
Abstract
Prophage enriched the prokaryotic genome, and their transcriptional factors improved the protein expression network of the host. In this study, we uncovered a new prophage-prophage interaction in E. coli JM83. The Rac prophage protein RacR (GenBank accession no. AVI55875.1) directly activated the transcription of φ80dlacZΔM15 prophage lysozyme encoding gene 19 (GenBank accession no. ACB02445.1, renamed it lysN, lysozyme nineteen), resulting in the growth defect of JM83. This phenomenon also occurred in DH5α, but not in BL21(DE3) and MG1655 due to the genotype differences. However, deletion of lysN could not completely rescued JM83 from the growth arrest, indicating that RacR may regulate other related targets. In addition, passivation of RacR regulation was found in the late period of growth of JM83, and it was transmissible to daughter cells. Altogether, our study revealed part of RacR regulatory network, which suggested some advanced genetic strategies in bacteria.
Similar content being viewed by others

Introduction
Generally, the genotypic diversity in different strains is mainly through mutation, rearrangement or horizontal gene transfer (HGT)1,2 and the latter plays a vital role3,4. In prokaryote, the pan-genome is the set of all genes5, which consists of two parts. The core genome exists in all related bacteria, while only a few bacteria contain the accessory genome6, which is acquired via HGT in most cases. Amounts of studies suggest that horizontally acquired DNA (known as mobile genetic elements) comprises transposons, plasmids, and prophages7,8, in which prophages are considered as the most significant factor. In the lifecycle of bacteria, some genes encoded by prophage are active and tightly associated with antibiotic resistance, virulence, or metabolism of the host9.
E. coli diverged from Salmonella 100 million years ago10, and the K-12 strain acquired nine defective prophages subsequently, including Rac, which was inserted over 4.5 million years ago11. The integration or excision of this lambda-like prophage has a great influence on the host12. Rac prophage conserves 40% of its original genes that are indispensable for K-12 in specific conditions. φ80 is also derived from lambdoid phage, and a large number of genes maintain the original function13. Its derivative φ80dlacZΔM15 is generated by aberrant excision and recombination during the integration of φ8014. Given their kinship, φ80dlacZΔM15 and Rac may be functionally linked, and this connection possibly be due to prophage-prophage interaction.
So far, a large number of studies have focused on the prophage-bacteria regulation, and a few cases in self-regulation of prophages were also reported. At specific time phase or environment, prophages regulators, either proteins or sRNAs, were expressed to control the behavior of the host. As currently reported, phage phi3T-encoded AimR activated the expression of its collinear gene aimX, which led to lysis of Bacillus cells15. And in E. coli O157: H7, a prophage CP-933H-encoded regulatory protein, PatE, upregulated the transcription of genes associated with acid resistance and also downregulated the expression of genes that belong to heat shock protein family and type III secretion pathways16. The third case is about sRNA DicF, which encoded by Qin prophage. DicF was confirmed to bind to ftsZ, xylR, and pykA mRNAs by directly base pairing and repress their translations. Hence, the cell division and metabolism of bacteria were significantly inhibited17.
In the process of transcription, cofactors are indispensable in some networks18,19. They usually bind directly to regulatory proteins or the promoter sequence, aimed to enhance binding stability20. While in other cases, the transcription can be switched on only when cofactors are modified or disappear because they exert the function as an inhibitor19,21. Once the concentration of transcription factors changes, the mRNA levels of target genes would be up-regulated or down-regulated immediately22. Occasionally, when the dynamic changes of transcription factors are unable to reserve the overreaction of cells in time, some drastic and irreversible pathways will be activated: (i) the methylation/semi-methylation of promoters can directly block downstream genes23. (ii) sRNA can degrade the formed mRNA rapidly, meanwhile, interrupt the extension of the polypeptide24. (iii) proteases can destroy the activity of effector proteins. In addition, mRNA methylation has also been revealed, which will alter the half-life of mRNA, or prevent the binding of ribosome25. In general, the flexible regulation of gene network has effect on the growth of cell anytime.
RacR is a Rac prophage protein and predicted as a transcriptional factor, which contains a conserved helix-turn-helix (HTH) motif. In this study, we reveal that RacR unexpectedly activates the expression of a distant gene, lysN, which is located in φ80dlacZΔM15 prophage, and lyses the JM83 cells eventually.
