
Abstract
Cholera is a life-threatening diarrhoeal disease caused by the human pathogen Vibrio cholerae. Infection occurs after ingestion of the bacteria, which colonize the human small intestine and secrete their major virulence factor – the cholera toxin (CT). The GM1 ganglioside is considered the primary receptor of the CT, but recent studies suggest that also fucosylated receptors such as histo-blood group antigens are important for cellular uptake and toxicity. Recently, a special focus has been on the histo-blood group antigen Lewisx (Lex), however, where and how the CT binds to Lex remains unclear. Here we report the high-resolution crystal structure (1.5 Å) of the receptor-binding B-subunits of the CT bound to the Lex trisaccharide, and complementary quantitative binding data for CT holotoxins. Lex, and also l-fucose alone, bind to the secondary binding site of the toxin, distinct from the GM1 binding site. In contrast, fucosyl-GM1 mainly binds to the primary binding site due to high-affinity interactions of its GM1 core. Lex is the first histo-blood group antigen of non-secretor phenotype structurally investigated in complex with CT. Together with the quantitative binding data, this allows unique insight into why individuals with non-secretor phenotype are more prone to severe cholera than so-called ‘secretors’.
Similar content being viewed by others

Introduction
Cholera is an acute and severe diarrhoeal disease that, if left untreated, can cause serious dehydration and death within hours1. The disease is a major burden, especially where proper sanitation is lacking, in war zones and after natural disasters. The socio-economic costs of cholera are significant, and a global road map to end cholera has been established2. The current outbreak in Yemen is the worst in recent history with more than one million reported cases3,4, further aggravating the ongoing malnutrition crisis.
Cholera is easily transmitted between humans through the faecal-oral route, e.g. by consuming food or water contaminated with the bacterium Vibrio cholerae. The bacteria colonize the small intestine and secrete the cholera toxin (CT)5,6. CT belongs to the protein family of AB5 toxins, which comprise one catalytically active A-subunit and a homopentamer of non-toxic B-subunits6,7 (Fig. 1a). The CT B-pentamer (CTB) facilitates binding to epithelial cells in the small intestine and subsequent cellular uptake of the holotoxin6,8. Once in the cytosol, the A-subunit hijacks the cells’ own signalling pathways, causing the secretion of chloride ions into the intestinal lumen and, due to the osmotic gradient, ultimately the severe watery diarrhoea typical of cholera8,9,10.

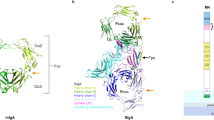
Schematic drawing of CT and its glycan receptors. (a) Toxin binding to host cell. The AB5 toxin CT consists of one catalytically active A subunit (CTA; dark blue) and a pentamer of the receptor-binding B subunits (CTB; light blue). The toxin’s primary and secondary binding sites (for GM1 and HBGAs, respectively) are indicated on one of the five B subunits with a green circle and an orange rectangle, respectively. (b) Structures of the oligosaccharides Lewisx triaose and tetraose, Lewisy tetraose and fucosyl-GM1 hexaose. Fucose residues are highlighted in red. Carbohydrate symbols follow the nomenclature of the Consortium for Functional Glycomics (Nomenclature Committee, Consortium for Functional Glycomics; d-galactose (Gal)–yellow circle, N-acetylgalactosamine (GalNAc)–yellow square, d-glucose (Glc)–blue circle, N-acetylglucosamine (GlcNAc)–blue square, l-fucose (Fuc)–red triangle, N-acetylneuraminic acid (Neu5Ac)–purple filled diamond.
Key aspects of cholera intoxication are still poorly understood, starting with the interplay between the toxins and their cellular receptors, and the cellular uptake mechanism. For decades, the GM1 ganglioside has been considered to be the only cellular receptor for the CT11,12,13. The binding of CT to the branched GM1 pentasaccharide is one of the strongest protein-carbohydrate interactions known (Kd = 43 nM)14. Target recognition is mediated mainly by the two terminal sugar residues galactose and sialic acid, via a “two-fingered grip”15. A large body of research points towards this high-affinity interaction being the main entryway for the CT12,16,17,18,19,20. For example, the incorporation of exogenously added GM1 in human intestinal mucosal cells or rabbit small bowel segments lead to a concentration-dependent increase in the number of toxin-binding structures and intoxication12,16. CT binds GM1 at the “bottom” surface of the B-pentamer, which serves as a landing platform on the cell membrane15 (Fig. 1a). More recently, CT was found to also bind histo-blood group antigens (HBGAs), at a secondary binding site on the lateral side of the B-pentamer21. There is now increasing evidence that GM1 is not the only CT receptor and that also fucosylated structures like HBGAs play an important role for toxin binding, internalization and toxicity13,21,22,23,24,25,26,27,28,29,30,31,32,33,34.
HBGAs are present on the surface of cells in the gastrointestinal tract as well as in bodily secretions, and their expression varies dependent on the individual’s genotype. Intriguingly, CT causes blood-group-dependent cellular intoxication31 despite having only low affinity for human HBGAs, with a Kd in the millimolar range21,27,28,29. Blood group H determinants, which are characteristic for blood group O, exhibit slightly higher affinity to CT than blood group A and B determinants (still in the millimolar range); and patients with blood group O are the most likely to get severe cholera symptoms35,36,37,38. Another group with increased risk of severe cholera are people with non-secretor phenotype. These individuals have a non-functional Se (FUT2) gene, resulting in reduced fucosylation and the absence of HBGAs from bodily secretions39. Compared to GM1, HBGAs are present in the intestine in high concentrations40, making them attractive targets for toxin entry. Indeed, Lex, a monofucosylated HBGA expressed on human granulocytes and intestinal cells, has been shown to specifically bind to CTB and was proposed to function as a cellular receptor32. Furthermore, cholera intoxication in mice can be independent of GM132, and fucose-based inhibitors are potent inhibitors of CT binding and internalization of intestinal cells33,34. It is, however, not clear how the CT binds to Lex and related structures. Recent experiments with CT variants and structure-activity relationship data were interpreted such that Lex binds to the primary binding site32. However, we find this unlikely since all CT-related crystal structures with HBGAs or HBGA-analogues to date feature these ligands in the secondary binding site21,25,26.
We hypothesized that Lex and related sugars interact solely with the secondary binding site of CTB based on their similarity to known toxin-bound HBGAs and human milk oligosaccharides (HMOs)21. The only fucosylated ligand that we deemed likely to bind to the primary binding site was fucosyl-GM1, due to its similarity with GM1. To test this hypothesis, we investigated the binding of various fucosylated structures, including the fucosyl-GM1 oligosaccharide (os), to CTB and CTB variants by X-ray crystallography and surface plasmon resonance (SPR) spectroscopy.
Results
To characterize the interaction of CT with different fucosylated ligands, we co-crystallized its receptor binding B subunits (classical CT biotype) with the Lex trisaccharide, l-fucose and the fucosyl-GM1 hexasaccharide (fucosyl-GM1os) (Fig. 1b). The crystal structures were solved to high resolution (1.5–2.0 Å), based on high-quality X-ray data sets (Table 1). As expected, Lex and l-fucose occupied the secondary binding sites of the CT, while we found fucosyl-GM1os in the primary toxin binding site, binding the CT with the same two-fingered grip as GM1. Its fucose residue contributes little to this interaction, serving more as decoration. Additionally, some of the secondary binding sites showed weak electron density for fucosyl-GM1os. In this case, the interaction was mediated via fucose.
In addition to the qualitative characterization of the CT-ligand interactions by X-ray crystallography, we acquired quantitative binding data by SPR spectroscopy (Table 2). Lex was found to bind more weakly to the CT than the difucosylated HBGA Ley 21, but still had an affinity in the millimolar range, as typical for many protein-carbohydrate interactions. These studies were done with CTB as well as with the CT holotoxin. We also tested several CT variants with mutations in the primary or secondary binding sites. Specifically, we chose CT mutant W88K to knock out the primary binding site, since Trp88 serves an important role for aromatic stacking interactions with the terminal galactose residue in GM1. In line with the crystallographic data, primary binding site knock-out hardly affected CT binding to Lex. In contrast, a mutation in the secondary binding site (H18A) resulted in reduced binding to Lex. Intriguingly, this toxin variant not only bound more weakly to Lex, but concomitantly showed enhanced binding to GM1os, suggesting allosteric cross-talk between the two binding sites.
