
Abstract
We investigate the formation of thin ionic layers driven by electro-osmotic forces, that are commonly found in micro- and nano-channels. Recently, multi-layers have been reported in the literature. However, the relation between classical Debye layers and multi-layers, which is a practically and fundamentally important question, was previously unexplained. Here, we fill this gap by using a continuum approach to investigate the flow of lithium ions inside double-layered graphene sheets. Fluid flow, charge conductivity and thermal stability will be investigated. We show that the separation and strength of forces between the sheets, the external electric field and thermal effects determine the topology of the ionic layers between the graphene sheets.
Similar content being viewed by others


Introduction
Double layers refer to the thin layers of charges, that form near the surface of objects, where the thickness of layers can be characterized by the Debye length. Such layers commonly occur in large surface area to volume ratio systems at the micro- and nano-scale accounting for some important phenomena including electrochemical behavior of electrodes1. Asymptotic matching is a common technique to solve the boundary layer problem for double layers1,2. Recently, multi-layers that are comprised of free ions, occupy the entire cavity were shown to occur at the nanoscale3,4,5. However, the relationship between the classical double layers and multi-layers is missing. Here, we adopt the Poisson-Nernst-Planck equation in conjunction with the mean field theory to investigate the ion transport inside nanomaterials including double-layered graphene sheets. Graphene sheets have a huge potential to be used as anodes for high capacity lithium-ion batteries. Graphene-related materials6,7,8 and graphene-based composite materials9,10,11 are also found to enhance charge storage of lithium-ion batteries.
Graphene sheets possess superior mechanical, electronic and magnetic properties to form promising materials for modern applications12. Due to these merits, atomistic models have been used to simulate lithium ion transport between two graphene sheets13. Density functional theory has also been employed to investigate the bonding and magnetic properties of metal atoms encapsulated inside graphite14. However, these first principle calculations are limited when analyzing the existence of multi-layers due to the heavy computational time needed for such methods. Therefore, a continuum approach turns out to be a computationally effective way to describe the ion transport in mesoscopic systems. Chan and Hill15 developed an empirical model to study the lithium storage and diffusion times between graphene. Diffusion and transport models16,17 have also been derived for depicting charge transport in lithium-ion batteries, where electro-chemical factors and boundary conditions for different components can be carefully addressed. Multi-scale modeling2 taking into account the merits of both atomistic and continuum approaches has also be used to tackle ion storage problems for lithium-ion batteries18. Shi et al.18 predicted the path-independent properties of lithium ion batteries in multiple spatial and temporal scales.
Amongst those continuum approaches, the Poisson-Nernst-Planck (PNP) equation19,20,21 has proven to be extremely useful in scrutinizing ion transport inside systems with larger length scales. Ernesto et al.16 adopted the PNP equation and took into account Faraday’s and Ohm’s laws for both the electrode and electrolyte to derive important quantities such as the cell voltage, and the electrode and electrolyte potentials for lithium-ion batteries16. Their research provides a vital theoretical background to determine certain electrical properties for lithium-ion batteries, which is crucial to supplement experimental efforts. More importantly, analytic and asymptotic solutions20,22 have been sought for the PNP equation for some simple geometries. However, the above continuum approaches using the PNP equation fail to address ion transport at the nanoscale as the molecular effects are significant in a porous cavity. Meng et al.3 employed the PNP equations in conjunction with a classical density functional theory to describe lithium ion flow and discover multi-layers at the nanoscale. The present authors have also successfully incorporated molecular and steric effects into the PNP equation to investigate particle-laden flow problems at the nanoscale4,5, where the force fields are approximated by the mean field theory23,24 and the steric effects are computed using the number density derived from the PNP equation. Mean field theory has been successfully employed in condensed matter physics25, ultra-filtration using carbon nanotube membranes26,27, axial buckling of nanopeapods28, nano drug delivery29 and nanoshuttle memory devices30.
In this paper, we show that the combination of the PNP equation and the mean field theory is a computationally effective method for describing the ionic flow inside regular nanomaterials, and can predict the double and multi-layers inside double-layered graphene. This provide a theoretical background for the establishment of the relation between different types of layers. In the following sections, numerical methods for solving the layering problem will be derived. These methods will be used to derive criteria for the occurrence and the properties of double and multi-layers. The transition between double and multi-layers will also be discussed. Some approximate analytical treatments are also provided to support our argument.