Results
Overexpression of RacR induces cell lysis
We found the growth of JM83 was significantly inhibited by overexpression of RacR, which was also observed in the case of DH5α. However, BL21(DE3) and MG1655 was not affected (Fig. 1a). Accordingly, it seems that the growth inhibition is not caused by toxicity of the protein but has possible genomic context connection. Viability test also suggests a lethal effect of RacR on JM83 (Fig. 1b), which could be further confirmed by serious cell debris in scanning electron microscope (SEM) results. While other strains like pBAD/JM83, pracR/DH10B, pracR/MG1655 seemed normal (Fig. 1c), and the pBAD/JM83 should be regarded as wild-type E. coli26. Given that RacR belongs to Rac prophage, which does not directly damage cells, we tested the phage bacteriolytic reaction by inoculating the culture of lytic JM83 strain into wild-type E. coli at the logarithmic stage, and we found that its growth was completely unaffected. Furthermore, there were no bacteriophage plaques on the LB plate. Altogether, it suggests that RacR probably leads to cell lysis by genome regulation.

RacR overexpression resulted in JM83 cells lysis. (a) Growth curves of JM83, BL21(DE3), DH5α, DH10B, and MG1655 transformed with pBAD or pBAD-racR (pracR) after induction (control [+] in blue and pracR[+] in red) and pracR without induction (pracR [−] in dark). (b) Living cell count of JM83 strain transformed with pBAD (control), pracR. 1 h, 3 h, and 5 h represent the time after induction. (c) SEM results of the morphology of cells after induction for 2 h or 5 h. Scale bar 5.0 µm. Data represent means ± standard deviations of results from three independent experiments.
The lysozyme protein LysN in φ80dlacZΔM15 is one of the potential targets of RacR
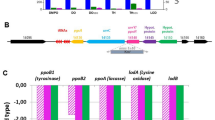
To ascertain the downstream regulatory target of RacR, we performed comparative genomics of the above five strains. Interestingly, we found that different with BL21(DE3) or MG1655, JM83 and DH5α both contain φ80dlacZΔM15 prophage but lack lacZYA-argF gene cluster that harbors CP4-6 prophage (Fig. 2), which may be responsible for lysis phenomenon of these two strains. In addition, overexpression of RacR in DH10B slightly inhibited cell growth (Fig. 1a), seems that lacZYA-argF (or maybe CP4-6) is a φ80dlacZΔM15 antagonistic cluster.

Comparative genomics determined genotype differences. Schematic representation of genotype difference among several E. coli strains, mainly depicting two regions, lac-proAB and φ80dlacZΔM15. The upper part roughly depicts the gene sites in φ80dlacZΔM15.
Recently, Revathy Krishnamurthi et al. uncover some possible binding motifs of RacR, like “GCCTAA” and “TTAGGC”, which lie in the region upstream of ydaS27. By genome research, we found four potential target genes, 19 (renamed lysN in this study), cI, cII, and 30 (cro) in φ80dlacZΔM15 contain these motifs (Table 1). Coincidentally, the candidate lysN (the black highlighted gene of φ80dlacZΔM15, shown on the top of Fig. 2) is annotated as a lysozyme gene, which is highly probable to participate in the process of cells lysis. Subsequently, LysN was overexpressed via pBAD-lysN (plysN) in JM83. The growth curve showed that the turbidity of culture decreased significantly, while the number of living cells only decreased slightly (Fig. 3a,b). In addition, LysN can also lead to lysis of the other four strains (Fig. 3a), suggesting that LysN may be indeed the direct functional protein associated with lysis. As expected, the fractal pattern due to LysN overexpression observed in JM83, DH10B, MG1655 cells was quite similar to pracR/JM83 overexpression case (Figs 1c and 3c). Combined together, we hypothesize that LysN is one of the direct effectors in the RacR regulatory loop to damage the cells.

The lysozyme gene lysN is a potential target of RacR. (a) Growth curves of JM83, BL21(DE3), DH5α, DH10B, and MG1655 transformed with pBAD or plysN after induction (control [+] in blue and pracR[+] in red) and pracR without induction (pracR [−] in dark). (b) Living cell count after induction. (c) SEM results of the morphology of control/JM83, plysN/JM83, plysN/DH10B, and plysN/MG1655 after induction. Scale bar 5.0 µm. Data represent means ± standard deviations of results from three independent experiments.
