
Abstract
Functioning as miRNA sponges, long non-coding RNA (lncRNA) exert its pharmacological action via regulating expression of protein-coding genes. However, the lncRNA-mediated ceRNA in cerebral Infarction (CI) remains unclear. In this study, the expression recordsets of mRNA, lncRNA and miRNA of CI samples were obtained from the NCBI GEO datasets separately. The differentially expressed lncRNAs (DELs), miRNAs (DEMis) and mRNAs (DEMs) were identified by limma package in R platform. A total of 267 DELs, 26 DEMis, and 760 DEMs were identified as differentially expressed profiles, with which we constructed the ceRNA network composed of DELs-DEMis-DEMs. Further, clusterProfiler package in R platform is employed for performing Gene Ontology (GO) and KEGG pathway analysis. An aberrant ceRNA network was constructed according to node degrees in CI, including 28 DELs, 19 DEMs and 12 DEMis, from which we extracted the core network, in which 9 nodes were recognized as kernel genes including Tspan3, Eif4a2, rno-miR-208a-3p, rno-miR-194-5p, Pdpn, H3f3b, Stat3, Cd63 and Sdc4. Finally, with the DELs-DEMis-DEMs ceRNA network provided above, we can improve our understanding of the pathogenesis of CI mediated by lncRNA.
Similar content being viewed by others

Introduction
Stroke is a very high risk factor for death and/or disability in the world,80% of which would be blamed for cerebral ischemic due to thromboembolic occlusion in the cerebral artery1,2. Half of the Cerebral Infarction(CI) affected individuals would suffer death or disability3,4. What’s more, the indirect cell death signals transmitted to heart would increase the risk of cardiovascular diseases roughly by three times5. Necropsy analyses of patients died of stroke indicate a high prevalence of coronary atherosclerosis and myocardial infarction6,7. However, it is still vague what the behind molecular progressions of CI are. It’s a milestone for understanding the molecular nature of CI to develop effective therapeutics.
More and more attention has been put into the regulatory network composed of long non-coding RNAs (lncRNAs), microRNAs (miRNAs) and messenger RNAs (mRNAs) to clarify the mechanisms underlying in CI. Several clinical and/or experimental studies have reported that some lncRNAs such as linc-DHFRL1-4, SNHG15, linc-FAM98A-3, SNHG12 and GAS5 take important part in the pathological development of CI8,9,10.
miRNA is a non-coding RNA composed of 22 nucleotides inhibiting the expression of a target gene by competitive binding to the response elements of microRNA. The regulating network between miRNAs and their target genes affected a variety of biological processes. miR-143-3p, miR-125a-5p and miR-125b-5p were evaluated as diagnostic biomarkers for acute CI of which the potential clinical use were also comprehensively investigated by Mahir Karakas et al.11 in 2017. Li et al.12 compiled miRNAs with the functions of regulating stroke and pre-disease mechanisms whose potential therapeutic value were further highlighted in clinical settings.
The development of CI has been proven to be involved in the competing endogenous RNA (ceRNA) hypothesis which was proposed by Salmena and colleagues in 2011. For example, Yan et al.13 uncovered the MEG3/miR-21/PDCD4 ceRNA strategy as a novel therapeutic intervention in regulating the molecular mechanisms of cerebral ischemic stroke. Chen et al.14 indicated that GAS5 acted as a ceRNA for miR-137/Notch1 signaling pathway to promote the progression of ischemic stroke form which an extensive understanding and novel therapeutic options for CI are provided.
In this paper, we retrieved RNA expression data from NCBI GEO datasets and analyzed the expression profiles between rats with middle cerebral artery occlusion (MCAO) and Sham operation. Following, we compared differentially expressed lncRNAs, miRNAs and mRNAs between the two groups. Finally, 12 miRNAs, 19 mRNAs and 28 lncRNAs were filtered out to build the lncRNA-miRNA-mRNA ceRNA network, from which we constructed a sub-network composed of 9 hub nodes including Tspan3, Eif4a2, rno-miR-208a-3p, rno-miR-194-5p, Pdpn, H3f3b, Stat3, Cd63 and Sdc4.