In the following, we give further technical details of our results.
Crystal structures
CTB complex with Lex
To study the interaction of CT with Lex, we crystallized purified CTB in complex with Lex triaose. Crystals were obtained in two different crystallization conditions. They belonged to space group P212121 and contained two CTB pentamers in the asymmetric unit. Therefore, we can observe a total of 20 crystallographically distinct primary and secondary binding sites (ten of each). The protein structures show the typical “doughnut-shaped” CTB structure of five symmetrically arranged B subunits, each consisting of two three-stranded antiparallel β-sheets with α-helices on both sides15. The structures were determined to 2.0 Å and 1.5 Å resolution, respectively. In both crystal forms, Lex is only observed in the secondary binding sites of the CT. The binding mode and interactions of Lex described here are based on the 1.5 Å resolution structure, which was refined to a high-quality model (Fig. 2a, Table 1; PDB ID: 6HJD, Rfree = 22.4%). Generally, the structure is well defined except for the flexible loop comprising residues 50–61 and the C-terminal asparagine residue, which exhibits some disorder. Inspection of the electron density maps revealed the presence of Lex triaose in eight of ten secondary binding sites (Fig. 2a,b). One additional binding site contained electron density of sufficient quality to place the fucose residue, and the last secondary binding site was blocked by crystal contacts. No ligand density was observed in any of the primary binding sites, even at low sigma cut-off levels.

Lewisx binds to the secondary binding site of the cholera toxin. (a) X-ray structure of CTB in complex with Lex (PDB ID: 6HJD, this work); side and top views (rotated by 90°). The toxin B-pentamer is shown in cartoon and transparent surface representation, and the ligands depicted in stick representation. Lex only binds to secondary binding sites on the lateral side of the toxin. (b) Close-up view of the secondary binding site (chain B), with σA-weighted Fo-Fc electron density map for Lex (grey mesh, contoured at 3.0 σ, generated before placing the ligand) and selected residues depicted in green stick representation. (c) Superimposition of Ley tetraose (blue sticks; PDB ID: 5ELB21) on CTB complex with Lex triaose (green sticks; PDB ID: 6HJD, this work). Carbohydrate residues are labelled in italics, and fucose carbons are numbered. (d) Stereo image of the carbohydrate-toxin interactions. Hydrogen bonds are shown as yellow dashed lines, amino acid residues are labelled with 3-letter code. A hash (#) identifies residues contributed by a neighbouring subunit in the toxin B-pentamer. The figure was prepared with MacPyMol (Schrödinger LLC (www.schrodinger.com/pymol); version 1.8.0.3).
Lex triaose superimposes well with the trisaccharide core of previously characterized HBGA determinants (Fig. 2c), and engages in similar H-bonding interactions (Fig. 2d, Supplementary Table S1)21. Its reducing end N-acetylglucosamine (GlcNAc) points towards residue 47. The secondary binding site of CT recognizes HBGAs mainly through the α1,3-linked fucose (although Ley can bind the CT in two orientations, where either Fucα2 or Fucα3 can serve as main anchoring points). As observed for CTB bound to Ley and A-Ley (5ELB21, 5ELD21), fucose forms hydrogen bonds to the side chain of Gln3# from the adjacent B-subunit and to the main chain of residues 47 and 94 (Fig. 2d, Supplementary Table S1). These data confirm our hypothesis that Lex binds to the secondary CT binding site, similarly to other HBGAs.
CTB complex with l-fucose
A recent study suggested that Lex binds to the GM1 binding site32, however, all relevant toxin crystal structures contain fucosylated molecules in the secondary binding site21,25. To alleviate concerns that protein purification by galactose affinity chromatography may hinder access to the GM1 binding site due to the presence of residual galactose (even though Gal is not observed in our crystal structures), we produced CTB by TALON affinity chromatography exploiting its known affinity to Ni2+ or Co2+ ions41. To directly test the hypothesis by Cervin et al. that fucosylated sugars may bind to the GM1 binding site, TALON-purified CTB was co-crystallized with l-fucose in a molar ratio of 1:500 (B-subunit to ligand). The crystals belonged to space group C2 and contained two CTB pentamers in the asymmetric unit. The structure was refined to 1.95 Å resolution and an Rfree of 23.7% (Table 1). The electron density of the loop regions and the C-terminal Asn103 were more disordered compared to the CTB-Lex structure, however, overall the structure is well defined. Electron density for β-l-fucose is observed in nine of ten secondary binding sites (Fig. 3a,b, Supplementary Table S2). We did not observe electron density corresponding to l-fucose in the GM1 binding site, confirming that fucose only binds to the secondary binding site. Additionally, l-fucose was found sandwiched between two B-pentamers and covalently attached to some of the N-terminal residues. The latter is likely due to non-enzymatic glycosylation42. These interactions are unlikely to be biologically relevant and probably caused by the high molar excess of l-fucose added before crystallization.

CTB complexes with l-fucose and fucosyl-GM1os. l-fucose binds to the secondary CT binding site, while fucosyl-GM1os binds to the primary binding site, facing the cell membrane. The cholera toxin B-pentamer is shown in cartoon and transparent surface representations, and the ligands depicted in stick representation. (a) X-ray structure of CTB in complex with l-fucose (orange sticks; PDB ID: 6HMW, this work); side view. (b) Close-up view of the secondary binding site, with σA-weighted Fo-Fc electron density map for l-fucose (grey mesh, contoured at 3.0 σ, generated before placing the ligand), and selected residues shown in stick representation, amino acid residues are labelled with 3-letter code. (c) X-ray structure of CTB in complex with fucosyl-GM1os (green sticks, with the terminal fucose highlighted in orange; PDB ID: 6HMY, this work). (d) Close-up view of the primary binding site, with σA-weighted Fo-Fc electron density map for fucosyl-GM1os (grey mesh, contoured at 3.0 σ, generated before placing the ligand) and Trp88 shown in stick representation. Carbohydrate residues are labelled in italics. The terminal fucose and glucose residues show weaker electron density compared to the four core residues of fucosyl-GM1os.
CTB complex with fucosyl-GM1os
Our data strongly suggest that the toxin secondary binding site is the sole binding site for Lex and similar sugars. There is, however, one fucosylated oligosaccharide that would be expected to bind to the primary binding site, namely fucosyl-GM1os. Fucosyl-GM1 binds CT almost as strongly as GM143. We crystallized TALON-purified CTB with fucosyl-GM1os in a 1:10 molar ratio (B-subunit to ligand), yielding crystals diffracting to 1.6 Å resolution. The structure was refined to an Rfree of 21.4%. Inspection of the electron density revealed that fucosyl-GM1os indeed binds to the primary binding site, similarly to GM1os, with the additional fucose residue facing outwards toward the solvent (Fig. 3c). The electron density for the fucose residue is less well defined compared to the other sugar residues (Fig. 3d), suggesting that it binds more weakly. It also has higher B-factors (average B-factors for sugar ring atoms in chain A: Fuc > GalNAc > Gal, 28.6 > 20.8 > 17.1 Å2). This explains the small difference in CT affinity between fucosyl-GM1 and GM1, as the fucose residue is not a major contributor to the interaction. In addition to the primary binding sites, we observe fucose binding in some of the toxin’s secondary binding sites. In these sites, fucosyl-GM1os is not well resolved, however, electron density extending from the fucose is compatible with a larger oligosaccharide like fucosyl-GM1os.
Protein interaction by SPR
CTB and Lex triaose
To determine the binding affinity of CTB to Lex, we performed SPR experiments, in which the toxin B-pentamer was immobilized on the SPR chip and the sugar was injected as the analyte over a range of different concentrations. Lex triaose was found to have a lower binding affinity to CTB than Ley tetraose or A-Ley pentaose, which both feature a second fucose residue (Kd = 10 ± 3 mM (Lex triaose), compared to 1.05 ± 0.04 mM (Ley) and 2.2 ± 0.1 mM (A-Ley)21; Supplementary Fig. S1).