Inactivation of lysN rescues JM83 from lysis
To further confirm whether lysN is under control of RacR and accordingly lead to cell lysis, the lysN mutant strain was tested, of which the region from +91 to +201 was replaced by a linker plus 3 × Flag-tag (111 bp) and ended with the stop codon TGA (Fig. 4a). Compared to JM83, cell density of lysN mutant strain (ΔlysN) stayed at a medium level (Fig. 4b), and SEM observation showed that cell debris almost disappeared (Fig. 4c). Taken together, these results showed that inactivation of lysN rescues JM83 from lysis. Meanwhile, when introducing the lysN expression cassette into pracR, as expected, cells would lyse, which indicated that RacR indeed regulates lysN in vivo (Fig. 4b and Supplementary Fig. S1).

The cell growth was partly recovered after lysN mutation. (a) Schematic representation of the mutation of lysN. The three boldface sequences are predicted binding motifs of RacR. (b) Growth curves of ΔlysN strain transformed with pBAD, pracR, plysN, and pracR-PlysN-lysN after induction. Right part were diagrams of these four plasmids, and PlysN represents the promoter regions of lysN. (c) SEM results of the morphology of pracR/ΔlysN after induction. Scale bar 5.0 µm. (d) Living cell count of ΔlysN strains. Data represent means ± standard deviations of results from three independent experiments.
We also noticed the cell elongation phenomenon by RacR overexpression in wild type, ΔlysN strains via SEM, no matter with or without lysN expression (Figs 1c and 4c). This suggests that there may also exist targets other than lysN regulated by RacR, which is responsible for cell elongation instead of cell lysis. While although the elongated cells overexpressing RacR didn’t show significant culture turbidity decrease (purple line in Fig. 4b) or cell debris (Fig. 4c) in the first 2–5 hours, the living cell number was decreased significantly (purple bars in Fig. 4d). The decrease of living cells overexpressing RacR was even more serious than lysN ovexpressing cells in the first 3 hours but ultimately became close after 5 hours. This may imply that the other regulatory pathway other than lysN by RacR is also quite important for cell survival.
RacR triggers transcription of the lysN directly
We then measured the transcription level of lysN in pracR/JM83. The accumulation of RacR led to a more than 4000-fold increase in lysN mRNA level, which is almost silent without RacR induced expression (Fig. 5a). It suggests that RacR has the ability to trigger the transcription of lysN. Then, electrophoretic mobility shift assay (EMSA) was performed with purified His6-RacR and the DNA probe (designated lysN*: from −235 to +7 relative to start codon of lysN). The top of Fig. 4a showed the potential binding motifs in lysN* that derived from half of the palindromic sequence 5′-GCCTAA-3′ and 5′-TTAGGC-3′27. As shown in Fig. 5b, His6-RacR was observed to bind to lysN* probe in a concentration-dependent manner. This interaction was nearly completely blocked by addition of 150-fold unlabeled lysN*, while not by addition of the 150-fold unlabeled unspecific DNA.

RacR directly activated the expression of lysN. (a) qRT-PCR analysis of lysN in JM83 with pracR. (b) EMSA analysis of His6-RacR and lysN*. The 150-fold unlabeled lysN* or unlabeled unspecific DNA was added as a control. (b) Relative activity of β-galactosidase in ΔlysN strain with the LacZ reporter plasmids. Data represent means ± standard deviations of results from three independent experiments. ***P < 0.001.
Furthermore, the upstream region and 90 bp sequence at the 5′ end of lysN was fused to the reporter gene lacZ, and the mutations were introduced into the potential binding motifs, as shown in Supplementary Fig. S2. The result showed the activity of controls were extremely low, while overexpression of RacR led to a 140-fold increase (Fig. 5c), which was highly consistent with the transcriptional levels of lysN in the genome (Fig. 5a). In addition, base substitution mutation significantly decreased the activity of LacZ, although not completely inhibited (Supplementary Fig. S2). These results demonstrated that RacR activates transcription of lysN by directly binding to the promoter region.
JM83 restored after 18 h of induction due to passivation of regulation
However, we were soon puzzled by the subsequent behavior of pracR/JM83, since the cell density of culture would begin to recover after 8 h of induction (Fig. 6a). What’s more, replenishment of strong inducer at 9 h point would not interfere with the original growth trend (Fig. 6a), indicated that the recovery of growth was not due to insufficiently induced expression of RacR. We then checked the morphology of pracR/JM83, plysN/JM83, and pracR/ΔlysN after 18 h of induction, as we expected, Fig. 6b confirmed the recovery of all these strains.