Methods
In this section, we present a general theoretical framework for ion transport inside nanomaterials such as double-layered graphene. Such methods have successfully been used in4,5. Both the approximate theoretical treatment and the numerical methods for the present problem will be discussed later, and the extension to other devices such as lithium-ion batteries, and photovoltaic and fuel cells can be found in16,17, where addition conditions such as the neutrality is assumed and the dynamics for each components is taken into account.
General theory
Suppose the flux of (any) ions inside a nanomaterial is driven by the diffusion generated by the concentration gradient, advection by the flow field, and molecular interactions among ions and between ions and the host material, and is given by
where c, u, D, kB, T and F denote the ion concentration, the fluid velocity, the diffusion coefficient, the Boltzmann constant, the temperature and the total intrinsic force between ions and between the ion and the host nanomaterial, respectively. The conservation of mass inside the host material gives the Nernst-Planck-type equation1
For constant D and T, Eq. (2) reduces to
The flow field u can be described by the Navier-Stokes equation for a Newtonian fluid and the incompressibility condition subject to the external electric field, E,
where ρ, η and ρE denote the fluid density, viscosity and charge density, respectively. Furthermore, the charge density can be derived from Poisson’s equation, which is given by
where ε and φ denote the electrical permittivity and the potential induced by the ions. We assume that the interaction of ions is subject to non-bonded electrical and van der Waals forces, which can be modeled by
where rij, qi, εij and σij denote the atomic distance between the i-th and j-th atoms, charge for i-th ion, the potential well depth between i and j atoms and the van der Waals diameter between i-th and j-th atoms. In addition, σij = (σi + σj)/2 and \({\varepsilon }_{ij}=\sqrt{{\varepsilon }_{i}{\varepsilon }_{j}}\). Following the account given by Cox et al., the total pairwise interaction, E between two ions/molecules with surface areas S1 and S2 can be approximated by23,24
where η1 and η2 denote the number density of atoms on the surface S1 and S2, respectively. Given U, the force field between two molecules can be easily computed by F = −▽U. We comment that Eqs (3–5) and (7) constitute the major equations for this paper.
Double-layered graphene
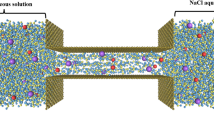
Here, we consider the transport of lithium ions inside double-layered graphene, where the coordinate system used in this paper and the direction of the electric field are shown in Fig. 1. To generate tractable numerical results, we only investigate finite systems with separation between graphene sheets, Ly and base area, Lx × Lz. But, the voltage difference of 5 V is applied into the system to produce physically realistic results. We also assume the periodic conditions in the x and z directions, and use Dirichlet condition for fluid flow and ionic concentration in the y direction.

Double-layered graphene sheets, where the coordinate system adopted in this paper and the direction of the external electric field are shown.
The electric field is applied parallel to the graphene sheets in the z direction. The x-axis is defined to be parallel to the graphene sheets but perpendicular to the field direction, while the y-axis is defined to be perpendicular to the graphene sheets (see Fig. 1). We smear the carbon atoms on the surface of graphene so that the interaction between lithium ions and graphene can be described by the 6–12 Lennard-Jones potential31. Given that, the total force acting on a lithium ion inside the cavity5 is approximated by Eq. (7), this is given by
where η, \(A=4{\varepsilon }_{ij}{\sigma }_{ij}^{6}\) and \(B=4{\varepsilon }_{ij}{\sigma }_{ij}^{12}\) denote the number density of carbon on the graphene, the attractive and repulsive constants for the lithium ions and carbon, respectively. Unlike the usual PNP formulation at the microscale32, ion-ion interactions are significant at the nanoscale. We describe these interactions by HH(r) at r, which is mediated by the average field generated by other ions from x positions, where x ≠ r. By considering both electric and ionic interactions, i.e. Eq. (6) and following the account given by33, we model such ion-ion interactions by the integral
where c(r), c(x), qr, qx, BI and dx denote the number density of ions at r, the number density of ions at x, charges at the site r, charges at the site x, the ion-ion repulsive constant and the volume element for x, respectively. Following33, cut-off lengths are introduced to obtain convergent results since singularities occur when x→r. We comment that graphene induces forces on ions only in the y direction, whereas steric effects act on the ions from all directions (see Eq. (8)).
