a concentration dependent manner. Small amounts of RNA, 50–100 times lower in concentration than rPrP dramatically enhance its aggregation – both in terms of the amount and the rate (Fig. 2A,B). Inversely, high concentrations of RNA, equimolar to rPrP (or higher) suppress rPrP aggregation (Figs 1A,B, 2, 3 and 4). This is a clear example of the bimodal action of RNA, observed for other prion-like systems such as p53C36, TDP43 and FUS25. In addition, SN from rPrP aggregation reactions with RNA in high concentration, promotes de novo rPrP aggregation, while SN from reactions with low RNA concentration does not (Fig. 2B). This result suggests that most likely oligomeric aggregates of rPrP emerge at high RNA conditions. Due to their sub-microscopic size, such species remain undetected by standard approaches such as LS, sedimentation and TEM. However, such oligomeric species are capable of nucleating de novo rPrP aggregation – a typical feature of prionogenic systems. This result further strengthens the proposed role of RNA in prion conversion and propagation44,45,46,58. In line with our observations we propose a schematic model of the bimodal role of RNA in rPrP aggregation, which is summarized in Fig. 7.

Schematic representation of the proposed effects of RNA on rPrP aggregation. Upon reaching the critical concentration rPrP forms very small aggregates in the absence of RNA. When the protein is in much excess over RNA (high rPrP to RNA ratio), rPrP aggregates profusely, sequestering the RNA. Alternatively, at low protein to RNA ratio, rPrP aggregation decreases and formation of oligomeric species, capable of nucleating de novo rPrP aggregation, prevails.
Another important finding of this work is that, irrespective of sequence, size or source RNA co-sediments with rPrP when added in low concentration (Fig. 5A). The RNA-rPrP association is such that a significant population of low molecular weight RNA remains protected even after extensive treatment of the aggregates with RNase (Fig. 5B). This result indicates a direct interaction between RNA and rPrP. The interaction appears to be associated with structural rearrangements in both macromolecules (Fig. 6), which supports our previously published observation that RNA alters the secondary structure of rPrP30. A similar result was recently reported for lysozyme aggregation in the presence of ribosomes59, where rRNA co-aggregated with lysozyme.
Since different RNAs tested in our study exerted analogous effect on rPrP aggregation, we speculate that the process depends more on the physical properties of RNA than on its sequence or structural motifs. As the process is virtually insensitive to free rNTPs and an inverse relationship between the size of the RNA and its effective concentration can be observed, our results highlight the significance of a defined polynucleotide surface for rPrP aggregation. On the other hand, our data suggest that rPrP interacts with RNA with a specific site. Since the N-terminally truncated rPrP variant (rPrP90–231) is completely insensitive to RNA (Fig. 1C), it seems likely that the unstructured N-terminal domain of rPrP is involved in RNA interaction. This is dissimilar to other proteins such as FUS and TDP-43, which interact with RNA through their prion-like domains25. The intrinsically disordered N-terminal domain of rPrP is referred to as the low-complexity region (LCR). Likely the LCR of rPrP has a hybrid function, acting as a mediator of RNA-based phase transition60. We have previously found that shortening the N-terminal domain destabilizes rPrP and promotes irreversible denaturation and aggregation61. The N-domain could, therefore, be a site for interaction with different ligands. While heparin binding has a stabilizing effect on it62, RNA binding leads to destabilization, condensation and aggregation. It remains to be established whether or not the rPrP-RNA complexes display any infectious behavior in vivo. There is mounting evidence showing that RNA, among other polyanions, can trigger the in vitro conversion of PrP into scrapie-like PrP species58,63,64,65. As long as PrPSc can be formed, it can amplify the infectious process by itself, but the role of the endogenous cofactors e.g. RNA in the propagation of infectivity of synthetic prion strains cannot be ruled out66. It has been described that different prion strains require different RNAs for efficient conversion27. However, demonstrating de novo infectivity using rPrP and RNA condensates is not straightforward44,64 and will require special laboratory set up beyond the scope of this work. One possibility would be to employ rPrP and RNA in the protein misfolding cyclic amplification (PMCA) assay under our reaction conditions, which will be a goal for future investigations.