JM83 restored after 18 h of induction. (a) Growth curves of JM83 strain transformed with pBAD or pracR after induction. The “0 h + Ara” strains were only added with 0.2% L-arabinose when OD600 reached 0.6, and the “0 h and 9 h + Ara” strains were added with enough L-Arabinose again at 9 h after induction (the arrows in green and red represent the time to add L-Arabinose). (b) SEM results of four strains after induction. Scale bar 5.0 µm. Data represent means ± standard deviations of results from three independent experiments.
To answer this question, we performed qRT-PCR. We found that the mRNA level of lysN decreased significantly at 18 h after induction (Fig. 7a). Hence, we inferred that there is a strong connection between lysN mRNA level and cell status. We assumed the possibility that the decline of RacR or the passivation of its regulation causes a lower transcriptional level of lysN, and detected cellular RacR protein level of each time points. As shown in Fig. 7a, the His6-RacR protein level did not change by culture, which excluded the possibility of cascaded decrease of RacR and LysN. Even so, we decided to introduce another plasmid in those recovered cells to produce RacR, in case there are any undetectable changes in pracR. We selected monoclonal recovered pracR/JM83 and renamed it JM83-Anti (resist the overexpression of RacR, Anti for short). After checked the availability in JM83 (Fig. 7b), pCA24N-racR was selected to produce pCA24N-racR/Anti (contain pracR and pCA24N-racR). We did not observe a obvious difference in the expression levels of RacR between JM83 and Anti strain, however, the latter cell growth was no longer inhibited (Fig. 7b). This result strengthened the hypothesis of passivation of RacR regulatory, and suggested this negative effect is permanent. Indeed, continuous cells culture imply the heritable peculiarity of passivation, since all daughter cells grew normally in the presence of L-Arabinose (Fig. 7c, G2 to G5). To sum up, although this particular third factor was not identified, we have uncovered the mechanism of cell growth recovery. In the following research, we will explore more deeply about the passivation of RacR regulatory.

The regulation of RacR is invalid in all daughter cells. (a,b) qRT-PCR analysis of pracR/JM83 and growth curves of JM83 and Anti after induction. Western blotting indicates the His6-RacR level in each strain, and the full-length blot is presented in Supplementary Fig. S4. The Anti strain was depicted in this study. (c) The continuous cell culture of pracR/JM83. The whole process contains five generations (G1, G2, G3, G4, and G5). Data represent means ± standard deviations of results from three independent experiments.
Discussion
RacR was previously predicted as a transcriptional repressor that belongs to MerR superfamily, whose inhibitory effect on Rac prophage toxin YdaS has been demonstrated recently27. In our study, RacR overexpression triggered φ80 lysozyme LysN and resulted in cell lysis. This kind of relationship is highly similar to the effect of RerR on the toxin genes in Clostridium difficile28. The phage ΦCD119 regulator RerR has been shown inhibit to the distant toxin genes tcdA and tcdR by directly binding of their promoter, and the PaLoc (pathogenicity locus) of these toxin genes is commonly thought to belong to mobile genetic elements29.
The activation and inhibition to LysN and YdaS mean that the host must provide a tight mechanism to adjust the concentration of RacR. In fact, the irregular palindrome in the racR-ydaS intergenic region raised the possibility that RacR might negatively regulate its own transcription (Supplementary Fig. S2). This assumption is consistent with the characteristic of regulators of MerR superfamily, which bind to palindrome sequences and were reported to regulate their own expression30. Another characteristic of these proteins is the N-terminal located HTH domain and irregular C-terminal domain, the latter is commonly used to bind metal ions, such as Hg2+ for Tn501 protein MerR31. The lysN transcription decreased significantly at 18 h, even reached the same level as the wild-type JM83, while the RacR was constant (Figs 5a and 7a). We propose a hypothesis that the N-terminal of RacR binding the promotor of lysN at the early stage, with the assistance of an unknown metal ion. While at the late stage, the concentration of these ions decreased and the free RacR no longer activates the expression of lysN. However, only three imperfect conserved motifs were found in the upstream region of lysN, and their base composition and spatial arrangement neither can be compared to the regulatory sequence of ydaS. More than that, obvious enzyme activity was detected when lacZ under the control of a mutant lysN promoter (Supplementary Fig. S2), which suggested that these “CCTA” containing motifs were not the key sequences for RacR regulation, and the promoter region of lysN contain some more important motifs.