Now, the flow field inside the cavity is driven by the external electric field, which is also affected by the feedback of electric field generated by ions and the base voltage (if applicable). We further assume that the flow is sufficiently slow at the nanoscale so that Eq. (4) reduces to Stoke’s flow, from which the steady solution can be obtained by solving
where ηv denotes the dynamic viscosity. Therefore, we can use Poisson’s equation, i.e. Eq. (5) and the potential distribution of ions to determine u.
Results and Discussions
Finite difference method can be used to solve the PNP equation and the general algorithm for the simulations is given as follow: Given the initial and boundary conditions, and a temperature, under an external electric field, we simulate the flow and force fields of the system, which can be used to update the number density using the Nernst-Planck Equation. The modified number density will then be used to update the velocity and force fields. This forms the basis of an iterative numerical scheme that is repeated until the numerical outcomes converge. We adopt the following non-dimensionalization
where C, \(\ell \), xi, ui and m denote the initial concentration, the material’s length in the z direction, {x, y, z} coordinates, velocities in the {x, y, z} directions and the mass of a single molecule, respectively. Using this non-dimensionalization, Eq. (3) becomes
where \(Pe=(U\ell )/D\) denotes the usual Peclet number and \({\rm{\Upsilon }}=(DmU)/({k}_{B}T\ell )\) denotes the comparison between convection and the thermal energy.
Here, we investigate lithium ion transport inside double-layered graphene sheets of the size Lx = Ly = Lz = 6 Å, 8 Å and 10 Å, where Ly denotes the separation of graphene sheets (see Fig. 1). Numerical values for all the constants adopted in this paper are provided in Table 1. As mentioned above, the periodic boundary conditions for c in the x and z directions, the Dirichlet boundary conditions, i.e. c(x, 0, z, t) = c(x, Ly, z, t) = 0 are applied in the y direction.
The spatial grid size is approximately the size of a single lithium ion and the initial uniform concentration of 0.1 is applied throughout the porous cavity. Three external applied electric fields of strength 1e4, 8e5 and 1e8 NC−1 are applied for each proposed systems at 300 K. The contour plot of the normalized concentrations in the mid-z plane are shown in Fig. 2 after the systems reach stable states.

Configurations for lithium ions inside double-layered graphene of various separations, where 1(a–c), 2(a–c), 3(a–c) represent graphene separation of Ly = 6 Å, 8 Å and 10 Å, respectively. In addition, a, b, c denotes the fields of 1e4, 8e5 and 1e8 NC−1, respectively.
For the separation of Ly = 6 Å, under the low electric field (see (1a) in Fig. 2), double layers are formed almost simultaneously between graphene sheets. As more ions are introduced into the system, resulting in the oscillation between the double layers and the completely filled cavity (see the double layers in (1b) of Fig. 2 and the completely filled cavity can be represented in (1c)). For even larger electric fields, the cavity is completely filled by ions since the molecular forces induced by the graphene sheets are strong enough to trap the ions (see (1c) in Fig. 2).
On the contrary, when Ly = 10 Å, double layers are formed in the low-field limit (see (3a) in Fig. 2). Multi-layers emerge spontaneously when the strength of electric fields reach a certain threshold (see (3b) in Fig. 2), where the two middle layers oscillate due to the attractive/repulsive forces generated by the layers and the graphene sheets so that the stable ionic structure is primarily maintained by the interactions between layers. However, under large electric fields (see (3c) in Fig. 2), thinner double layers are initially formed. As the repulsive forces between ionic layers cannot be fully counteracted by the weak ion-graphene forces in the central domain, ions are pulled onto the surface of graphene sheets and distinctive multi-layers are unable to form. Instead, the void region forms and few ions are encapsulated in the channel leading to thicker double layers in the vicinity of sheets’ surface. Intriguingly, when we scrutinize the ionic concentrations of the central region, multi-layers still exist but are fluctuating with the lower concentrations (see Fig. 3). This phenomenon is expected to happen for even larger separations under large electric fields but the layers are oscillating with much lower ionic concentrations and are diminished for large separations. The numerical results suggest the transition from the thinner double layers to multi-layers and then into the thicker double layers so that the optimal storage capacity exists for the double-layered graphene of separation 10 Å at 300 K (see Fig. 4 for further details).