CT holotoxin and Lex triaose and tetraose
Hitherto all crystal structures of CTB complexes with fucosylated receptors, including the structures reported here, suggest that Lex-related antigens bind exclusively to the secondary binding site of CT. To verify this binding mode in solution, we performed SPR experiments with holotoxin variants harbouring mutations in the primary binding site (W88K) or the HBGA binding site (H18A, H18AH94A; with residue 94 linking the primary and secondary binding sites) (Fig. 4, Table 2). Native folding of these variants was confirmed by circular dichroism (CD) spectroscopy (Supplementary Fig. S2). Toxin variants or wild-type CT were immobilized on SPR chips, before injecting oligosaccharides at different concentrations. First, we showed that substitution of W88A in the primary binding site prevented GM1os binding (Fig. 4). The other toxin variants bound GM1os similarly to wild-type CT, indicating intact primary binding sites. We then compared binding of Lex oligosaccharides to wild-type CT and CT variants.

SPR sensorgrams and affinity plots for cholera toxin variants. SPR experiments were performed with CT holotoxins coupled to the sensor chip and using Lex tetraose (sensorgrams and corresponding plots of steady state response against concentration) or GM1os (sensorgrams with model fit) as analytes, as indicated in the panel legends. Kd values were calculated from the steady state plots (Lex) or using a Langmuir 1:1 model (GM1os). The Kd value for H18A + Lex could not be calculated since saturation was not reached upon addition of 40 mM ligand. H18AH94A only showed weak interaction for the highest Lex concentrations used; therefore, the affinity plot does not show a saturation-binding curve.
Lex triaose binds equally well to CTB and CT (Kd = 10 ± 3 mM for CTB, 8 ± 3 mM for CT) (Supplementary Fig. S1). This result was expected, since there is no major structural difference between free CTB and the B-pentamer in the holotoxin15,44. In our crystal structures, Lex and related sugars pre-dominantly bind with the α-anomer of the reducing end GlcNAc, whereas relevant glycoconjugates on the cell surface are β-glycosidically linked to proteins or lipids. Lex tetraose, with a fixed β-anomer, and Lex triaose bound equally well to CT (Kd = 8.1 ± 3.2 mM for triaose, 7.7 ± 0.5 mM for tetraose), suggesting that the triaose core is mainly responsible for binding and that the linkage does not significantly affect binding affinity.
Next, we determined binding affinities for all toxin variants using Lex tetraose (Fig. 4, Table 2). The primary binding site variant W88K was found to bind Lex tetraose with an affinity comparable to wild-type CT (Kd = 8.6 ± 0.1 mM for W88K, 7.7 ± 0.5 mM for CT), whereas H18A, which features a mutation in the secondary binding site, exhibited significantly reduced binding to Lex tetraose (Kd > 50 mM for H18A, 7.7 ± 0.5 mM for CT), in agreement with our structural data. This reduced binding affinity is not due to disruption of the primary binding site, since GM1os binding was found to be even stronger than for the wild-type protein (Kd = 39.5 ± 0.9 nM for H18A, 57.7 ± 0.3 nM for CT; Fig. 4). The double mutant H18AH94A, which was designed to disrupt the water network in the binding site and additionally precludes H-bond formation to the His94 side chain, only bound Lex tetraose at the highest analyte concentration applied (40 mM), but bound to GM1os almost as strongly as wild-type CT (Kd = 80.0 ± 0.4 nM for H18AH94A, 57.7 ± 0.3 nM for CT). Our results confirm Lex binding to the secondary binding site of CT and suggest cross-talk between the primary and secondary binding sites, since mutations in the latter affect GM1 binding, reducing the Kd by one third.
Discussion
Lex and similar fucosylated structures bind to the secondary binding site of CT
We set out to explore the molecular interaction of CT with fucosylated receptors, in particular Lex. Already twenty years ago, CTB was shown to bind fucose45. More recently, evidence was presented that HBGAs may act as functional toxin receptors for the related LTB46,47, and fucosylated structures were shown to be functionally active and cause cellular uptake of CT30,32,33,34. For example, CTB binding to jejunal epithelial cells can be blocked by the Lex trisaccharide or a monoclonal antibody against Lex 32. Crosslinking and immunoprecipitation of CTB-bound cellular proteins revealed that CTB binds to glycoproteins modified with Lex 32. It was, however, unclear how Lex binds to CT. Since G33D, a primary site CTB variant with greatly reduced affinity to GM117,48, showed weaker binding to Lex 32, it was suggested that also Lex might bind to the primary binding site32. To explore this possibility, Cervin et al. performed competition experiments with plate-bound GM1 or Lex and CTB pre-incubated with the free sugars32. Competition experiments were also performed with human granulocytes, T84 and Colo205 cells. Despite the fact that these cell lines only express low levels of GM1, pre-incubation of CTB with GM1os had an inhibitory effect. Taken together, these results were interpreted to indicate that GM1 competitively blocks fucosylated sugars from binding to the CT primary binding site32. However, it should be noted that even at high concentrations, GM1os could not block CTB binding completely and Lex could not block binding to plate-bound GM1.
Here we show that Lex binds solely to the secondary binding site of CTB, and even l-fucose alone (when co-crystallized in 500-fold molar excess) did not bind to the primary binding site. Moreover, SPR experiments with toxin variants confirmed that disruption of the secondary binding site, but not the primary binding site, lowered the affinity to Lex (Fig. 4, Table 2). We also found that Lex bound more weakly to CTB than Ley or A-Ley, which is in good agreement with inhibitor studies identifying Ley as the most potent small-molecule inhibitor of CTB33. The binding mode of Lex is highly similar to that of Ley tetraose and A-Ley pentaose21, which both feature an additional fucose residue (Fig. 2c). The partial competition of GM1 and Lex observed by Cervin et al.32 is most likely due to allosteric cross-talk, in line with recent findings49,50. Likewise, we found that the CT secondary binding site variant H18A exhibits increased GM1os binding.
Comparison of ligand structures to structure–activity relationship data
l-fucose has been shown to block CTB binding in cell culture, serving as a promising starting point for the design of novel cholera inhibitors33. Recently, Wands et al. described important molecular determinants of l-fucose by competition binding assays of CTB to various intestinal epithelial cells33. Their structure–activity relationship (SAR) studies showed that the stereochemical positioning of the terminal fucose is crucial for CTB binding. While the removal of hydroxyl groups on C3 or C4 (OH3, OH4) had a significant effect on CTB binding, substitution of the hydroxyl group at C2 (OH2) did not result in reduced binding (compare Fig. 2c). The addition of a hydroxyl group to C6 was tolerated, but the removal of C6 resulted in a complete loss of CTB binding suggesting the importance of a hydrophobic patch within the binding pocket. Addition of a methyl group to OH1, locking the anomeric hydroxyl group in the β-configuration, resulted in less efficient CTB binding compared to l-fucose or α-locked fucose. Finally, the SAR study showed increased binding of α1,2-linked fucose compared to α1,3-linked fucose33.
All crystal structures of CTB with Lex (6HJD; this work) and related molecules, such as Ley or A-Ley (5ELB21 and 5ELD21), show that fucose OH3 can form one or two hydrogen bonds to His94 and to a conserved water molecule (not shown in Fig. 2), explaining why removing OH3 in the SAR study caused reduced CTB binding. OH4 was reported to contribute the strongest to CTB binding33, which is in good agreement with the fact that OH4 forms two hydrogen bonds to backbone atoms of residues 47 and 94 (Fig. 2d). The crystal structures also identify a match for the proposed key hydrophobic residues interacting with C6 in Phe48, possibly together with Ala46 (Fig. 2d).