Materials and Methods
Collection of raw data
The expression recordsets of Rat mRNAs were downloaded from NCBI GEO (GSE97537) of platform GPL1355 containing 7 Sprague-Dawley rats with MCAO and 5 with Sham operation. Rat miRNAs expression data were downloaded from NCBI GEO (GSE97532) of platform GPL21572 of which containing 3 MCAO operated rats and 3 Sham operated rats. Rat lncRNAs microarray data containing 5 MCAO operated rats and 5 Sham operated rats were collected from NCBI GEO (GSE78200) of platform GPL18694. The approval from the Ethics Committee is exempt for the data deriving from the GEO database.
Screening strategy for differentially expressed lncRNAs, miRNAs and mRNAs
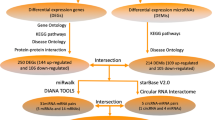
The differentially expressed lncRNAs (DELs), miRNAs (DEMis) and mRNAs (DEMs) between the Sham operated and MCAO groups were determined by the two-class differential examination. The t-test was applied to filter the differentially expressed genes. The DELs, DEMis and DEMs were selected according to the P-values < 0.05 and fold change (log FC) > Mean (log FC) + 2*SD (log FC). In order to visualize the DELs, DEMis and DEMs, heat maps and volcano maps were generated by employing the ggplot215 and pheatmap16 packages in the R platform.
Prediction of target lncRNAs and mRNAs of DEMis
Firstly, the UCSC Genome Browser(http://genome.ucsc.edu/) which is proud to visualize interactions between regions of the genome were employed to annotate the lncRNAs17. The interaction between lncRNAs and miRNAs were predicted by LncBase Predicted v.2 of DIANA Tools18 and then validated by the RNAhybrid program19. The predicted lncRNAs of lncRNA-miRNA pairs was further filtered by matching the DELs selected before, then we can get the information of DELs-DEMis pairs.
Next, The targeted mRNA of DEMis were retrieved from MiRBase20, MirTarBase21 and Targetscan22. All these three miRNA references databases were highly reliable. The predicted mRNAs of mRNA-miRNA pairs was further filtered by matching the DEMs selected before, then we got the information of DEMis-DEMs.
Finally, The pairs of DELs-DEMis and DEMis-DEMs were certified.
The construction of DELs-DEMis-DEMs network
The DELs-DEMis-DEMs network was reconstructed by aggregating all co-expression competing triplets identified above, and was visualized using Cytoscape software at the same time. All node degrees of the DELs-DEMis-DEMs network were calculated simultaneously.
Functional enrichment analysis
Gene Ontology (GO) Biological Processes term and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway were analyzed using clusterProfiler package23 in R platform to make a better understanding of the behind biological mechanisms of DEMs in the DELs-DEMis-DEMs network. Then the topGO package24 of R platform was employed to reconstruct the GO interaction network.
Data acquisition
The datasets analyzed in this study are available in the GEO datasets, https://www.ncbi.nlm.nih.gov/gds.
Results
Screening results of DELs in CI
The expression levels of lncRNAs in 5 MCAO operated rats and 5 Sham operated rats were investigated in this study. According to the screening criterion described above, the cutoff for log FC of lncRNAs was 43.713, 177 (66.29%) up-regulated lncRNAs and 90 (33.71%) down-regulated lncRNAs were identified by using the limma package25 of R platform. In Table 1, we provided the top 25 up-regulated and 25 down-regulated ones including their symbol, logFC value, P-value together with FDR values. We also provided a complete file of DELs in appendix 1. In Fig. 1, a volcano map illustrating the distribution of all the DELs on the correlation of –log10 (p-value) and log2FC was exhibited. All the expression levels of lncRNAs were normalized to the sample mean. The heat map of DELs was also generated by pheatmap package in R platform as shown in Fig. 2 from which the difference between MCAO and Sham groups was visually displayed. As we can see in Table 1 and Fig. 1, the up-regulated lncRNAs are more significant than the down-regulated ones while the logFC value is relatively close.