Although we have uncovered the causes of RacR overexpression leading to JM83 lysis, the growth of ΔlysN strain cannot recover completely (Fig. 4b), indicated that RacR may also influence other genes. In the early stages of our research, the famous phage regulators CI, CII, and Cro were also considered as potential targets for RacR. We detected their mRNA in ΔlysN after 2 h and 18 h of RacR overexpression, as shown in Supplementary Fig. S3. It seems that RacR tried to break the balance of these three regulators in E. coli to establish a phage lytic state32, since the concentration of primary repressor CI was reduced and its negative regulator Cro was increased. However, we did not find active phages in our solid or liquid medium, and the cultures containing broken JM83 cells were not infectious. We speculate that the defects of φ80dlacZΔM15 prophage prevent the assembly and packaging of φ80, but whether CII or Cro plays a role in lysN expression and growth defect of ΔlysN is unknown. On the other hand, the elongation of cells is probably related to division genes, as the proteins that constitute the divisome33, the ZapA-ZapB complex34, and the Tol-Pal system35. It has been reported that the interference with cell division leads to an elongation phenotype in E. coli36,37, which is extremely similar to the cell morphology of RacR overexpressing JM83 (Figs 1c and 4c).
In summary, the regulation of RacR to lysN is special, since they belong to two different prophages. While in the typical phage lytic cycle, the S holin and R transglycosylase are under the strictly controlled of their own major phage regulators, which activated in the late stage to release the phage38. Although the physiological significances for lysozyme activated by foreign regulator still unclear, the model of the cell response to LysN and eventual recovery (Fig. 6a) reveals a diversity of bacterial genetic strategies. We attempted to find clues of the temporal expression of LysN, by introducing a Flag-tag in JM83 genome (Fig. 4a). However, the target protein (LysN_30N-Flag, 8.2 kDa) was not detected successfully, and we supposed that the short half-life period of a small artificial protein affects its detection, since the mRNA of LysN_30N-Flag was transcribed (Supplementary Fig. S5).
Materials and Methods
Bacterial strains and growth conditions
Bacterial strains and the plasmids used in this study are listed in Supplementary Table S1. Unless indicated otherwise, E. coli were grown aerobically at 37 °C in liquid or on solid Luria-Bertani (LB) media. To better observe the lysis process, the E. coli strains harboring pBAD, pCA24N plasmid and their derivatives were cultured at 28 °C after induction. The PCR products of primer pair RacR-1/RacR-2, 19-1/19-2, and RacR-fwd/RacR-rev were ligated into the NdeI/HindIII site of mCherry-pBAD and pCA24N respectively, to forming the pracR, plysN, and pCA24N-racR plasmids. After constructed pracR, the (P19-19)-1/(P19-19)-2 primer pair is used to amplify the lysN expression cassette, which ligated into the NdeI site of pracR to yield pracR-PlysN-lysN plasmid. Antibiotics were added at the following concentrations: kanamycin (50 μg/mL), chloramphenicol (30 μg/mL) and/or ampicillin (100 μg/mL) as appropriate.
Growth curves and spotting assay
Growth was monitored by measuring the optical density (OD) at 600 nm (OD600). A single colony of each strain was inoculated in LB and grown at 37 °C overnight. Then, the strains were transferred to 500 mL flasks containing 100 mL of LB medium and were cultured at 37 °C in a shaking incubator (190 rpm). 0.2% L-Arabinose and/or 1 mM IPTG was added when OD600 reached about 0.6. We recorded the optical density of these strains at an hour intervals for 8 hours at 28 °C. Meanwhile, 100 μL bacterial suspensions after 1 hour, 3 hours and 5 hours of induction were harvested, 10-fold gradient diluted in fresh LB medium and spread on LB agar plates. The plates were incubated at 28 °C for 24 hours followed by calculating the average colony-forming units (CFU) per milliliter according to the formula [(viable count from each concentration × dilution fold × 10)/n]. Above assays were repeated in triplicate.