CTB binds l-fucose with its free anomeric hydroxyl group in the β-configuration (Fig. 3b), thus free fucose is not limited to bind in the α-configuration. However, locking the fucose in the β-configuration by the addition of a methyl group would cause steric clashes with residues Thr47 and Gly45, explaining why α-linked fucose bound stronger to CTB than β-linked fucose33.
According to Wands et al., OH2 does not contribute significantly as a hydrogen bond acceptor33, whereas we observe a hydrogen bond to Gln3# from the adjacent B-subunit (Fig. 2d). However, their conclusion was based on a compound with a fluorine replacing the OH2 group (rather than with OH2 removed), and it is still under debate if fluorine can form hydrogen bonds51,52.
Finally, Wands et al. found increased inhibition of CTB binding by fucosyllactose with α1,2-linked compared to α1,3-linked fucose residues33. The latter showed no difference compared to l-fucose alone. The crystal structure of CTB in complex with Ley (5ELB21) clearly shows that both α1,2-linked and α1,3-linked fucose can bind to CT. However, the preferred orientation appears to be highly influenced by the precise details of ligands and toxins. For example, the structure of a CTB-LTB chimera in complex with a human milk oligosaccharide similar to A-Ley exhibited exclusive binding of Fucα2 in the fucose binding pocket of the toxin25.
In conclusion, the structural data presented here are a very good match to the reported structure–activity relationship data, pointing to the same binding site identified in both studies, i.e. the secondary binding site of the toxin.
Roles of GM1 and fucosylated receptors
A large body of research points to GM1 and fucosylated structures facilitating toxin binding and uptake12,16,17,18,19,20,30,32,33,34, however, little is known about the exact localization and expression of specific fucosylated structures on healthy intestinal cells and tissues32,53,54,55,56,57. It is not clear which fucosylated structures are the major cellular attachment sites besides GM1. However, all evidence points towards glycoproteins with type-2 core structures similar to Lex, Ley and related HBGAs21,27,28,29,30,32,33. In particular, Lex has been shown to allow CTB binding to intestinal cells32.
Whether the binding of GM1 and fucosylated structures happens simultaneously or sequentially is also unknown, nor what triggers or causes toxin uptake. Recent crystal structures from our lab suggest that CTB can bind sugars at both binding sites at the same time, specifically Ley tetraose and galactose21. However, it has not yet been shown if this also holds true for GM1os. A recently published hetero-multivalent binding model suggests that CTB first binds to a high-affinity ligand such as GM1. Subsequent binding occurs much more readily (up to 10,000-fold faster) also for low-affinity ligands like GM2, since binding is then confined to the 2D membrane surface58. In fact, cooperativity was found to be enhanced for a heterogeneous mixture of ligands58. In a biological context, it seems more likely that the toxins first bind to the broadly available fucosylated structures on the cell surface or mucus layer until they can bind to the few available GM1 receptors, preventing the flow in the small intestine to remove unbound toxins from the intoxication site21. In addition, V. cholerae has other virulence factors that may influence receptor distribution and availability, e.g. neuraminidase VcN59. It has been shown recently that VcN can remodel intestinal polysialylated gangliosides into GM1 to provide an increased numbers of cellular receptors for CT59, and it is well conceivable that fucosylated glycoconjugates serve as intermediate anchoring points until VcN unveils enough GM1 receptors. Indeed, several studies showed that the exogenous addition of GM1 to cells markedly increases CT binding and intoxication12,34,60,61. Remarkably, a new study by Sethi et al. indicates that fucosylated glycoconjugates play a functional role in the process, in the presence or absence of GM134.
Correlation with epidemiological data: Why are secretors protected?
Many diseases show blood group association62, and cholera is the textbook example. Individuals with blood group O experience the most severe cholera symptoms35,36,37,38. Similarly, individuals with non-secretor phenotype are at increased risk of severe cholera, whereas so-called ‘secretors’, who secrete HBGAs and display these structures in their small intestinal mucosa, enjoy some protection63,64. Recently, progress has been made in the understanding of cholera ABO(H) blood group dependence21, however, it remains unclear why secretors are protected from severe cholera compared to non-secretors. The work presented here for Lex represents the first relevant structural and affinity data for a non-secretor HBGA, bringing us in a unique position to shed light on this phenomenon.
Non-secretors have a non-functional Se (FUT2) gene, which codes for an α1,2-fucosyltransferase that is important for the generation of HBGAs on epithelial and endothelial cells and most body fluids39. Non-secretors therefore display a distinct set of receptors on their intestinal cells that is enriched in Lewisa (Lea) and Lex antigens, and characterized by limited fucosylation39,55,65. Interestingly, when probing jointly for secretor status and ABO(H) blood group, only secretors with blood group A or B showed less severe cholera symptoms, whereas the disease burden for blood group O individuals was equally high independent of secretor status64, which could be explained if the H-antigen characteristic of blood group O itself is a risk factor for severe cholera.
The H-antigen exists in two types, type-1 and type-2, with different linkages, both of which contain an α1,2-linked fucose. Addition of a second fucose residue (Fucα4 or Fucα3) then gives rise to Leb/y determinants, respectively. If the same fucose is added to HBGA precursors instead, this yields Lea/x epitopes, which in contrast to H-antigens and Leb/y cannot be further modified by A or B glycosyltransferases, since they lack the α1,2-linked fucose. So far, all evidence points towards only type-2 antigens being able to bind to the CT13,21,25, focusing the attention on Ley and Lex. The lower affinity of Lex compared to the other HBGAs makes it unlikely that Lex binding is the cause for more efficient CT uptake in non-secretors. There are only limited data regarding the expression of fucosyltransferases and the distribution of glycoconjugates in human tissues32,53,54,55,56,66. Moreover, it is unknown which fucosylated CT receptors are relevant in vivo. The available data, however, suggest that compared to the villi, the deeper parts of the intestinal membranes are unaffected by the non-functional FUT2 of non-secretors, due to expression of an alternative 1,2-fucosyltransferase encoded by the H gene65,67. Non-secretors cannot secrete soluble ABO(H) glycans, but might still present suitable docking sites (i.e., Ley) on these deeper membrane regions, independent of their secretor status, and cooperative binding is expected to be enhanced for hetero-multivalent binding including GM158. Indeed, Cervin et al. observed tighter binding of CTB to the crypts than to the villi32. Due to the lack of soluble HBGAs, non-secretors would moreover lack competition by soluble and mucin-bound CT receptors, leading to more efficient CT uptake and more severe cholera symptoms.
Conclusions and perspective
The importance of fucosylated sugars for cholera intoxication has recently been in the limelight21,27,28,29,30,31,32,33,34, but there has been limited structural information on their interaction with the CT. Here we show that fucosylated receptors, except fucosyl-GM1, bind solely to the secondary CT binding site, in contrast to recent suggestions32. We provide detailed information on how Lex and l-fucose bind to CTB, and corroborate our findings with quantitative binding data of holotoxin variants. In addition, we present the first CT variant – H18A – with reduced affinity to fucosylated structures, but slightly increased affinity to GM1os. This not only lends further support to a possible cross-talk between the primary and secondary CT binding sites, but may also have practical value. Currently CT is widely used as a marker of GM1 and lipid rafts68. We suggest to replace wild-type CTB with variant H18A as more specific GM1 marker, to limit the number of false-positive results caused by the interaction with secondary binding site receptors. Furthermore, CT variants H18A and H18AH94A, being devoid of a functional secondary binding site, are predicted to facilitate the analysis of cellular uptake in cellular and organoid models31,69 by allowing discrimination of the effects caused by interaction with primary and secondary sites.
Materials and Methods
Mutagenesis
Vector pARCT5, which was kindly provided by Professor Randall K. Holmes, contains an arabinose-inducible CT operon, with signal sequences derived from the LT-IIb B gene70. Site-directed mutagenesis was performed using the manufacturer’s protocol (Q5® Site-Directed Mutagenesis Kit, NEB) and DNA oligo nucleotides (Eurofins Genomics) shown in Table 3. Successful mutagenesis was verified by Sanger sequencing using primers Seq 1–3 (Eurofins Genomics, GATC).