The role of RNA in formation of membraneless structures, such as stress granules and speckles is well-established. In case of the prion-like RNA binding proteins TDP43 and FUS, it has been demonstrated that RNA induces phase separation of the proteins in a concentration dependent manner25. These proteins remain soluble in the nucleus – where RNA is readily available, but have higher tendency to undergo phase separation in the cytoplasm – where RNA is present in much lower concentration25. The significance of our data could be discussed in similar terms when asked for in vivo implication of the observed RNA effect on rPrP (Fig. 7). Although cellular PrP is primarily GPI-anchored to the plasma membrane, certain cell tissues appear to retain a significant fraction of endoplasmic reticulum with non-translocated PrP in the cytoplasm67,68,69. In that respect, it was already demonstrated that cytosolic PrP interacts with RNA in vivo, leading to the formation of large membraneless aggresomes30,43,70. Nonetheless, the true nature, functional or pathogenic, of cytosolic PrP-RNA condensates remains to be elucidated. In conclusion, our demonstration of the bimodal action of RNA on rPrP aggregation provides strong reinforcement and mechanistic insight to the proposed role of RNA in the processes of prion conversion44,45,46,52 and membraneless compartmentalization of the prion-like proteins39,71.
Methods
Protein expression and purification
Heterologous expression in E. coli and consecutive purification of full-length murine rPrP (rPrP23–231) and its N-terminally deleted variant rPrP90–231 was conducted by high affinity column refolding in a ÄKTA Prime Plus (GE Healthcare) as previously described72.
In vitro transcription and extraction of RNAs
Almost all RNAs included in this study were transcribed in vitro with T7 RNA polymerase using PCR amplified DNA templates as described in73. These include mRNAs for DHFR (E. coli) and GFP + , and domains II, IV and V from 23S rRNA (E. coli). The pGEM4Z plasmid encoding the complete 23S rRNA sequence of E. coli K1274 was used as the template for different domains of rRNA. The templates for DHFR and GFP+ mRNAs were generated from their respective DNA clones in pET-24b (Novagen). The bulk-tRNA was extracted from E. coli MRE600 cells by phenol-chloroform treatment using laboratory protocol. All RNAs were quantified using Nanodrop™ 1000 spectrophotometer (Thermo Scientific V3.6).
Equilibrium aggregation assays
rPrP (final concentration 5 μM unless otherwise mentioned) was diluted in 10 mM Tris-HCl (pH 7.5) and 100 mM NaCl (buffer A) prior to addition of the RNAs or rNTPs. For all equilibrium aggregation measurements such as LS, DLS, TEM and sedimentation, the reactions were incubated 10 minutes at room temperature. The rPrP stock was centrifuged at 20,000 g for 30 minutes prior to sample preparation. Aggregation assays were done without agitation or stirring.
Light scattering – equilibrium and fast kinetics measurements
Equilibrium aggregation of rPrP and rPrP90–231 proteins (5 μM) was followed by Rayleigh light scattering (both excitation and emission at 400 nm) in an Infinite M200 PRO multimode microplate reader (Tecan Trading AG, Switzerland). Each experiment was repeated three times and averaged for presentation in the figures, where background LS for the various RNA concentrations was subtracted. Further, fast kinetics of rPrP (2 μM) aggregation without or with various RNAs were recorded under the same conditions in a SFM-3000 stopped-flow instrument (BioLogic Science Instruments, France) by monitoring the increase in LS for up to 150 seconds with 500 μs increments. The data were fitted with single exponential function for rate estimation.
Seeding experiments
For seeding, rPrP (2 μM) samples at varying rPrP to DHFR mRNA ratios were incubated at room temperature for 10 minutes and then treated with 25 μg/mL RNase A (ThermoFisher Scientific) for 2 hours at 37 °C. Larger aggregates were removed by centrifugation at 20,000 g for 30 minutes. A fraction of the resulting supernatant (10% v/v) was rapidly added to a fresh preparation of rPrP (2 μM) in a stopped-flow instrument and the fast kinetics of aggregation were recorded as described above. Rates were estimated by fitting the data with a single-exponential function.