Volcano map of DELs. Up-regulated genes are represented by red spots while down-regulated genes by blue spots.

Heat map of DELs. The left 5 samples were from Sham group, the right 5 samples were from MCAO group. The color from blue to red shows the progression from low expression to high expression.
The DEMis screening results in CI
The expression levels of miRNAs in 3 MCAO operated rats and 3 Sham operated rats were investigated. The cutoff for log FC of miRNAs was 0.524, 13 (50.00%) up-regulated miRNAs and 13 (50.00%) down-regulated ones were identified. Table 2 with their symbol, logFC, P-value and FDR values of all DEMis were provided. A complete file of DEMis was also settled and uploaded in appendix 2. In Fig. 3, the distribution of miRNAs on the correlation of –log10 (p-value) and log2FC was displayed by a volcano map, together with a heat map of DEMis as shown in Fig. 4 presenting the difference between MCAO and Sham groups directly. All the expression levels of miRNAs were normalized to the sample mean. There is no obvious difference between up-regulated miRNAs and down-regulated ones, as we can tell from Table 2 and Fig. 3.

Volcano map of DEMis. Up-regulated genes are represented by red spots while down-regulated genes by blue spots.

Heat map of DEMis. The left 3 samples were from Sham group, the right 3 samples were from MCAO group. The color from blue to red shows the progression from low expression to high expression.
Results of the DEMs screening in CI
7 MCAO operated rats and 5 Sham operated rats, whose expression levels of mRNAs were investigated in this study. The cutoff for log FC of mRNAs is 0.671, 563 (74.08%) up-regulated mRNAs and 197 (25.92%) down-regulated ones were identified. The top 25 of each are exhibited in Table 3, accompanying with their symbol, logFC, P-value and FDR values. We also provided a complete file of DEMs in appendix 3. Figure 5, a volcano map, illustrated the distribution of all the mRNAs on the correlation of –log10 (p-value) and log2FC vividly, based on the premise that all the expression levels of mRNAs were normalized to the sample mean. A heat map as shown in Fig. 6 was plotted to exhibit the difference between MCAO and Sham group. we can tell that the difference of up-regulated mRNAs is more significant than the down-regulated ones, reports Table 3 and Fig. 5, but the FDR value between them is fundamentally close.

Volcano map of DEMs. Up-regulated genes are represented by red spots while down-regulated genes by blue spots.

Heat map of DEMs. The left 5 samples were from Sham group, the right 7 samples were from MCAO group. The color from blue to red shows the progression from low expression to high expression.
Functional enrichment analysis of DEMs in CI
The ClusterProfiler package in the R platform was employed to execute KEGG and GO (Biological Process) analysis of DEMs in the ceRNA network, illuminating the mechanisms involved in the development of CI. 154 KEGG pathways were enriched, of which the top 20 ones were outlined in Table 4. The most important 10 pathways were shown in Fig. 7, for which bearing the most significant p-values. As we can see, Calcium signaling pathway, MAPK signaling pathway, Ras signaling pathway, Phospholipase D signaling pathway, PI3K-Akt signaling pathway, Endocrine resistance, Propanoate metabolism, cGMP-PKG signaling pathway, Glycine, serine and threonine metabolism and Neuroactive ligand-receptor interaction were involved in the pathological development of CI.

The first 10 Enriched KEGG pathways of DEMs in Cerebral Infarction. The x-axis indicates the number of DEMs participated in the pathway.
The 235 enriched GO terms in the “Biological Process (BP)” were revealed by GO analysis, including response to molecule of bacterial origin, negative regulation of immune system process, response to lipopolysaccharide, regulation of leukocyte activation, and so forth. The first 10 terms were considered as the most important ones for the most significant p-values they bearing, as shown in Fig. 8. In order to reflect the inner interactions among these GO terms, we reconstructed the GO interaction network as shown in Fig. 9.