Production of classical CTB, standard protocol
Expression was performed essentially as described previously21. Briefly, the gene for CTB (Uniprot: Q57193; classical biotype) was heterologously expressed in E. coli BL21 (DE3) using a CTB-pET21b+ construct. For protein production, cells were grown at 37 °C in LB medium containing ampicillin until OD600 nm of 0.5 was reached. The temperature was reduced to 25 °C and isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM to start CTB production. Cells were harvested after 14–18 h by centrifugation (6900 × g, 20 min, 4 °C) and the pellet was re-suspended in ice-cold sucrose buffer (20 mM Tris/HCl, 25% (w/v) sucrose, 5 mM EDTA at pH 8.0). After 15 min on ice, the solution was centrifuged (8000 × g, 20 min, 4 °C) and the pellet was re-suspended in periplasmic lysis buffer (5 mM MgCl2, 150 μg/mL lysozyme, DNase). The solution was kept cold for 30 min, centrifuged (8000 × g, 20 min, 4 °C) and dialyzed for several hours against PBS in a Snakeskin tube (Thermo Scientific, 3500 MWCO).
Production of CTB and CT holotoxins for TALON chromatography
The gene for CTB (Uniprot: Q57193) was heterologously expressed in E. coli BL21 (DE3) using a CTB-pET21b +construct. For protein production, cells were grown at 37 °C in LB medium containing ampicillin until OD600 nm of 0.5 was reached. The temperature was reduced to 25 °C and IPTG was added to a final concentration of 0.5 mM to start CTB production.
The genes for CT and CT variants (W88K, H18A, H18AH94A) were heterologously expressed in OverExpress™ C43 (DE3) cells (Sigma) using pARCT5 or pARCT5 derivatives. For protein production, cells were grown at 37 °C in TB medium containing chloramphenicol until OD600 nm of 2.0 was reached. l-arabinose was added to a final concentration of 0.2% (w/v) to start holotoxin production.
Cells were harvested after 14–18 h (CTB) or 3 h (holotoxin) by centrifugation (6900 × g, 20 min, 4 °C) and the pellet was re-suspended in 1/60th volume of TALON A buffer (50 mM sodium phosphate, 300 mM NaCl, pH 8) with 1 mg of polymyxin B (Sigma Aldrich) per mL, cOmplete™ Protease Inhibitor (Roche) and benzonase (EMD Millipore) and shaken at 37 °C for 15 min. Insoluble debris and cells were removed from the periplasmic extracts by centrifugation (8000 × g, 20 min, 4 °C). The filtered supernatant was directly applied to the TALON affinity column.
Purification of CTB using D-galactose affinity chromatography
The protein solution was loaded onto a d-galactose-sepharose affinity column (Thermo Scientific) and eluted using 300 mM galactose in PBS. Fractions containing pure CTB were pooled and concentrated using Vivaspin 20 mL concentrator tubes (5000 MWCO, PES membrane, Sartorius). The protein was subjected to size-exclusion chromatography using PBS and a Superdex 75 column mounted on an ÄKTA purifier (GE Healthcare). Fractions with toxin were pooled, dialyzed over night against Tris buffer (20 mM Tris/HCl, 200 mM NaCl, pH 7.5), concentrated to 3–9 mg/mL, snap-frozen in liquid nitrogen and stored at −80 °C.
Purification of CTB and CT holotoxins using TALON affinity chromatography
As alternative to the d-galactose-sepharose affinity purification (to preclude contamination with galactose), the periplasmic extract of CTB or CT (wild-type and variants of classical biotype) was loaded onto a pre-equilibrated HiTrap TALON crude column (GE Healthcare) and eluted with TALON B buffer (50 mM sodium phosphate, 300 mM NaCl, 50 mM imidazole at pH 8) in order to avoid residual D-galactose bound to the protein. The protein solution was concentrated using Vivaspin concentrator tubes (10000 MWCO, PES membrane, Sartorius) and loaded onto a Superdex 75 (CTB) or Superdex 200 column (holotoxins) mounted on an ÄKTA purifier (GE Healthcare) equilibrated with Tris buffer (20 mM Tris/HCl, 200 mM NaCl at pH 7.5) or PBS. Fractions with CTB pentamer (Tris buffer) were pooled, concentrated to 3 mg/mL, snap-frozen in liquid nitrogen and stored at −80 °C. Fractions with holotoxins (PBS) were pooled and stored at 4 °C.
CD spectroscopy and MS
Correct folding of all CT variants was verified by CD spectroscopy (J-810 CD spectrometer, JASCO). Prior to the measurement, buffer exchange to 10 mM sodium phosphate buffer pH 7.5 was performed using Vivaspin concentrator tubes. All CT variants were characterized by SDS-PAGE analysis, tryptic digestion and mass spectrometry (performed in house). The amino acid substitution H94A was only confirmed indirectly by DNA sequencing, since tryptic digestion and MS did not yield a suitable C-terminal peptide.
Co-crystallization of CTB complexes
CTB (Gal-affinity) and Lex triaose (Gly049, Elicityl) or CTB (TALON) and fucosyl-GM1os (GLY103, Elicityl) were mixed at a molar ratio of 1:10 (B-subunit to ligand). CTB (TALON) and l-fucose (F2252, Sigma Aldrich) were mixed at a molar ratio of 1:500 (B-subunit to ligand), corresponding to a final concentration of 0.14 M of l-fucose, for a protein concentration of 3.3 mg/ml. All samples were incubated at RT for 2 h. Initially, crystals were obtained by sitting-drop vapour-diffusion using a crystallization robot (Oryx4, Douglas Instruments) at 20 °C in the Morpheus screen71 condition A12 (Molecular Dimensions). For CTB and Lex triaose another crystal form was obtained in Morpheus screen condition A4. Crystals from condition A12 were optimized for all ligands by several rounds of hanging-drop vapour-diffusion experiments. Final crystals for the CTB complex with Lex were obtained by mixing protein-ligand solution (1 µL, 3.3 mg/mL) 1∶1 with crystallization buffer (0.1 M Tris (base)/BICINE pH 8.5, 8% PEG1000, 8% PEG3350, 8% MPD, 0.03 M MgCl2, 0.03 M CaCl2) and 0.2 µL seed solution (microseeding, Seed bead kit, Hampton Research). CTB l fucose co-crystals were obtained by mixing protein-ligand solution (1 µL, 3 mg/mL) 1:1 with crystallization buffer (0.1 M Tris (base)/BICINE pH 8.7, 10% PEG1000, 10% PEG3350, 10% MPD, 0.03 M MgCl2, 0.03 M CaCl2). Final crystals for the CTB complex with fucosyl-GM1os were obtained by mixing protein-ligand solution (1.5 µL, 1.5 mg/mL) 1:1 with crystallization buffer (0.1 M Tris (base)/BICINE at pH 8.7, 6% PEG1000, 6% PEG3350, 6% MPD, 0.03 M MgCl2, 0.03 M CaCl2) and cryoprotection was achieved by transferring this crystal to crystallization buffer with higher PEG and MPD concentrations (0.1 M Tris (base)/BICINE pH 8.7, 10% PEG1000, 10% PEG3350, 10% MPD, 0.03 M MgCl2, 0.03 M CaCl2. Crystals were harvested, mounted in loops, and flash-cooled in liquid nitrogen (CTB l fucose, CTB fucosyl GM1os) or a nitrogen cryo-stream (CTB-Lex), respectively.