Sedimentation assays
rPrP at a final concentration of 10 μM was incubated at room temperature without and with RNAs at various ratios for sedimentation analysis using SDS-PAGE. After 10 min, the reactions were centrifuged at 20,000 g at 4 °C and the supernatants were removed in fresh tubes. Pellets were then solubilized in buffer A and the volume was made equal to the corresponding supernatant. Further, fixed volumes of pellet and supernatant from each sample were run in 15% SDS-PAGE. After completion, gels were visualized by Coomassie blue staining and the protein bands were analyzed in pairs by Image J 1.51j8 software75. The bands in each supernatant-pellet pair, corresponding to each rPrP:RNA ratio, were assumed to have a combined intensity of 100%.
For quantitative estimation of RNA in pellet and supernatant, a sedimentation assay was conducted with fixed concentration of RNA (1 μM) and rPrP concentration was varied to achieve various ratios. After 10 min incubation the pellet and the supernatant were separated in the same way as described above. The pellets were re-solubilized with buffer A supplemented with 0.1% SDS and then subjected to 1% agarose gel electrophoresis. The band intensities were quantified with Image J 1.5 software75.
RNase protection assay
rPrP (20 μM) was mixed with DHFR mRNA (1 μM) or domain IV rRNA (1 μM) to achieve 1:0.05 (rPrP:RNA) ratio and incubated at room temperature for 10 minutes. After that, the reactions (along with RNA only controls) were subjected to RNase digestion by incubation with 25 μg/mL RNase A (ThermoFisher Scientific) at 37 °C for 30 minutes. Then the samples were supplemented with 1 U/μL of RiboLock RNase inhibitor (ThermoFisher Scientific) and RNA was extracted by phenol-chloroform to ensure complete nuclease inactivation. The RNAs were run in 1% agarose gel and quantified with Image J 1.5 software75.
Transmission electron microscopy
For TEM measurements, rPrP (5 µM) was incubated with GFP+ mRNA and domain IV rRNA to achieve rPrP:RNA ratios of 1:0.02 and 1:1. All samples were incubated at room temperature for 10 minutes and then 20 μL of each sample were applied to a carbon coated copper grid. After 5 min the grids were washed with sterile water, stained with 2% (w/v) uranyl acetate for 1 minute and washed again with water. Images were collected on a Jeol 1200 microscope (Boston, MA, USA) operating at 80 kV.
Dynamic light scattering
The hydrodynamic radius (Rh) of samples at a rPrP to RNA ratio of 20:1, aggregated in buffer A for 10 minutes at room temperature was evaluated using DynaPro Nanostar (Wyatt, USA), equipped with a 662 nm laser in quartz cuvette at 25 °C. The DLS data for each sample were collected one to three times, with ten cumulates in each measurement, the mean of which lead to respective Rh values. Population distributions were analyzed by correlation function graphs.
Circular dichroism
rPrP (5 μM) was mixed with domain V rRNA, DHFR mRNA and GFP+ mRNA to 1:0.05 ratio (rPrP:RNA) in buffer A and incubated from 5 to 10 min at room temperature before each measurement. CD spectra were collected with a Chirascan CD Spectrometer (Applied Photophysics, Surrey, UK) from 250 to 200 nm at 70 nm/min rate using a 1 mm-path length quartz cuvette. The spectra for protein and RNAs alone at their respective final concentrations were recorded separately.
Data Availability
The data used in this manuscript are added in the result section, especially in the figures and tables.
References
Prusiner, S. B. Prions. Proc Natl Acad Sci USA 95, 13363–13383, https://doi.org/10.1073/pnas.95.23.13363 (1998).
Aguzzi, A. & Polymenidou, M. Mammalian prion biology: one century of evolving concepts. Cell 116, 313–327 (2004).
Prusiner, S. B. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47, 601–623, https://doi.org/10.1146/annurev-genet-110711-155524 (2013).