Scanning electron microscope (SEM)
Equivalent cell densities of different E. coli strains were collected through centrifugation (2300 × g for 5 min at 4 °C) and washed three times with phosphate buffer (PBS, 0.1 M, pH 7.5). Then, the cell pellets were fixed with 2.5% glutaraldehyde at 4 °C for 5 hours. After washing three times at 4 °C, these samples were dehydrated for 10 min each in increasing concentrations of ethanol (30%, 50%, 70%, 80%, and 90% (V/V)). Subsequently, the samples were frozen at −80 °C for about 24 hours, dried with a vacuum freeze dryer, and then observed with S-3400N scanning electron microscopy.
Construction of lysN in-frame deletion mutant ∆lysN
All primers used in mutant construction are listed in Supplementary Table S2. PCR amplifications were performed to generate the upstream fragment of lysN with primer pair 19SY-1/19SY-2 and the downstream part with primer pair 19XY-1/19XY-2. Otherwise, we introduced a linker plus 3 × Flag-tag sequences which replaced the in-frame deletion region from +91 to +201 in lysN (Fig. 4a). The PCR product containing a site-directed deletion of lysN was obtained via overlap PCR with primer pair 19SY-1/19XY-2 and ligated into the NheI/XbaI site of the suicide vector pDMKE (the insB deleted derivative of pDMK39). The resulting plasmid, pDMKE-lysN, were duplicated in E. coli DH5α(λpir) and then electrotransformed into E. coli JM83. Single colonies selected on LB plate with kanamycin and chloramphenicol suggest that the plasmid was integrated into the chromosome by homologous recombination. The double-crossover recombination was selected on LB plate with 10% sucrose. The lysN in-frame deletion mutant was designated as ∆lysN and confirmed via PCR and sequencing.
Quantitative real-time PCR (qRT-PCR)
RNA from E. coli JM83 or ∆lysN strains frozen at −80 °C was extracted using Pure RNA Isolation Kit according to the manufacturer’s protocols. For removal of the remaining DNA, total RNA was incubated with RNase-free DNase I at 28 °C for 1 hour. 1 μg total RNA was used to generate cDNA using Reverse Transcription M-MLV (RNase H-) kit. Subsequently, quantitative real-time PCR was performed according to SYBR Green PCR Master Mix and each sample was made in triplicate. rpoD acts as the internal reference gene. To normalize data, transcription levels of the rpoD gene in all samples were set to 1.0. Relative mRNA levels were analyzed using the 2−ΔCt (ΔCt = Cttested genes − Ct rpoD) method. The primers for qRT-PCR are listed in Supplementary Table S2.
Overexpression and purification of RacR protein
The racR gene was PCR-amplified from E. coli JM83 and cloned into the NdeI/EcoRI site of the pET-28a (+) vector to yield pET28a-racR with an N-terminal His6-tag. The RacR expression plasmid was transformed into E. coli BL21(DE3). The E. coli strain was induced with 1 mM IPTG until OD600 reached about 0.6 and grown at 16 °C for 16 hours. Then, the strain was harvested by centrifugation (5900 × g for 5 min at 4 °C) and washed three times with phosphate buffer. Pellets were resuspended to a final concentration of 10 OD/mL, sonicated on ice, and centrifuged at 5900 × g for 5 min at 4 °C. The protein was then purified via nickeliminodiacetic acid–agarose chromatography and desalinated into 1 × binding buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.1 M NaCl, 0.1 mM dithiothreitol, 5% glycerol, and 10 μg/mL bovine serum albumin27). Purified protein was analyzed by 12% SDS-PAGE, and the protein concentration was determined by the Bradford assay.
Electrophoretic mobility shift assay (EMSA)
EMSAs were carried out using the purified His6-RacR and PCR-amplified DNA probes. The biotin-labeled probes were obtained by PCR with primer head-biotin in Supplementary Table S2, then purified and quantified. Increasing amounts of RacR were added to the 1 × binding buffer that containing target lysN* probes (5 ng) and 50 μg/mL poly(dI·C), and incubated at 28 °C for 40 min. Samples were run on a 6% polyacrylamide gel in 0.5 × TBE buffer at 130 V for 1 hour, then transferred to a nylon membrane at 380 mA for 55 min, subsequently analyzed using chemiluminescent EMSA kits.
β-galactosidase assays
The target fragment was amplified by PCR with primer pair 19_30Z-fwd/19_30Z-rev, 19_30Z-fwd/19M123-4, and 19M123-3/19_30Z-rev respectively, and cloned into the HindIII/PstI of the LacZ reporter vector pXG40. The E. coli strains containing resulting plasmid were grown at 37 °C in LB and induced by adding 0.2% L-Arabinose when OD600 reached about 0.6. After 5 h of induction, the cells were harvested and disrupted in cold phosphate buffer by sonication. The cell debris was removed after centrifugation. Subsequently, β-Galactosidase activity was measured as described previously41.