The first 10 Enriched GO BP terms of DEMs in Cerebral Infarction.

The GO interaction network of DEMs in Cerebral Infarction.
Reconstruction of a ceRNA network in CI
A ceRNA regulatory network of lncRNA-miRNA-mRNA was constructed to further illustrate the interaction between DELs, DEMis and DEMs which is helpful for understanding the role of lncRNAs in CI betterly.
First, LncBase Predicted v.2 of DIANA Tools was used to predict the interaction between lncRNAs and miRNAs, the interactions were further validated by the RNAhybrid program. Among all the 23 DEMis, there was 12 miRNAs interacting with 28 DELs identified by limma.
Next, the targeted mRNA of 12 DEMis in miRNA-lncRNA pairs were retrieved from MiRBase, MirTarBase and Targetscan. We predicted that the 12 miRNAs could interact with 19 differentially expressed mRNAs identified above. Following, a ceRNA regulatory network of CI was reconstructed by incorporating 28 DELs, 19 DEMs and 12 DEMis, as shown in Fig. 10A.

DELs mediated ceRNA regulatory network in Cerebral Infarction. (A) DELs mediated ceRNA network; (B) The sub-network; (C) The cnetplot of mRNAs in the ceRNA regulatory network. The red nodes indicate up-regulation expression while blue down-regulation. DELs, DEMis and DEMs are indicated by diamonds, ellipses, and rectangle, respectively.
The hub genes in the ceRNA network were recognized in the engaged of Cytoscape plug-in MCODE.A total of 9 nodes, including Tspan3, Eif4a2, rno-miR-208a-3p, rno-miR-194-5p, Pdpn, H3f3b, Stat3, Cd63 and Sdc4, could be selected hub nodes. The sub-network was shown in Fig. 10B. Two lncRNAs (Tspan3, Eif4a2) were found that not only had higher node degrees, but also had a higher number of lncRNA-miRNA and miRNA-mRNA pairs. This suggests that the two lncRNAs may play crucial roles in the origin and development of CI, which could be selected as the key lncRNAs. A cnetplot of hub genes indicated that the sub-network could participate in the pathological development process of CI via cell-substrate adhesion, positive regulation of cell adhesion, regulation go cell-substrate adhesion, multicellular organism growth and cell-matrix adhesion biological processes, as shown in Fig. 10C.
Validation of key genes in the ceRNA sub-network
Different modeling platforms were employed to verify the validation of the key genes in the sub-network of CI. lncRNA Hif1a and Fam98a were reported down-regulated significantly in rat cerebral cortex and mice brain endothelium26,27. Tspan3 and Eif4a2 are the two lncRNAs with up-regulated expression in the current network. A Pearson and Spearman correlation analysis on the expression among them in GSE78200 was executed to determine the validity of key lncRNAs in our finding. As shown in Fig. 11A, Tspan3 and Eif4a2 were positively correlated with each other and both negatively correlated with Hif1a and Fam98a (P < 0.05, P < 0.01). GSE46266, a GEO dataset emphasizing on the microRNAs involved in regulating embolic stroke recovery following spontaneous reperfusion in rat, and GSE86291, another GEO dataset emphasizing on microRNAs expression in Homo sapiens of hyperacute cerebral infarction were selected to determine the validity of key microRNAs. Compared to normal groups, the expression of rno-miR-208a of MCAO in GSE46266 was up regulated and hsa-miR-194 of MCAO in GSE86291 was down regulated, respectively, which is consistent with the expression patterns of the very two microRNAs in the current study, as shown in Fig. 11B,C. GSE119121 dataset was employed to verify the mRNAs, from which we can tell that Stat3, Cd63, H3f3b and Pdpn of MCAO were significantly up regulated than normal group, as shown in Fig. 11D–G, in keeping with our finding and an important backing up was provided.