Si, K., Lindquist, S. & Kandel, E. R. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 115, 879–891 (2003).
Wickner, R. B., Edskes, H. K., Shewmaker, F. & Nakayashiki, T. Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol 5, 611–618, https://doi.org/10.1038/nrmicro1708 (2007).
Brettschneider, J., Del Tredici, K., Lee, V. M. & Trojanowski, J. Q. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16, 109–120, https://doi.org/10.1038/nrn3887 (2015).
Silva, J. L., De Moura Gallo, C. V., Costa, D. C. & Rangel, L. P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem Sci 39, 260–267, https://doi.org/10.1016/j.tibs.2014.04.001 (2014).
Maury, C. P. Self-propagating beta-sheet polypeptide structures as prebiotic informational molecular entities: the amyloid world. Orig Life Evol Biosph 39, 141–150, https://doi.org/10.1007/s11084-009-9165-6 (2009).
Si, K. Prions: what are they good for? Annu Rev Cell Dev Biol 31, 149–169, https://doi.org/10.1146/annurev-cellbio-100913-013409 (2015).
Eisenberg, D. & Jucker, M. The amyloid state of proteins in human diseases. Cell 148, 1188–1203, https://doi.org/10.1016/j.cell.2012.02.022 (2012).
Caughey, B. W. et al. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 30, 7672–7680, https://doi.org/10.1021/bi00245a003 (1991).
Groveman, B. R. et al. Parallel in-register intermolecular beta-sheet architectures for prion-seeded prion protein (PrP) amyloids. J Biol Chem 289, 24129–24142, https://doi.org/10.1074/jbc.M114.578344 (2014).
Requena, J. R. & Wille, H. The structure of the infectious prion protein: experimental data and molecular models. Prion 8, 60–66, https://doi.org/10.4161/pri.28368 (2014).
Pan, K. M. et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 90, 10962–10966, https://doi.org/10.1073/pnas.90.23.10962 (1993).
Chien, P., Weissman, J. S. & DePace, A. H. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem 73, 617–656, https://doi.org/10.1146/annurev.biochem.72.121801.161837 (2004).
Griffith, J. S. Self-replication and scrapie. Nature 215, 1043–1044, https://doi.org/10.1038/2151043a0 (1967).
Prusiner, S. B. Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144, https://doi.org/10.1126/science.6801762 (1982).
Weissmann, C. A. Unified Theory of prion propagation. Nature 352, 679–683, https://doi.org/10.1038/352679a0 (1991).
Fernandez-Borges, N. et al. Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol 135, 179–199, https://doi.org/10.1007/s00401-017-1782-y (2018).
Caughey, B. & Kocisko, D. A. Prion diseases: a nucleic-acid accomplice? Nature 425, 673–674, https://doi.org/10.1038/425673a (2003).
Botsios, S. & Manuelidis, L. CJD and Scrapie Require Agent-Associated Nucleic Acids for Infection. J Cell Biochem 117, 1947–1958, https://doi.org/10.1002/jcb.25495 (2016).
Manuelidis, L. Nuclease resistant circular DNAs copurify with infectivity in scrapie and CJD. J Neurovirol 17, 131–145, https://doi.org/10.1007/s13365-010-0007-0 (2011).
Cordeiro, Y. & Silva, J. L. The hypothesis of the catalytic action of nucleic acid on the conversion of prion protein. Protein Pept Lett 12, 251–255 (2005).
Bera, A. & Biring, S. A quantitative characterization of interaction between prion protein with nucleic acids. Biochem Biophys Rep 14, 114–124, https://doi.org/10.1016/j.bbrep.2018.04.006 (2018).
Maharana, S. et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921, https://doi.org/10.1126/science.aar7366 (2018).
Polymenidou, M. The RNA face of phase separation. Science 360, 859–860, https://doi.org/10.1126/science.aat8028 (2018).
Saa, P. et al. Strain-specific role of RNAs in prion replication. J Virol 86, 10494–10504, https://doi.org/10.1128/JVI.01286-12 (2012).