Residual ions in the central cavity of 3(c) in Fig. 2.

Relative concentration versus external electric fields for the three proposed separations.
To illustrate the transition from Ly = 6 Å to Ly = 10 Å, the results for a separation of 8 Å are shown in 2(a, b, c) of Fig. 2. Results in the low-field limit are similar to those for Ly = 6 Å and 10 Å. While the results for the mid-field limit are similar to those for Ly = 10 Å, significant ionic residuals remain in the middle of channel due to the attraction of some tiny body forces under the large-field limit.
To generalize the results for the above analysis, the effect of the external electric fields on the relative concentration (normalized concentration per unit volume of lithium ions) at 300 K for the three proposed systems is investigated and the numerical results are shown in Fig. 4. We comment that in order to obtain smooth results, only results arising from stable states are used and the relative concentration is calculated by taking the average of those stable results. The relative concentration for all three cases is zero when the external field is zero. When the external field is smaller than 0.6e6 NC−1, thinner double layers are formed for all the proposed systems. However, the relative concentration for Ly = 6 Å is lower than that of Ly = 8 Å or 10 Å simply due to the small cavity volume in the case of Ly = 6 Å. Spontaneous emergence of layers occurs around 0.6e6 NC−1, where multi-layers occur for Ly = 8 Å and 10 Å whereas the oscillation between the double layers and the completely filled cavity is observed for Ly = 6 Å. For even higher fields, the central cavity of Ly = 6 Å is completely filled with ions whereas thicker double layers form with the fluctuation of lithium ions in between occurring for Ly = 8 and 10 Å.
Approximate Analytical Solution
Here, we investigate analytically the existence of double layers and multi-layers inside the double layered graphene. In order to perform eigenfunction expansion and solve analytically for c, we assume that F is constant and u to be incompressible so that Eq. (3) admits a neat compact from, which is the usual convection-diffusion equation:
where V = u/D + F/(kBT) so that all the information of force and velocity fields are absorbed in V. Due to the symmetry of the present problem, c is assumed to be homogeneous in the x direction. c can be determined using the eigenfunction expansion, where ct = −dn,mc for some dn,m ∈ R and n, m ∈ Z, and the solution form of c is given by \(c(t,y,z)=\sum _{n,m}\,{c}_{n,m}\{{T}_{n,m}(t){Y}_{n}(y){Z}_{m}(z)\}\), where cn,m can be determined by an initial condition, and Yn(y) and Zm(z) are obtained from solving the following ordinary differential equations:
where k and \(\ell \) are the separation constants, and Ly and Lz are the separation and the maximum length in the z direction, respectively. Due to the boundary conditions for Y, Y admits the oscillatory solution \(Y=\sum _{n}\,{Y}_{n}\,\sin ({\lambda }_{n}y)\), where λn = (nπ)/Ly, n ∈ Z. In addition, Z is periodic with the period Lz so that Z can be written as \(Z=\sum _{m}\,{Z}_{m}\,\cos ({K}_{m}z)\), where Km = (2πm)/Lz, m ∈ Z. Upon substituting the trial solutions of Y and Z into Eq. (14), we obtain two algebraic equations for the separation constants in terms of the eigenvalues λn and Km.