Validation of key genes of ceRNA sub-network in Cerebral Infarction. (A) Correlation of Tspan3, Eif4a2 with Hif1a, Fam98a; (B) The expression value of rno-miR-208a between MCAO and Normal group in GSE_46266; (C) The expression value of rno-miR-194 between MCAO and Normal group in GSE_86291; (D,E,F,G)The expression value of Stat3, Cd63, H3f3b and Pdpn between MCAO and Normal group in GSE_119121. The red color in A indicate negative correlation while blue positive correlation, size of circle indicates correlation value. *P < 0.05, **P < 0.01
Discussion
Stroke ranks only second to heart disease for death and adult disability worldwide28. Ischemic stroke accounts for approximately 85% of acute cerebral vascular diseases29. To provide more timely reporting, only the datasets published after 2017 in GEO were included to construct the ceRNA network for illuminating the behind mechanism of CI, considering the data homogeneity requirement, GSE78200, GSE97537 and GSE97532 were filtered out to construct the ceRNA network of lncRNA-miRNA-mRNA incorporating 28 DELs, 19 DEMs and 12 DEMis. Further, a sub net-work including 9 nodes was reconstructed to propose a deeper understanding for the development of CI.
Tspan3 and Eif4a2 are the two lncRNAs in the ceRNA network. The former is a member of tetraspanin family which is widely expressed in oligodendrocytes, which forms tight junctions (TJs) of myelin sheaths in central nervous system30.
In this paper, we filtered miR-194 as a hub node in the sub- ceRNA regulatory of CI. Ayako Takuma et al.31 found that mir-194-1 in whole blood was down regulated significantly in their analysis of the effect of ischemic infarction. Sen Matsumoto et al.32 demonstrated that in the serum of patients after acute myocardial infarction(AMI) onset, miR-194 combined with miR-192 and miR-34a were unregulated as early as a median of 18 days. They came to the conclusion that miR-194 could serve as predictive indicators of HF. miR-208a, another microRNA in the ceRNA network and also an important member in the miR-208 family, takes important part in the development of cardiac diseases, such as myocardial infarction, hypertrophy, cardiac fibrosis and heart failure33. Several studies on the distribution of miRNAs in the heart, brain, kidney, lung, liver etc.34,35,36 revealed that miR-208a is exclusively expressed in heart, but here interestingly in this paper, we found the up regulation of miR-208a in blood of CI rat model which may serves as a backing for the brain-heart interaction theory.
Given the evidence of the cross regulation including hypothalamic-pituitary-adrenal axis, sympathetic and parasympathetic regulation, mircoRNAs and systemic inflammation7 in the brain-heart interaction after stroke, here we believe that miR-194 and miR-208a have special significance for the occurrence and development of CI which need further validation of related experiments.
Pdpn, H3f3b, Stat3, Cd63 and Sdc4 are the selected mRNAs in the ceRNA network. Kolar et al.37 suggested Pdpn as a novel cell surface marker for brain lesions with gliomas and non-neoplastic, which prevents brain injury and gliomas via normal host response. In the conclusion of Cimini et al.38, the expression of Pdpn in the infarcted myocardium were useful for identifying different cell categories, epitopes of fibrogenic and endothelial commitment. H3f3b, in charge of encoding the variant histone H3.3, is mutated in pediatric brain and bone malignancies at high frequency39.
Stat3, a signal transduction and transcriptive activation factor known as the signal transducers and activators of transcription family 3 protein, is, is easily activated by cerebral ischemic injury reported by several studies, implicating its vital role in the pathophysiological process of cerebral ischemia and reperfusion injury as well40. Endothelial Stat3 is essential for long-term recovery after stroke for its regulations on angiogenesis, axon growth and ECM-remodeling which might serve as a potential target for stroke treatment via fostering angiogenesis and neuroregeneration41. Phosphorylation of Stat3 at tyrosine Y705 residue is involved in microglial-mediated inflammatory processes. Pro-inflammatory cytokines after brain injury would trigger JAK kinase-induced phosphorylation of Stat3 and further regulate inflammatory process of many CNS diseases by JAK2/Stat3 pathways29,42,43. CRYAB/Stat3 pathway could adjust neuroinflammation, which takes important part in ischemic stroke-induced secondary cerebral injury44. Stat3/VEGF signaling pathway is an important pathway that affects angiogenesis and cognitive deficits in the cerebral small vessel disease45.