Silva, J. L., Gomes, M. P., Vieira, T. C. & Cordeiro, Y. PrP interactions with nucleic acids and glycosaminoglycans in function and disease. Front Biosci (Landmark Ed) 15, 132–150 (2010).
Silva, J. L., Lima, L. M., Foguel, D. & Cordeiro, Y. Intriguing nucleic-acid-binding features of mammalian prion protein. Trends Biochem Sci 33, 132–140, https://doi.org/10.1016/j.tibs.2007.11.003 (2008).
Gomes, M. P. et al. Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J Biol Chem 283, 19616–19625, https://doi.org/10.1074/jbc.M802102200 (2008).
Macedo, B. et al. Nonspecific prion protein-nucleic acid interactions lead to different aggregates and cytotoxic species. Biochemistry 51, 5402–5413, https://doi.org/10.1021/bi300440e (2012).
Silva, J. L. et al. Experimental approaches to the interaction of the prion protein with nucleic acids and glycosaminoglycans: Modulators of the pathogenic conversion. Methods 53, 306–317, https://doi.org/10.1016/j.ymeth.2010.12.002 (2011).
Lima, L. M. et al. Structural insights into the interaction between prion protein and nucleic acid. Biochemistry 45, 9180–9187, https://doi.org/10.1021/bi060532d (2006).
Marques, A. F., Cordeiro, Y., Silva, J. L. & Lima, L. M. Enhanced prion protein stability coupled to DNA recognition and milieu acidification. Biophys Chem 141, 135–139, https://doi.org/10.1016/j.bpc.2008.12.011 (2009).
Cordeiro, Y., Vieira, T., Kovachev, P. S., Sanyal, S. & Silva, J. L. Modulation of p53 and prion protein aggregation by RNA. Biochim Biophys Acta Proteins Proteom. https://doi.org/10.1016/j.bbapap.2019.02.006 (2019).
Kovachev, P. S. et al. Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain. J Biol Chem 292, 9345–9357, https://doi.org/10.1074/jbc.M116.762096 (2017).
Ishimaru, D. et al. Cognate DNA stabilizes the tumor suppressor p53 and prevents misfolding and aggregation. Biochemistry 48, 6126–6135, https://doi.org/10.1021/bi9003028 (2009).
Vieler, M. & Sanyal, S. p53 Isoforms and Their Implications in Cancer. Cancers (Basel) 10, https://doi.org/10.3390/cancers10090288 (2018).
Silva, J. L. & Cordeiro, Y. The “Jekyll and Hyde” Actions of Nucleic Acids on the Prion-like Aggregation of Proteins. J Biol Chem 291, 15482–15490, https://doi.org/10.1074/jbc.R116.733428 (2016).
Voisset, C. et al. The double life of the ribosome: When its protein folding activity supports prion propagation. Prion 11, 89–97, https://doi.org/10.1080/19336896.2017.1303587 (2017).
Grenier, C., Bissonnette, C., Volkov, L. & Roucou, X. Molecular morphology and toxicity of cytoplasmic prion protein aggregates in neuronal and non-neuronal cells. J Neurochem 97, 1456–1466, https://doi.org/10.1111/j.1471-4159.2006.03837.x (2006).
Ma, J. & Lindquist, S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science 298, 1785–1788, https://doi.org/10.1126/science.1073619 (2002).
Beaudoin, S. et al. A large ribonucleoprotein particle induced by cytoplasmic PrP shares striking similarities with the chromatoid body, an RNA granule predicted to function in posttranscriptional gene regulation. Biochim Biophys Acta 1793, 335–345, https://doi.org/10.1016/j.bbamcr.2008.10.009 (2009).
Simoneau, S. et al. Synthetic scrapie infectivity: interaction between recombinant PrP and scrapie brain-derived RNA. Virulence 6, 132–144, https://doi.org/10.4161/21505594.2014.989795 (2015).
Deleault, N. R., Lucassen, R. W. & Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 425, 717–720, https://doi.org/10.1038/nature01979 (2003).