X-ray data collection and refinement
Synchrotron data collection for the CTB complex with Lex was performed at ID23–1, ESRF, Grenoble, France (100 K, 0.9770 Å). Data collection for CTB complexes with l-fucose or fucosyl-GM1os was performed at BioMAX, Max IV, Lund, Sweden (100 K, 0.9795 Å and 0.9808 Å, respectively). Data were processed with XDS72 and NEGGIA/DECTRIS (CTB l fucose, CTB fucosyl GM1os) or xia2 and DIALS (CTB-Lex)73,74 and AIMLESS from the CCP4 software suite75,76 (Table 1). Diffraction data cut-offs were chosen based on the assessment of the anisotropic CC1/2 and the quality of the electron density. Both crystal forms of CTB-Lex belong to space group P212121 and contain two B-pentamers in the asymmetric unit, but have different cell parameters. For the CTB-Lex model, we used the highest resolution data set (1.5 Å) from an optimized Morpheus A12 condition, which crystallized under similar conditions, and with the same space group and pentamer arrangement (“top-to-top”) as the CTB complex with Ley tetraose reported previously (5ELB21). CTB l fucose and CTB fucosyl GM1os crystals belong to space group C2 and contain two B-pentamers in the asymmetric unit. The structures were solved by molecular replacement using Phaser77 from the CCP4 software suite75,76 and search model 5ELB21, from which ligands and water molecules had been manually removed. To avoid potential model bias, five cycles of refinement including two cycles with simulated annealing (starting temperature of 5000 K) were carried out with the Phenix software suite78. The final model was obtained after several cycles of manual building with Coot79, followed by refinement with REFMAC580. Initial refinement steps involved local NCS restraints, while final refinement steps involved TLS parameterization (REFMAC5, automatic, 5 cycles)81. Water molecules were placed using COOT:Find_waters and then manually inspected for several criteria, including distances from hydrogen-bond donors/acceptors and quality of the electron-density. Most of the disulfide bridges are partly reduced, due to minor radiation damage. Lex triaose, GM1os and fucosyl-GM1os were built using MAKE LIGAND (AceDRG)82 from the CCP4 software suite75,76 and isomeric SMILES strings. The restraints for a Thr-Fuc bond were generated using JLigand83. Lex triaose, fucosyl-GM1os and α-l-fucose or β-l-fucose were included last to avoid model bias. To improve the density for the terminal fucose residue, GM1os was included prior to fucosyl-GM1os. For the CTB complex with fucose, additional elongated electron density was found in two of the primary binding sites, however, the origin of the density could not be identified, even with Polder84 maps calculated with the Phenix software suite78 (the density was clearly not compatible with a sugar ring). PDB_REDO85 was used to evaluate the models before final refinement steps. Occupancies were refined by evaluating the difference Fourier maps and by comparing the B-factors of the ligands with interacting protein atoms (exception: fucose residues in the CTB-fucosyl-GM1os secondary sites, which were modelled at full occupancy). The final models were analysed using the Analyse geometry task of the CCP4 software suite75,76. The percentages of amino acid residues occupying the favoured, allowed and outlier regions in the Ramachandran plot are 97.5/2.5/0.0% for CTB-Lex, 97.4/2.4/0.2% for CTB l fucose, and 97.7/2.3/0.0% for CTB-fucosyl-GM1os, respectively. Figures were generated using PyMol (Schrödinger LLC), α-helices and β-strands were assigned using STRIDE86.
Surface plasmon resonance spectroscopy
SPR analyses were performed using Series S CM5 sensor chips and a Biacore T100 biosensor system (Biacore Life Sciences, GE Healthcare). Due to the high cost of the oligosaccharides combined with the relatively low affinity typical for carbohydrate-protein interactions, experiments were performed in duplicates or triplicates. Proteins were immobilized on the surface of the sensor chip to yield a signal of approximately 4200 response units, as described previously21. Lex tetraose (GLY050, Elicityl) and triaose were dissolved in the running buffer (10 mM HEPES/NaOH, 140 mM NaCl, 3 mM EDTA, 0.005% P20, pH 7.4) and injected in concentrations from 78.125 μM up to 40 mM for 45 s. Dissociation was monitored for additional 15 s. The regeneration was not needed for Lex tetraose, whereas for Lex triaose, a short pulse (6 s) of 3 mM NaOH was injected to completely remove it from the surface. Binding of 3.125–200 nM GM1os (GM1a, GLY096, Elicityl) was monitored for 240 s, with additional 420 s for dissociation. The regeneration of the chip was the same as for Lex triaose. All binding steps were performed at 30 μL/min and 25 °C. The data were evaluated using Biacore T100 Evaluation software. For GM1os, data were fitted to the Langmuir 1:1 interaction model, since responses did not reach the equilibrium during the association phase. Other binding affinities were determined from plots of steady-state analyte binding levels against the concentration.
Data Availability
The coordinates and structure factors have been deposited with the Protein Data Bank with accession codes: 6HJD, 6HMW and 6HMY.
References
Sack, D. A., Sack, R. B., Nair, G. B. & Siddique, A. K. Cholera. Lancet 363, 223–233 (2004).
WHO. Ending cholera: a global roadmap to 2030, http://www.who.int/cholera/publications/global-roadmap-summary.pdf (Date of access: 22/11/2018) (2018).
The Lancet Gastroenterology, H. Health catastrophe: the toll of cholera in Yemen. Lancet Gas troenterol Hepatol 2, 619, https://doi.org/10.1016/S2468-1253(17)30224-8 (2017).
WHO. Yemen, Annual report 2017, https://reliefweb.int/report/yemen/world-health-organization-yemen-annual-report-2017 (Date of access: 29/09/2018) (2018).
Sanchez, J. & Holmgren, J. Cholera toxin - a foe & a friend. Indian J Med Res 133, 153–163 (2011).
Heggelund, J. E., Bjørnestad, V. A. & Krengel, U. Vibrio cholerae and Escherichia coli heat-labile enterotoxins and beyond. In The Comprehensive Sourcebook of Bacterial Protein Toxins (eds Alouf, J., Landant, D. & Popoff, M. R.) 195–229 (Elsevier, 2015).
Merritt, E. A. & Hol, W. G. J. AB5 toxins. Curr Opin Struct Biol 5, 165–171, https://doi.org/10.1016/0959-440X(95)80071-9 (1995).
Chinnapen, D. J., Chinnapen, H., Saslowsky, D. & Lencer, W. I. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol Lett 266, 129–137, https://doi.org/10.1111/j.1574-6968.2006.00545.x (2007).
Gill, D. M. & Meren, R. ADP-ribosylation of membrane proteins catalyzed by cholera toxin: basis of the activation of adenylate cyclase. Proc Natl Acad Sci USA 75, 3050–3054 (1978).
Field, M., Rao, M. C. & Chang, E. B. Intestinal electrolyte transport and diarrheal disease (1). N Engl J Med 321, 800–806, https://doi.org/10.1056/NEJM198909213211206 (1989).
Holmgren, J. Comparison of the tissue receptors for Vibrio cholerae and Escherichia coli enterotoxins by means of gangliosides and natural cholera toxoid. Infect Immun 8, 851–859 (1973).
Holmgren, J., Lönnroth, I., Mansson, J. & Svennerholm, L. Interaction of cholera toxin and membrane GM1 ganglioside of small intestine. Proc Natl Acad Sci USA 72, 2520–2524 (1975).
Ångström, J. et al. Novel carbohydrate binding site recognizing blood group A and B determinants in a hybrid of cholera toxin and Escherichia coli heat-labile enterotoxin B-subunits. J Biol Chem 275, 3231–3238 (2000).
Turnbull, W. B., Precious, B. L. & Homans, S. W. Dissecting the cholera toxin-ganglioside GM1 interaction by isothermal titration calorimetry. J Am Chem Soc 126, 1047–1054, https://doi.org/10.1021/ja0378207 (2004).
Merritt, E. A. et al. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci 3, 166–175, https://doi.org/10.1002/pro.5560030202 (1994).
Fishman, P. H., Moss, J. & Vaughan, M. Uptake and metabolism of gangliosides in transformed mouse fibroblasts. Relationship of ganglioside structure to choleragen response. J Biol Chem 251, 4490–4494 (1976).
Jobling, M. G. & Holmes, R. K. Analysis of structure and function of the B subunit of cholera toxin by the use of site-directed mutagenesis. Mol Microbiol 5, 1755–1767 (1991).
Wolf, A. A. et al. Ganglioside structure dictates signal transduction by cholera toxin and association with caveolae-like membrane domains in polarized epithelia. J Cell Biol 141, 917–927 (1998).