Western blotting
The target proteins from E. coli strains were separated with 12% SDS-PAGE and transferred to PVDF membranes. The samples were blocked in TBS (2% pH 7.5 Tris HCl, 0.8% NaCl) containing 5% skimmed milk overnight at 4 °C. The membranes were probed with anti-His tag primary antibody (1:2000) at room temperature for 2 hours, washed three times with TBS, then incubated with 1:2000 dilution of horseradish peroxidase-conjugated goat anti-mouse antibody at room temperature for 2 hours. Subsequently, the blot was detected using chemiluminescent with TMB.
Data Availability
All data generated or analysed during this study are included in this article (and its Supplementary Information Files).
References
Juhas, M. et al. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33, 376–393 (2009).
Thomas, C. M. & Nielsen, K. M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol 3, 711–721 (2005).
Gogarten, J. P. & Townsend, J. P. Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol 3, 679–687 (2005).
Ochman, H., Lawrence, J. G. & Groisman, E. A. Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304 (2000).
Mira, A., Martín-Cuadrado, A. B., D’Auria, G. & Rodríguez-Valera, F. The bacterial pan-genome: a new paradigm in microbiology. Int Microbiol 13, 45–57 (2010).
Soucy, S. M., Huang, J. & Gogarten, J. P. Horizontal gene transfer: building the web of life. Nat Rev Genet 16, 472–482 (2015).
Heuer, H. & Smalla, K. Horizontal gene transfer between bacteria. Environ Biosafety Res 6, 3–13 (2007).
Jackson, R. W., Vinatzer, B., Arnold, D. L., Dorus, S. & Murillo, J. The influence of the accessory genome on bacterial pathogen evolution. Mobile Genetic Elements 1, 55–65 (2011).
Liu, X. et al. Physiological function of rac prophage during biofilm formation and regulation of rac excision in Escherichia coli K-12. Sci Rep 5, 16074 (2015).
Lawrence, J. G. & Ochman, H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA 95, 9413–9417 (1998).
Wang, X. et al. Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1, 147 (2010).
Casjens, S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol 49, 277–300 (2003).
Rotman, E., Kouzminova, E., Plunkett, G. & Kuzminov, A. Genome of enterobacteriophage Lula/phi80 and insights into its ability to spread in the laboratory environment. J Bacteriol 194, 6802–6817 (2012).
Durfee, T. et al. The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. J Bacteriol 190, 2597–2606 (2008).
Erez, Z. et al. Communication between viruses guides lysis-lysogeny decisions. Nature 541, 488–493 (2017).
Bender, J. K. et al. Involvement of PatE, a prophage-encoded AraC-like regulator, in the transcriptional activation of acid resistance pathways of enterohemorrhagic Escherichia coli strain EDL933. Appl Environ Microbiol 78, 5083–5092 (2012).
Balasubramanian, D., Ragunathan, P. T., Fei, J. & Vanderpool, C. K. A prophage-encoded small RNA controls metabolism and cell division in Escherichia coli. mSystems 1, e00021–15 (2016).
Reiter, F., Wienerroither, S. & Stark, A. Combinatorial function of transcription factors and cofactors. Curr Opin Genet Dev 43, 73–81 (2017).
Schumacher, M. A., Chinnam, N. B., Cuthbert, B., Tonthat, N. K. & Whitfill, T. Structures of regulatory machinery reveal novel molecular mechanisms controlling B. subtilis nitrogen homeostasis. Gene Dev 29, 451–464 (2015).
Zabidi, M. A. & Stark, A. Regulatory enhancer-core-promoter communication via transcription factors and cofactors. Trends Genet 32, 801–814 (2016).
Bass, S. H. & Yansura, D. G. Application of the E. coli trp promoter. Mol Biotechnol. 16, 253–260 (2000).
Johnson, M. M., Michelhaugh, S. K., Bouhamdan, M., Schmidt, C. J. & Bannon, M. J. The transcription factor NURR1 exerts concentration-dependent effects on target genes mediating distinct biological processes. Front Neurosci 5, 135 (2011).
Low, D. A. & Casadesús, J. Clocks and switches: bacterial gene regulation by DNA adenine methylation. Curr Opin Microbiol 11, 106–112 (2008).