Supattapone, S. Synthesis of high titer infectious prions with cofactor molecules. J Biol Chem 289, 19850–19854, https://doi.org/10.1074/jbc.R113.511329 (2014).
Gomes, M. P., Cordeiro, Y. & Silva, J. L. The peculiar interaction between mammalian prion protein and RNA. Prion 2, 64–66, https://doi.org/10.4161/pri.2.2.6988 (2008).
Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 357, https://doi.org/10.1126/science.aaf4382 (2017).
Toretsky, J. A. & Wright, P. E. Assemblages: functional units formed by cellular phase separation. J Cell Biol 206, 579–588, https://doi.org/10.1083/jcb.201404124 (2014).
Langdon, E. M. et al. mRNA structure determines specificity of a polyQ-driven phase separation. Science 360, 922–927, https://doi.org/10.1126/science.aar7432 (2018).
Prions and Diseases. Vol. 1 (Springer Science+Business Media New York, 2013).
Tribouillard-Tanvier, D. et al. Protein folding activity of ribosomal RNA is a selective target of two unrelated antiprion drugs. PLoS One 3, e2174, https://doi.org/10.1371/journal.pone.0002174 (2008).
Geoghegan, J. C. et al. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem 282, 36341–36353, https://doi.org/10.1074/jbc.M704447200 (2007).
Banerjee, D. & Sanyal, S. Protein folding activity of the ribosome (PFAR)–a target for antiprion compounds. Viruses 6, 3907–3924, https://doi.org/10.3390/v6103907 (2014).
Reis, S. D. et al. Mode of action of the antiprion drugs 6AP and GA on ribosome assisted protein folding. Biochimie 93, 1047–1054, https://doi.org/10.1016/j.biochi.2011.03.002 (2011).
Pang, Y. et al. The antiprion compound 6-aminophenanthridine inhibits the protein folding activity of the ribosome by direct competition. J Biol Chem 288, 19081–19089, https://doi.org/10.1074/jbc.M113.466748 (2013).
Voisset, C., Saupe, S. J. & Blondel, M. The various facets of the protein-folding activity of the ribosome. Biotechnol J 6, 668–673, https://doi.org/10.1002/biot.201100021 (2011).
Deleault, N. R., Harris, B. T., Rees, J. R. & Supattapone, S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 104, 9741–9746, https://doi.org/10.1073/pnas.0702662104 (2007).
Pathak, B. K., Mondal, S., Banerjee, S., Ghosh, A. N. & Barat, C. Sequestration of Ribosome during Protein Aggregate Formation: Contribution of ribosomal RNA. Sci Rep 7, 42017, https://doi.org/10.1038/srep42017 (2017).
Franzmann, T. M. & Alberti, S. Prion-like low-complexity sequences: Key regulators of protein solubility and phase behavior. J Biol Chem 294, 7128–7136, https://doi.org/10.1074/jbc.TM118.001190 (2019).
Cordeiro, Y. et al. The amino-terminal PrP domain is crucial to modulate prion misfolding and aggregation. Biophys J 89, 2667–2676, https://doi.org/10.1529/biophysj.105.067603 (2005).
Vieira, T. C. et al. Heparin binding by murine recombinant prion protein leads to transient aggregation and formation of RNA-resistant species. J Am Chem Soc 133, 334–344, https://doi.org/10.1021/ja106725p (2011).
Kim, C. et al. Artificial strain of human prions created in vitro. Nat Commun 9, 2166, https://doi.org/10.1038/s41467-018-04584-z (2018).
Timmes, A. G., Moore, R. A., Fischer, E. R. & Priola, S. A. Recombinant prion protein refolded with lipid and RNA has the biochemical hallmarks of a prion but lacks in vivo infectivity. PLoS One 8, e71081, https://doi.org/10.1371/journal.pone.0071081 (2013).
Wang, F., Wang, X., Yuan, C. G. & Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135, https://doi.org/10.1126/science.1183748 (2010).
Abid, K., Morales, R. & Soto, C. Cellular factors implicated in prion replication. FEBS Lett 584, 2409–2414, https://doi.org/10.1016/j.febslet.2010.04.040 (2010).