Furthermore, \({T}_{n,m}(t)={e}^{-{d}_{n,m}t}={e}^{-({k}_{n}^{2}-{\ell }_{m})t}\). If c is independent of t, we obtain \({k}_{n}^{2}={\ell }_{m}\), and the following equality holds
Some observations are provided below for interpreting Eq. (16). Under low external electric fields, without loss of generality, suppose the fundamental mode in z, i.e. m = 0 and Vz ≈ 0, Eq. (16) reduces to Fy/(kbT) = (nπ)/Ly so that when Fy < < (πkBT)/Ly, only double layers are formed leading to the classical case of Debye/double layers (This condition will be automatically satisfied when Fy ≈ 0 that automatically happens in micro-channels). Besides, the higher the temperature, the easier for Fy to satisfy such inequality; For small separations and the external filed is not too large, usually ions are oscillating in the y direction due to the confinement of the system so that Vy ≈ 0. Therefore, the higher the mode in n, the larger Vz is. Hence, the larger electric field is needed to induce multi-layers; Under large external fields, both Vy and Vz come into effect and the results are inconclusive. For small separations, Vy can not be ignored and we expect that modes are divided between y and z-directions leading to multi-layers; while for large separations, Vy ≈ 0, modes are concentrated in the z-direction leading to the classical Debye layers. These observations are consistent with the above numerical results.
Flow, Conductivity and Thermal Stability
In the following, we investigate the fluid flow, charge conductivity and the thermal stability for the present problem. Fluid flow is ready to be extracted from Eq. (10) while the conductivity can be computed using the following formula
where J denotes the ionic flux inside the graphene sheets3. Using Ly = 10 Å as an example, the corresponding contour plots of fluid flow and conductivity when E = 8e8 NC−1 in the mid-z is shown below in Fig. 5.

(Left) Velocity and (Right) Conductivity for Ly = 10 Å and E = 8e8 NC−1.
Fluid velocity is almost constant in the central cavity implying that ionic layers are moving almost at the same speed as the velocity driven by the external field, since the applied voltage difference between the graphene dominates the ionic velocity. On the other hand, conductivity correlates exactly with the position of the multi-layers revealing that the higher charge conductivity occurs in the regime of ionic layers. Finally, we investigate the thermal stability for the three proposed cases when E = 1.5e6 NC−1, where stable multi-layers are formed for Ly = 8 Å and 10 Å at 300 K (see Fig. 4). The relative concentrations versus temperatures are shown in Fig. 6.

(Left) Relative concentrations for different graphene sheets with E = 1.5e6 NC−1. (Right) Relative concentrations for four different external electric fields for Ly = 10 Å.
We comment that the relative concentrations for all the proposed systems fall sharply when T < 50K and stay almost constant for normal daily operational temperatures. We suspect that the strength of external field might also cause the sharp drop. To test such a hypothesis, both field and temperature effects on the relative concentrations are investigated. We fix Ly = 10 Å and propose four distinctive field strengths, i.e. 0.2e6, 1.5e6, 1e7 and 8e7 NC−1, where the effect of fields on the ionic concentration for different temperatures is shown in Fig. 6. The relative concentrations decline for E = 0.2e6 and 1.5e6 NC−1 but decline less sensitively for E = 1.5e6 NC−1 in comparison to that for E = 0.2e6 NC−1 due to the fact that more ions have intruded into the system and the more stable multi-layers are formed for the case of E = 1.5e6 NC−1. On the contrary, a surprising rise of relative concentrations happens under the large-field case such as E = 1e7 and 8e7 NC−1 when the temperature increases but eventually reaches a plateau. We suspect that such systems saturate under the large-field limit and the extra increase in thermal energies raises the chance of lithium ions anchoring to the surface of graphene sheets. In addition, the more rapid rise for E = 1e7 NC−1 in comparison to E = 8e7 NC−1 due to the more stable and thinker double layers are formed for E = 8e7 NC−1. In conclusion, the thermal stability of relative concentrations appears to correlate with the stability of systems.
We comment that while double Debye layers always occur in ionic channels with sufficiently large separation1, multi-layers have been observed in several works3,5. However, prior to this work, the transition from double layers to multi-layers and vice versa had not been studied. We have performed a detailed parametric study of this transition and presented a simple theory that captures many of the key features of the dynamics. These issues were not address in5. We end by noting that multi-layers are found to accommodate significantly more ions than double layers and hence have the potential to be of significant practical importance in applications such as lithium-ion batteries in which high-density charge storage is a major issue.