Feng, L. et al. A Qrr noncoding RNA deploys four different regulatory mechanisms to optimize quorum-sensing dynamics. Cell 160, 228–240 (2015).
Peer, E., Rechavi, G. & Dominissini, D. Epitranscriptomics: regulation of mRNA metabolism through modifications. Curr Opin Chem Biol 41, 93–98 (2017).
Xia, H. et al. A yigP mutant strain is a small colony variant of E. coli, and shows pleiotropic antibiotic resistance. Can J Microbiol 63, 961–969 (2017).
Krishnamurthi, R., Ghosh, S., Khedkar, S. & Seshasayee, A. S. N. Repression of YdaS toxin is mediated by transcriptional repressor RacR in the cryptic rac prophage of Escherichia coli K-12. mSphere 2, e00392–17 (2017).
Govind, R. et al. Bacteriophage-mediated toxin gene regulation in Clostridium difficile. J Virol 83, 12037–45 (2009).
Eklund, M. W., Poysky, F. T., Reed, S. M. & Smith, C. A. Bacteriophage and the toxigenicity of Clostridium botulinum type C. Science 172, 480–2 (1971).
Brown, N. L., Stoyanov, J. V., Kidd, S. P. & Hobman, J. L. The MerR family of transcriptional regulators. FEMS Microbiol Rev 27, 145–63 (2003).
Brown, N. L., Pridmore, R. D. & Fritzinger, D. C. The mercury-resistance genes of transposon Tn501: nucleotide sequence of the mer operon and a possible mechanism for mercury detoxification. Biochem Soc Trans 12, 276–277 (1984).
Court, D. L., Oppenheim, A. B. & Adhya, S. L. A new look at bacteriophage lambda genetic networks. J Bacteriol 189, 298–304 (2007).
Du, S. & Lutkenhaus, J. Assembly and activation of the Escherichia coli divisome. Mol Microbiol 105, 177–187 (2017).
Buss, J. A., Peters, N. T., Xiao, J. & Bernhardt, T. G. ZapA and ZapB form an FtsZ-independent structure at midcell. Mol Microbiol 104, 652–663 (2017).
de Boer, P. A. Advances in understanding E.coli cell fission. Curr Opin Microbiol 13, 730–737 (2010).
Krupka, M. et al. Escherichia coli FtsA forms lipid-bound minirings that antagonize lateral interactions between FtsZ protofilaments. Nat Commun 8, 15957 (2017).
Sánchez-Gorostiaga, A. et al. Life without division: physiology of Escherichia coli FtsZ-deprived filaments. mBio 7, e01620–16 (2016).
Węgrzyn, G., Licznerska, K. & Węgrzyn, A. Phage λ-new insights into regulatory circuits. Adv Virus Res 82, 155–78 (2012).
Xiao, J. et al. Characterization of Edwardsiella tarda rpoS: effect on serum resistance, chondroitinase activity, biofilm formation, and autoinducer synthetases expression. Appl Microbiol Biotechnol 83, 151–160 (2009).
Xia, H. et al. EsrE-A yigP locus-encoded transcript-is a 3′ UTR sRNA involved in the respiratory chain of E. coli. Front Microbiol 8, 1658 (2017).
Wang, Y., Ye, J. & Zhang, H. Identification of transcriptional regulatory sequences of yigP gene in Escherichia coli. Wei Sheng Wu Xue Bao 52, 566–72 (2012).
Acknowledgements
We thank Xiaoxue Wang from the South China Sea Institute of Oceanology for providing the pCA24N plasmid used in this work. We are grateful to members of our lab, and specifically to Siyuan Chen and Yajing Kang for their help in this study. National Natural Science Foundation of China (Grant Nos 31372550, 3120026, and 31070073) financially supported this study.
Author information
Authors and Affiliations
Contributions
Q.T., M.F., H.W. and H.Z. designed the experiments. Q.T. and M.F. performed the experiments. Q.T., M.F., B.H. and H.Z. analyzed the data. J.Y. contributed reagents/materials/analysis tools. Q.T. and M.F. wrote the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, Q., Feng, M., Hou, B. et al. Prophage protein RacR activates lysozyme LysN, causing the growth defect of E. coli JM83. Sci Rep 9, 12537 (2019). https://doi.org/10.1038/s41598-019-48690-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48690-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