CD63 is one of platelet activation markers (CD62P, CD63, and CD40L). Tsai et al.46 demonstrated that the expression of CD63 and CD62P which were mainly enhanced in large-vessel cerebral infarction was significantly higher in acute stroke patients than in convalescent stroke and control subjects. The enhanced platelet activity would be blamed for the poor outcome and high recurrent stroke rate in large artery cerebral infarction. CD63 is also one of exosomes markers (CD63, HSP70 and TSG101) protecting remote ischemic postconditioning (RIP) on neurological damage in femoral arteries. Xiao et al.47 highlighted the importance of CD63 for CI based on their finding that CD63 was increased significantly in plasma of rat model with RIP. In the opinion of Bielecka-Dabrowa et al.48, Sdc4 serves as the only biomarkers independently distinguishing HF pts with preserved ejection fraction from reduced ejection fraction.
Based on the results of ceRNA network pharmacology analysis, we constructed a core network composed of 9 key genes including Tspan3, Eif4a2, rno-miR-208a-3p, rno-miR-194-5p, Pdpn, H3f3b, Stat3, Cd63 and Sdc4 that were thought to participate the key pathological progress of CI.
Combined with reports from existing literatures, Tspan3, a member in tetraspanin superfamily widely expressed in central nervous system, were verified to be the most important functional lncRNA regulating Tspan3/miR-194/Cd63 and Tspan3/miR-208a/Stat3 signaling pathways in CI. However, due to the lacking of direct experimental validation, the hypothesis generated above should be handled cautiously.
Conclusion
Taken together, all the nodes in the sub-ceRNA network affect the pathological process of CI directly or indirectly. Tspan3 is the key functional lncRNA in CI regulating Tspan3/miR-194/Cd63 and Tspan3/miR-208a/Stat3 signaling pathways. However, systematic and rigorous experiments are needed to verify our findings.
References
Tian, T. et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 150, 137–149 (2018).
Kelly, P., Kavanagh, E. & Murphy, S. Stroke: New Developments and Their Application in Clinical Practice. Semin Neurol 36, 317–323 (2016).
Kamp, M. et al. A Systematic and Meta-Analysis of Mortality in Experimental Mouse Models Analyzing Delayed Cerebral Ischemia After Subarachnoid Hemorrhage. Transl Stroke Res 8, 206–219 (2017).
Rohlwink, U. et al. Biomarkers of Cerebral Injury and Inflammation in Pediatric Tuberculous Meningitis. Clin. Infect. Dis. 65, 1298–1307 (2017).
Chen, J. et al. MiR-126 Affects Brain-Heart Interaction after Cerebral Ischemic Stroke. Transl Stroke Res 8, 374–385 (2017).
Gongora-Rivera, F. et al. Autopsy prevalence of coronary atherosclerosis in patients with fatal stroke. Stroke 38, 1203–1210 (2007).
Chen, Z. et al. Brain-Heart Interaction: Cardiac Complications After Stroke. Circ. Res. 121, 451–468 (2017).
Deng, Q. et al. Differential long noncoding RNA expressions in peripheral blood mononuclear cells for detection of acute ischemic stroke. Clin. Sci. 132, 1597–1614 (2018).
Zhao, M., Wang, J., Xi, X., Tan, N. & Zhang, L. SNHG12 Promotes Angiogenesis Following Ischemic Stroke via Regulating miR-150/VEGF Pathway. Neuroscience 390, 231–240 (2018).