Badizadegan, K. et al. Floating cholera toxin into epithelial cells: functional association with caveolae-like detergent-insoluble membrane microdomains. Int J Med Microbiol 290, 403–408, https://doi.org/10.1016/S1438-4221(00)80052-1 (2000).
Fujinaga, Y. et al. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to endoplasmic reticulm. Mol Biol Cell 14, 4783–4793, https://doi.org/10.1091/mbc.e03-06-0354 (2003).
Heggelund, J. E. et al. High-resolution crystal structures elucidate the molecular basis of cholera blood group dependence. PLoS Pathog 12, e1005567, https://doi.org/10.1371/journal.ppat.1005567 (2016).
Morita, A., Tsao, D. & Kim, Y. S. Identification of cholera toxin binding glycoproteins in rat intestinal microvillus membranes. J Biol Chem 255, 2549–2553 (1980).
Monferran, C. G., Roth, G. A. & Cumar, F. A. Inhibition of cholera toxin binding to membrane receptors by pig gastric mucin-derived glycopeptides: differential effect depending on the ABO blood group antigenic determinants. Infect Immun 58, 3966–3972 (1990).
Balanzino, L. E., Barra, J. L., Galvan, E. M., Roth, G. A. & Monferran, C. G. Interaction of cholera toxin and Escherichia coli heat-labile enterotoxin with glycoconjugates from rabbit intestinal brush border membranes: relationship with ABH blood group determinants. Mol Cell Biochem 194, 53–62, https://doi.org/10.1023/A:1006971913175 (1999).
Holmner, Å. et al. Novel binding site identified in a hybrid between cholera toxin and heat-labile enterotoxin: 1.9 Å crystal structure reveals the details. Structure 12, 1655–1667, https://doi.org/10.1016/j.str.2004.06.022 Erratum in: Structure 15, 253 (2007). (2004).
Holmner, Å., Askarieh, G., Ökvist, M. & Krengel, U. Blood group antigen recognition by Escherichia coli heat-labile enterotoxin. J Mol Biol 371, 754–764, https://doi.org/10.1016/j.jmb.2007.05.064 (2007).
Heggelund, J. E. et al. Both El Tor and classical cholera toxin bind blood group determinants. Biochem Biophys Res Commun 418, 731–735, https://doi.org/10.1016/j.bbrc.2012.01.089 (2012).
Mandal, P. K. et al. Towards a structural basis for the relationship between blood group and the severity of El Tor cholera. Angew Chem Int Ed Engl 51, 5143–5146, https://doi.org/10.1002/anie.201109068 (2012).
Vasile, F. et al. Comprehensive analysis of blood group antigen binding to classical and El Tor cholera toxin B-pentamers by NMR. Glycobiology 24, 766–778, https://doi.org/10.1093/glycob/cwu040 (2014).
Wands, A. M. et al. Fucosylation and protein glycosylation create functional receptors for cholera toxin. Elife 4, e09545, https://doi.org/10.7554/eLife.09545 (2015).
Kuhlmann, F. M. et al. Blood group O-dependent cellular responses to cholera toxin: parallel clinical and epidemiological links to severe cholera. Am J Trop Med Hyg 95, 440–443, https://doi.org/10.4269/ajtmh.16-0161 (2016).
Cervin, J. et al. GM1 ganglioside-independent intoxication by cholera toxin. PLoS Pathog 14, e1006862, https://doi.org/10.1371/journal.ppat.1006862 (2018).
Wands, A. M. et al. Fucosylated molecules competitively interfere with cholera toxin binding to host cells. ACS Infect Dis, https://doi.org/10.1021/acsinfecdis.7b00085 (2018).
Sethi, A. et al. Cell type and receptor identity regulate cholera toxin subunit B (CTB) internalization. Interface Focus 9, 20180076, https://doi.org/10.1098/rsfs.2018.0076 (2019).
Glass, R. I. et al. Predisposition for cholera of individuals with O blood group. Possible evolutionary significance. Am J Epidemiol 121, 791–796 (1985).
Swerdlow, D. L. et al. Severe life-threatening cholera associated with blood group O in Peru: implications for the Latin American epidemic. J Infect Dis 170, 468–472 (1994).
Harris, J. B. et al. Blood group, immunity, and risk of infection with Vibrio cholerae in an area of endemicity. Infect Immun 73, 7422–7427, https://doi.org/10.1128/IAI.73.11.7422-7427.2005 (2005).
Harris, J. B. et al. Susceptibility to Vibrio cholerae infection in a cohort of household contacts of patients with cholera in Bangladesh. PLoS Negl Trop Dis 2, e221, https://doi.org/10.1371/journal.pntd.0000221 (2008).
Henry, S., Oriol, R. & Samuelsson, B. Lewis histo-blood group system and associated secretory phenotypes. Vox Sang 69, 166–182, https://doi.org/10.1111/j.1423-0410.1995.tb02591.x (1995).
Breimer, M. E., Hansson, G. C., Karlsson, K. A., Larson, G. & Leffler, H. Glycosphingolipid composition of epithelial cells isolated along the villus axis of small intestine of a single human individual. Glycobiology 22, 1721–1730, https://doi.org/10.1093/glycob/cws115 (2012).
Dertzbaugh, M. T. & Cox, L. M. The affinity of cholera toxin for Ni2+ ion. Protein Eng 11, 577–581 (1998).
Furth, A. J. Methods for assaying nonenzymatic glycosylation. Anal Biochem 175, 347–360, https://doi.org/10.1016/0003-2697(88)90558-1 (1988).
Masserini, M., Freire, E., Palestini, P., Calappi, E. & Tettamanti, G. Fuc-GM1 ganglioside mimics the receptor function of GM1 for cholera toxin. Biochemistry 31, 2422–2426, https://doi.org/10.1021/bi00123a030 (1992).
O’Neal, C. J., Amaya, E. I., Jobling, M. G., Holmes, R. K. & Hol, W. G. Crystal structures of an intrinsically active cholera toxin mutant yield insight into the toxin activation mechanism. Biochemistry 43, 3772–3782, https://doi.org/10.1021/bi0360152 (2004).
Mertz, J. A., McCann, J. A. & Picking, W. D. Fluorescence analysis of galactose, lactose, and fucose interaction with the cholera toxin B subunit. Biochem Biophys Res Commun 226, 140–144, https://doi.org/10.1006/bbrc.1996.1323 (1996).
Galvan, E. M., Diema, C. D., Roth, G. A. & Monferran, C. G. Ability of blood group A-active glycosphingolipids to act as Escherichia coli heat-labile enterotoxin receptors in HT-29 cells. J Infect Dis 189, 1556–1564, https://doi.org/10.1086/383349 (2004).
Galvan, E. M., Roth, G. A. & Monferran, C. G. Functional interaction of Escherichia coli heat-labile enterotoxin with blood group A-active glycoconjugates from differentiated HT29 cells. FEBS J 273, 3444–3453, https://doi.org/10.1111/j.1742-4658.2006.05368.x (2006).
Merritt, E. A. et al. Structural studies of receptor binding by cholera toxin mutants. Protein Sci 6, 1516–1528, https://doi.org/10.1002/pro.5560060716 (1997).
Hatlem, D., Heggelund, J. E., Burschowsky, D., Krengel, U. & Kristiansen, P. E. 1H, 13C, 15N backbone assignment of the human heat-labile enterotoxin B-pentamer and chemical shift mapping of neolactotetraose binding. Biomol NMR Assign 11, 99–104, https://doi.org/10.1007/s12104-017-9728-9 (2017).
Heggelund, J. E. et al. Specificity of Escherichia coli heat-labile enterotoxin investigated by single-site mutagenesis and crystallography. Int J Mol Sci 20, https://doi.org/10.3390/ijms20030703 (2019).
Howard, J. A. K., Hoy, V. J., O’Hagan, D. & Smith, G. T. How good is fluorine as a hydrogen bond acceptor? Tetrahedron 52, 12613–12622, https://doi.org/10.1016/0040-4020(96)00749-1 (1996).