Rane, N. S., Yonkovich, J. L. & Hegde, R. S. Protection from cytosolic prion protein toxicity by modulation of protein translocation. EMBO J 23, 4550–4559, https://doi.org/10.1038/sj.emboj.7600462 (2004).
Kikuchi, Y. et al. Hypoxia induces expression of a GPI-anchorless splice variant of the prion protein. FEBS J 275, 2965–2976, https://doi.org/10.1111/j.1742-4658.2008.06452.x (2008).
Mironov, A. Jr. et al. Cytosolic prion protein in neurons. J Neurosci 23, 7183–7193 (2003).
Goggin, K., Beaudoin, S., Grenier, C., Brown, A. A. & Roucou, X. Prion protein aggresomes are poly(A)+ ribonucleoprotein complexes that induce a PKR-mediated deficient cell stress response. Biochim Biophys Acta 1783, 479–491, https://doi.org/10.1016/j.bbamcr.2007.10.008 (2008).
Lin, Y., Protter, D. S., Rosen, M. K. & Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell 60, 208–219, https://doi.org/10.1016/j.molcel.2015.08.018 (2015).
Macedo, B., Sant’Anna, R., Navarro, S., Cordeiro, Y. & Ventura, S. Mammalian prion protein (PrP) forms conformationally different amyloid intracellular aggregates in bacteria. Microb Cell Fact 14, 174, https://doi.org/10.1186/s12934-015-0361-y (2015).
Banerjee, D. et al. Spectroscopic and DFT studies on 6-aminophenanthridine and its derivatives provide insights in their activity towards ribosomal RNA. Biochimie 97, 194–199, https://doi.org/10.1016/j.biochi.2013.10.012 (2014).
Samanta, D. et al. Protein folding by domain V of Escherichia coli 23S rRNA: specificity of RNA-protein interactions. J Bacteriol 190, 3344–3352, https://doi.org/10.1128/JB.01800-07 (2008).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675 (2012).
Acknowledgements
This work is supported by research grants from the Swedish Research Council [2016-06264 and 2018-05498]; Knut and Alice Wallenberg Foundation (KAW 2017.0055), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT- Joint Brazilian-Swedish Research Collaboration) (BR2013-5223) and Wenner-Gren Foundation (UPD2017-0238, UPD2018-0306) to S.S.; and Carlos Chagas Filho Foundation for Research Support in the State of Rio de Janeiro (FAPERJ) (170.027/2008); Coordination for the Improvement of Higher Education Personnel (CAPES) (1698/2012); the National Council of Technological and Scientific Development (CNPq) (573767/2008-4, 467500/2014-2, 470994/2012-6) to J.S. Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
S.S., J.S. and Y.C. conceived the project idea. P.K. and M.G. performed the main biochemical and microscopy experiments. Y.C., N.F., L.F.V., L.A. and L.R. conducted the supporting experiments. P.K. wrote the manuscript draft and prepared the figures. S.S., J.S., Y.C. and P.K. edited and finalized the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kovachev, P.S., Gomes, M.P.B., Cordeiro, Y. et al. RNA modulates aggregation of the recombinant mammalian prion protein by direct interaction. Sci Rep 9, 12406 (2019). https://doi.org/10.1038/s41598-019-48883-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48883-x
This article is cited by
-
The ALS/FTD-related C9orf72 hexanucleotide repeat expansion forms RNA condensates through multimolecular G-quadruplexes
Nature Communications (2023)
-
(Dys)functional insights into nucleic acids and RNA-binding proteins modulation of the prion protein and α-synuclein phase separation
Biophysical Reviews (2023)
-
CAPRIN1P512L causes aberrant protein aggregation and associates with early-onset ataxia
Cellular and Molecular Life Sciences (2022)
-
Tau protein- induced sequestration of the eukaryotic ribosome: Implications in neurodegenerative disease
Scientific Reports (2020)
-
Effect of additives on liquid droplets and aggregates of proteins
Biophysical Reviews (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