Zheng, Z., Liu, S., Wang, C. & Han, X. A Functional Polymorphism rs145204276 in the Promoter of Long Noncoding RNA GAS5 Is Associated with an Increased Risk of Ischemic Stroke. J Stroke Cerebrovasc Dis (2018).
Karakas, M. & Zeller, T. A Biomarker Ocular: Circulating MicroRNAs Toward Diagnostics for Acute Ischemic Stroke. Circ. Res. 121, 905–907 (2017).
Li, G. et al. Impact of microRNAs on ischemic stroke: From pre- to post-disease. Prog. Neurobiol. 163–164, 59–78 (2018).
Yan, H. et al. Long non-coding RNA MEG3 functions as a competing endogenous RNA to regulate ischemic neuronal death by targeting miR-21/PDCD4 signaling pathway. Cell Death Dis 8, 3211 (2017).
Chen, F., Zhang, L., Wang, E., Zhang, C. & Li, X. LncRNA GAS5 regulates ischemic stroke as a competing endogenous RNA for miR-137 to regulate the Notch1 signaling pathway. Biochem. Biophys. Res. Commun. 496, 184–190 (2018).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York (2016).
Kolde, R. pheatmap: Pretty Heatmaps. R package version 1.0.10., https://CRAN.R-project.org/package=pheatmap (2018).
Rosenbloom KR et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41(Database issue), D56–63 (2013 Jan).
Maria D. Paraskevopoulou et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucl. Acids Res, gkv1270 (2016).
Marc, R., Peter, S., Matthias, H. & Robert, G. Fast and effective prediction of microRNA/target duplexes RNA. RNA (2004).
Kozomara, A. S. G.-J. miRBase: annotating high confidence microRNAs using deep sequencing data. NAR 42, D68–D73 (2014).
Chou, C. H. et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Research (2018).
Agarwal, V., Bell, G. W., Nam, J. & DP, B. Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005 (2015).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A. Journal of Integrative Biology 16, 284–287 (2012).
Alexa, A. & Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R package version 2.32.0. (2016).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research 43, e47 (2015).
Dharap, A., Nakka, V. P. & Vemuganti, R. Effect of focal ischemia on long noncoding RNAs. Stroke 43, 2800–2802 (2012).
Zhang, J. et al. Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia. Experimental neurology 277, 162–170 (2016).
Feigin, V., Lawes, C., Bennett, D., Barker-Collo, S. & Parag, V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol 8, 355–369 (2009).
Meng, H. et al. Neuronal Soluble Fas Ligand Drives M1-Microglia Polarization after Cerebral Ischemia. CNS Neurosci Ther 22, 771–781 (2016).
Tiwari-Woodruff, S. et al. OSP/claudin-11 forms a complex with a novel member of the tetraspanin super family and beta1 integrin and regulates proliferation and migration of oligodendrocytes. J. Cell Biol. 153, 295–305 (2001).
Takuma, A. et al. Gene Expression Analysis of the Effect of Ischemic Infarction in Whole Blood. Int J Mol Sci 18 (2017).
Matsumoto, S. et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ. Res. 113, 322–326 (2013).
Huang, Y. & Li, J. MicroRNA208 family in cardiovascular diseases: therapeutic implication and potential biomarker. J. Physiol. Biochem. 71, 479–486 (2015).
Callis, T. et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Invest. 119, 2772–2786 (2009).
Ji, X. et al. Plasma miR-208 as a biomarker of myocardial injury. Clin. Chem. 55, 1944–1949 (2009).
van Rooij, E. et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 17, 662–673 (2009).
Kolar, K. et al. Podoplanin: a marker for reactive gliosis in gliomas and brain injury. J. Neuropathol. Exp. Neurol. 74, 64–74 (2015).
Cimini, M., Cannatá, A., Pasquinelli, G., Rota, M. & Goichberg, P. Phenotypically heterogeneous podoplanin-expressing cell populations are associated with the lymphatic vessel growth and fibrogenic responses in the acutely and chronically infarcted myocardium. PLoS ONE 12, e0173927 (2017).