Dalvit, C., Invernizzi, C. & Vulpetti, A. Fluorine as a hydrogen-bond acceptor: experimental evidence and computational calculations. Chem. Eur. J. 20, 11058–11068, https://doi.org/10.1002/chem.201402858 (2014).
Björk, S., Breimer, M. E., Hansson, G. C., Karlsson, K. A. & Leffler, H. Structures of blood group glycosphingolipids of human small intestine. A relation between the expression of fucolipids of epithelial cells and the ABO, Le and Se phenotype of the donor. J Biol Chem 262, 6758–6765 (1987).
Finne, J. et al. Novel polyfucosylated N-linked glycopeptides with blood group A, H, X, and Y determinants from human small intestinal epithelial cells. J Biol Chem 264, 5720–5735 (1989).
Ravn, V. & Dabelsteen, E. Tissue distribution of histo-blood group antigens. APMIS 108, 1–28, https://doi.org/10.1034/j.1600-0463.2000.d01-1.x (2000).
Henry, S. M. Molecular diversity in the biosynthesis of GI tract glycoconjugates. A blood-group-related chart of microorganism receptors. Transfus Clin Biol 8, 226–230, https://doi.org/10.1016/S1246-7820(01)00112-4 (2001).
Robbe, C. et al. Evidence of regio-specific glycosylation in human intestinal mucins: presence of an acidic gradient along the intestinal tract. J Biol Chem 278, 46337–46348, https://doi.org/10.1074/jbc.M302529200 (2003).
Krishnan, P. et al. Hetero-multivalent binding of cholera toxin subunit B with glycolipid mixtures. Colloids Surf B Biointerfaces 160, 281–288, https://doi.org/10.1016/j.colsurfb.2017.09.035 (2017).
Alisson-Silva, F. et al. Human evolutionary loss of epithelial Neu5Gc expression and species-specific susceptibility to cholera. PLoS Pathog 14, e1007133, https://doi.org/10.1371/journal.ppat.1007133 (2018).
Cuatrecasas, P. Gangliosides and membrane receptors for cholera toxin. Biochemistry 12, 3558–3566 (1973).
Moss, J., Fishman, P. H., Manganiello, V. C., Vaughan, M. & Brady, R. O. Functional incorporation of ganglioside into intact cells: induction of choleragen responsiveness. Proc Natl Acad Sci USA 73, 1034–1037, https://doi.org/10.1073/pnas.73.4.1034 (1976).
Heggelund, J. E., Varrot, A., Imberty, A. & Krengel, U. Histo-blood group antigens as mediators of infections. Curr Opin Struct Biol 44, 190–200, https://doi.org/10.1016/j.sbi.2017.04.001 (2017).
Chaudhuri, A. & DasAdhikary, C. R. Possible role of blood-group secretory substances in the aetiology of cholera. Trans R Soc Trop Med Hyg 72, 664–665 (1978).
Arifuzzaman, M. et al. Individuals with Le(a+b−) blood group have increased susceptibility to symptomatic Vibrio cholerae O1 infection. PLoS Negl Trop Dis 5, e1413, https://doi.org/10.1371/journal.pntd.0001413 (2011).
Glynn, L. E., Holborow, E. J. & Johnson, G. D. The distribution of blood-group substances in human gastric and duodenal mucosa. Lancet 273, 1083–1088 (1957).
Torrado, J., Blasco, E., Cosme, A., Gutierrez-Hoyos, A. & Arenas, J. I. Expression of type 1 and type 2 blood group-related antigens in normal and neoplastic gastric mucosa. Am J Clin Pathol 91, 249–254 (1989).
Mollicone, R., Bara, J., Le Pendu, J. & Oriol, R. Immunohistologic pattern of type 1 (Lea, Leb) and type 2 (X, Y, H) blood group-related antigens in the human pyloric and duodenal mucosae. Lab Invest 53, 219–227 (1985).
Chiricozzi, E., Mauri, L., Ciampa, M. G., Prinetti, A. & Sonnino, S. On the use of cholera toxin. Glycoconj J, https://doi.org/10.1007/s10719-018-9818-7 (2018).
Zomer-van Ommen, D. D. et al. Functional characterization of cholera toxin inhibitors using human intestinal organoids. J Med Chem 59, 6968–6972, https://doi.org/10.1021/acs.jmedchem.6b00770 (2016).
Jobling, M. G., Palmer, L. M., Erbe, J. L. & Holmes, R. K. Construction and characterization of versatile cloning vectors for efficient delivery of native foreign proteins to the periplasm of Escherichia coli. Plasmid 38, 158–173, https://doi.org/10.1006/plas.1997.1309 (1997).
Gorrec, F. The MORPHEUS protein crystallization screen. J Appl Crystallogr 42, 1035–1042, https://doi.org/10.1107/S0021889809042022 (2009).
Kabsch, W. XDS. Acta Crystallogr D Biol Crystallogr 66, 125–132, https://doi.org/10.1107/S0907444909047337 (2010).
Parkhurst, J. M. et al. Robust background modelling in DIALS. J Appl Crystallogr 49, 1912–1921, https://doi.org/10.1107/S1600576716013595 (2016).
Winter, G. et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr D Struct Biol 74, 85–97, https://doi.org/10.1107/S2059798317017235 (2018).
Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760–763, https://doi.org/10.1107/S0907444994003112 (1994).
Potterton, L. et al. CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr D Struct Biol 74, 68–84, https://doi.org/10.1107/S2059798317016035 (2018).
McCoy, A. J. et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674, https://doi.org/10.1107/S0021889807021206 (2007).
Adams, P. D. et al. The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106, https://doi.org/10.1016/j.ymeth.2011.07.005 (2011).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, https://doi.org/10.1107/S0907444910007493 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367, https://doi.org/10.1107/S0907444911001314 (2011).
Winn, M. D., Isupov, M. N. & Murshudov, G. N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr 57, 122–133 (2001).
Long, F. et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr D Struct Biol 73, 112–122, https://doi.org/10.1107/S2059798317000067 (2017).
Lebedev, A. A. et al. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D Biol Crystallogr 68, 431–440, https://doi.org/10.1107/S090744491200251X (2012).
Liebschner, D. et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr D Struct Biol 73, 148–157, https://doi.org/10.1107/S2059798316018210 (2017).
Joosten, R. P., Long, F., Murshudov, G. N. & Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220, https://doi.org/10.1107/S2052252514009324 (2014).
Heinig, M. & Frishman, D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res 32, W500–502, https://doi.org/10.1093/nar/gkh429 (2004).
Acknowledgements
We are grateful to our colleagues Per Eugen Kristiansen for help with CD spectroscopy, Bernd Thiede for MS measurements, Kaare Bjerregaard-Andersen for synchrotron data collection for CTB-fucosyl-GM1 crystals, Gregor Bajc for SPR support, and Steven R. H. Wilson and Nikolina Sekulic for constructive comments on the manuscript. We would further like to thank Professor Randall K. Holmes for the generous gift of plasmid pARCT5, and the staff at the ESRF and Max IV for assistance and support using beamlines ID23-1 and BioMAX, respectively. This work was funded by the University of Oslo (position of J.B.H.) and the Norwegian Research Council (Grant 247730). Further support was by iNEXT and the Norwegian Graduate School in Biocatalysis (BioCat).
Author information
Authors and Affiliations
Contributions
U.K. conceived the research; J.B.H. designed the experiments; J.B.H. and V.H. performed the experiments; J.B.H., J.E.H., V.H., G.A. and U.K. analysed the data; U.K. validated the crystal structures, J.B.H. and U.K. wrote the manuscript, with input from all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heim, J.B., Hodnik, V., Heggelund, J.E. et al. Crystal structures of cholera toxin in complex with fucosylated receptors point to importance of secondary binding site. Sci Rep 9, 12243 (2019). https://doi.org/10.1038/s41598-019-48579-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48579-2
This article is cited by
-
Holotoxin disassembly by protein disulfide isomerase is less efficient for Escherichia coli heat-labile enterotoxin than cholera toxin
Scientific Reports (2022)
-
Glycan-dependent cell adhesion mechanism of Tc toxins
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