Conclusion
Layer analysis is performed for the three proposed double-layered graphene sheets. For graphene sheets with extremely small separations, only double layers can be formed in the small and mid-range electric fields due to the repulsive force between ions. However, due to the strength of molecular forces inside the cavity, the cavity is completely filled with lithium ions under the large-field limit. On the other hand, the transition of the thinner double layers into the multi-layers and then into the thicker double layers (with fluctuating ions in between) occurs for relatively large separations. Only double layers form under large electric fields as the weak molecular force are not able to maintain the structural stability of multi-layers. The relative concentrations versus the applied electric fields are reported, and the fluid flow and charge conductivity are also analyzed. Finally, the thermal stability is discussed and we discover that the relative concentrations decline under the low- and mid-field limits, whereas the relative concentrations raise over the temperatures under the large-field limit as more ions adhere with the graphene sheets due to thermal fluctuation. The thermal stability of the relative concentration is thus closely related to the stability of the system. The present results bridge the gap between the classical theory for Debye layers and the multi-layers that occur at the nanoscale.
References
Kirby, B. J. Micro- and Nanoscale Fluid Mechanics Transport in Microfluidic Devices (Cambridge University Press, Cambridge, 2010).
Weinan, E. Principles of Multiscale Modeling. (Cambridge University Press, Cambridge, 2011).
Meng, D., Zheng, B., Lin, G. & Sushko, M. L. Numerical solution of 3d Poisson-Nernst-Planck Equations coupled with classical density functional theory for modeling ion and electron transport in confined environment. CiCP. 16, 1298–1322 (2014).
Chan, Y., Wylie, J. J., Xia, L., Ren, Y. & Chen, Y.-T. Modelling of particle-laden flow inside nanomaterials. Proc. R. Soc. A 472, 20160289 (2016).
Chan, Y. & Wylie, J. J. A continuum model of lithium ion transport inside graphene and nanotubes. EPL. 123, 14002 (2018).
Yoo, E. J., Kim, J., Hosono, E., Zhou, H. S. & Kudo, T. Large reversible Li storage of grapheme nanosheet families for use in rechargeable lithium ion batteries. Nano Lett. 8, 2277–2282 (2008).
Wang, G., Shen, X. P., Yao, J. & Park, J. Graphene nanosheets for enhanced lithium storage in lithium ion batteries. Carbon 47, 2049–2053 (2009).
Bhardwaj, T., Antic, A., Pavan, B., Barone, V. & Fahiman, B. D. Enhanced electrochemical lithium storage by graphene nanoribbons. JACS 132, 12556–12558 (2010).
Wang, D. et al. Self-assembled TiO2-graphene hybrid nanostructures for enhanced Li-ion insertion. ACS Nano 4, 907–914 (2009).
Wang, G. et al. Sn/graphene nanocomposite with 3D architecture for enhanced reversible lithium storage in lithium ion batteries. J. Mater. Chem. 19, 8378–8384 (2009).
Lee, J. K., Smith, K. B., Hayner, C. M. & Kung, H. H. Silicon nanoparticles-graphene paper compsites for Li ion battery anodes. Chem. Commun. 46, 2025–2027 (2010).
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–9 (2004).
Suzuki, T., Hasegawa, T., Mukai, S. R. & Tamon, H. A theoretical study on storage states of Li ions in carbon anodes of Li ion batteries using molecular orbital calculations. Carbon 41, 1933–1939 (2003).
krasheninnikov, A. V., Lehtinen, P. O., Foster, A. S., Pyykko, P. & Nieminen., R. M. Embed-ding transition-metal atoms in graphene: structure, bonding and magnetism. Phys. Rev. Lett. 102, 126807 (2009).
Chan, Y. & Hill, J. M. Lithium ions storage between two graphene sheets. Nanoscale Res. Lett. 6, 203 (2011).
Ernesto, M. R., Ruben, V. M. & Antonio, F. T. Modeling and simulation of lithium-ion batteries. Comput. Chem. Eng. 35, 1937–1948 (2011).
Katharina, B. S., Stefan, F. & Manuel, L. A mathematical model for all solid-state lithium ion batteries. ECS Trans. 25, 285–296 (2010).
Shi, S. et al. Modeling and simulation of lithium-ion batteries. Chinese Phys. B 35, 1937–1948 (2015).
Rademaker, T. J., Akkermans, G. R. A., Danilov, D. L. & Notten, P. H. L. On the deviation of electro-neutrolity in Li-Ion battery. J. Electrochem. Soc. 161, E3365–E3372 (2014).