Shi, L., Shi, J., Shi, X., Li, W. & Wen, H. Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis. J. Mol. Biol. 430, 1562–1565 (2018).
Chen, S. et al. Homocysteine induces mitochondrial dysfunction involving the crosstalk between oxidative stress and mitochondrial pSTAT3 in rat ischemic brain. Sci Rep 7, 6932 (2017).
Hoffmann, C. et al. Vascular signal transducer and activator of transcription-3 promotes angiogenesis and neuroplasticity long-term after stroke. Circulation 131, 1772–1782 (2015).
Tian, Y. et al. Upregulation of miR-216a exerts neuroprotective effects against ischemic injury through negatively regulating JAK2/STAT3-involved apoptosis and inflammatory pathways. J. Neurosurg., 1–12 (2018).
Zhang, Y. et al. Extract Inhibits Astrocytic Lipocalin-2 Expression and Alleviates Neuroinflammatory Injury via the JAK2/STAT3 Pathway After Ischemic Brain Stroke. Front Pharmacol 9, 518 (2018).
Qiu, J. et al. Sinomenine activates astrocytic dopamine D2 receptors and alleviates neuroinflammatory injury via the CRYAB/STAT3 pathway after ischemic stroke in mice. J Neuroinflammation 13, 263 (2016).
Wang, W. & Hu, W. Salvianolic acid B recovers cognitive deficits and angiogenesis in a cerebral small vessel disease rat model via the STAT3/VEGF signaling pathway. Mol Med Rep 17, 3146–3151 (2018).
Tsai, N. et al. Levels and value of platelet activation markers in different subtypes of acute non-cardio-embolic ischemic stroke. Thromb. Res. 124, 213–218 (2009).
Xiao, B. et al. Endothelial cell-derived exosomes protect SH-SY5Y nerve cells against ischemia/reperfusion injury. Int. J. Mol. Med. 40, 1201–1209 (2017).
Bielecka-Dabrowa, A. et al. Differences in biochemical and genetic biomarkers in patients with heart failure of various etiologies. Int. J. Cardiol. 221, 1073–1080 (2016).
Acknowledgements
This work was supported by Chinese Medicine Pharmaceutical Key Discipline of Shaanxi province [Grant Number 303061107]; Key research and development plan of Shaanxi province [Grant Number 2018SF-314]; Natural Science Foundation of China [Grant Number 81703720]; Discipline Innovation team Project of Shaanxi University of Chinese Medicine(2019-YL11).
Author information
Authors and Affiliations
Contributions
J.-b.Z., H.-b.C. and X.-f.Z. wrote the main manuscript text. D.-y.G., J.T., and Y.W. analyzed the data. Y.-l.L., F.W., J.-x.C. and J.W. performed the data extraction and prepared all the figures and tables. Y.-j.S. designed the study and all the authors amended the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zou, JB., Chai, HB., Zhang, XF. et al. Reconstruction of the lncRNA-miRNA-mRNA network based on competitive endogenous RNA reveal functional lncRNAs in Cerebral Infarction. Sci Rep 9, 12176 (2019). https://doi.org/10.1038/s41598-019-48435-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48435-3
This article is cited by
-
Molecular docking, network pharmacology and experimental verification to explore the mechanism of Wulongzhiyangwan in the treatment of pruritus
Scientific Reports (2023)
-
Construction of a lncRNA-associated competing endogenous RNA regulatory network after traumatic brain injury in mouse
Molecular Brain (2022)
-
Integrated analysis of lncRNA–miRNA–mRNA ceRNA network and the potential prognosis indicators in sarcomas
BMC Medical Genomics (2021)
-
LncRNA TINCR favors tumorigenesis via STAT3–TINCR–EGFR-feedback loop by recruiting DNMT1 and acting as a competing endogenous RNA in human breast cancer
Cell Death & Disease (2021)
-
A bioinformatics investigation into molecular mechanism of Yinzhihuang granules for treating hepatitis B by network pharmacology and molecular docking verification
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