Berg, P. & Findlay, J. Analytical solution of the Poisson-Nernst-Planck-Stokes equations in a cylindrical channel. Proc. R. Soc. A 467, 3157–3169 (2011).
Ciucci, F. & Lai, W. Derivation of micro/macro lithium battery models from homogenization. Transp. Porous Med. 88, 249–270 (2011).
Lenzi, E. K., Zola, R. S., Rossato, R., Vieira, D. S. & Evangelista, L. R. Asymptotic behaviors of the Poisson-Nernst-Planck model, generalziations and best adjust of experimental data. Electrochim. Acta 226, 40–45 (2017).
Cox, B. J., Thamwattana, N. & Hill, J. M. Mechanics of atoms and fullerenes in single-walled carbon nanotubes. I. Acceptance and suction energies. Proc. R. Soc. London, Ser. A 463, 461 (2007).
Cox, B. J., Thamwattana, N. & Hill, J. M. Mechanics of atoms and fullerenes in single-walled carbon nanotubes. II. Oscillatory behaviour. Proc. R. Soc. London, Ser. A 463, 477 (2007).
Han, F. Problems in Solid State Physics with Solutions. (World Scientific, Singapore, 2011).
Chan, Y. & Hill, J. M. A mechanical model for single-file transport of water through carbon nanotube membranes. J. Membr. Sci. 372, 57–65 (2011).
Chan, Y. Modelling and md simulations on ultra-filtration using graphene sheet. J. Math. Chem. 54, 1041–1056 (2016).
Chan, Y., Thamwattana, N. & Hill, J. M. Axial buckling of multi-walled carbon nanotubes and nanopeapods. Eur. J. Mech. A-Solid 30, 794–806 (2011).
Hilder, T. A. & Hill, J. M. Modeling the loading and unloading of drugs into nanotubes. Small 5, 300–8 (2008).
Chan, Y., Lee, R. K. & Hill, J. M. Metallofullerenes in composite carbon nanotubes as a nanocomputing memory device. IEEE Trans. Nanotechnol. 10, 947–952 (2010).
Jones, J. E. On the determination of molecular fileds. I. From the variation of the viscosity of a gas with temperature. Proc. R. Soc. 106A, 441 (1924).
Yu, R. J., Su, Y.C. & Chieng, C.C. Transport of nanoparticles inside microchannels with nano-sized cavities. 16 th International Symposium on Transport Phenomena, pages 1–6 (2005).
Horng, T.-L., Lin, T.-C., Liu, C. & Eisenberg, B. Pnp equations with steric effects: A model of ion flow through channels. J. Phys. Chem. B 116, 11422–41 (2012).
Maitland, G. C., Rigby, M., Smith, E. B. & Wakeham, W. A. Intermolecular forces – Their origin and determination. (Clarendon Press, Oxford, 1981).
Bouazizi, S. & Nasr, S. Local order in aqueous lithium chloride solutions as studied by x-ray scattering and molecular dynamics simulations. J. Mol. Struct. 837, 206–213 (2007).
Acknowledgements
J.J.W. was supported by the Research Grants Council of Hong Kong Special Administrative Region, China [CityU 11303314]. We also acknowledge the GuangDong Natural Science Foundation: 2018A030313467, Shenzhen City Science and Technology Department (1. the oversea talent set-up funding; 2. JCYJ20180305124732178) and Ningbo Science and Technology Bureau Technology Innovation Team under Grant No. 2016B10010.
Author information
Authors and Affiliations
Contributions
Y.C. and J.J.W. developed the theoretical frameworks for the present paper; M.S. carried out numerical solutions; S.L.L. contributed the chemical analysis; Y.C. wrote the paper; J.J.W. proofread the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chan, Y., Saeed, M., Lee, SL. et al. A continuum study of layer analysis for single species ion transport inside double-layered graphene sheets with various separations. Sci Rep 9, 11712 (2019). https://doi.org/10.1038/s41598-019-48101-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48101-8
This article is cited by
-
Time-dependent Langevin modeling and Monte–Carlo simulations of diffusion in one-dimensional ion channels
Journal of Mathematical Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
