
Abstract
Plant roots are vital for acquiring nutrients and water from soil. However, the mechanisms regulating root growth in hexaploid wheat remain to be elucidated. Here, an integrated comparative proteome study on the roots of two varieties and their descendants with contrasting root phenotypes was performed. A total of 80 differentially expressed proteins (DEPs) associated with the regulation of primary root growth were identified, including two plant steroid biosynthesis related proteins and nine class III peroxidases. Real-time PCR analysis showed that brassinosteroid (BR) biosynthesis pathway was significantly elevated in long-root plants compared with those short-root plants. Moreover, O2.− and H2O2 were distributed abundantly in both the root meristematic and elongation zones of long root plants, but only in the meristematic zone of short-root plants. The differential distribution of reactive oxygen species (ROS) in the root tips of different genotypes may be caused by the differential expression of peroxidases. Taken together, our results suggest that the regulation of wheat primary root growth is closely related to BR biosynthesis pathway and BR-mediated ROS distribution.
Similar content being viewed by others

Introduction
Plant breeding is an important way to increase crop yield. Currently, breeding selection is mainly based on aerial traits, and genetic improvement for root traits is still relatively rare1,2,3,4. In many regions of the world, current crop production still depends on excessive application of fertilizer and water, which is detrimental to sustainable agricultural development5,6,7. Due to environmental pressures and resource shortages, improvements in resource use efficiency should be prioritized in future crop breeding programmes8. Roots are the main organs for plants to uptake nutrients and water. Genetic improvement of root traits is one of the important strategies to increase nutrient and water use efficiency of crops2,7,9,10,11. Therefore, it is important to study the molecular mechanisms that control the growth and development of plant roots in food crops to enable genetic improvement of root traits.
Previous studies in the model plant Arabidopsis demonstrated that plant root growth and development are regulated by phytohormones12. PLETHORA (PLT) transcription factors are indispensable to maintain the niche of root stem cell and function as principal regulators of the root apical meristem (RAM). The expression of PLT genes is induced by auxin accumulation13,14. Cytokinin controls root growth and development by balancing the division and differentiation of stem cells15. The balance between auxin and cytokinin in root tips plays a crucial role in determining the size of RAM16. The expression of PLT genes is also regulated by local brassinosteroids (BRs) signalling in the stele17. Mutants with weakened BR biosynthesis or signal transduction show a short-root phenotype18. Moreover, supplementing low concentration of BRs can promote root growth19.
Wheat (Triticum aestivum L.) is an important food crop. Several studies have reported a number of important quantitative trait loci (QTLs) for root traits in wheat20,21,22,23,24,25. He et al. conducted a proteomic study using 2D-DIGE (Two-Dimensional Difference Gel Electrophoresis) and identified 16 DEPs involved in the regulation of wheat root development26. Li et al. identified a plant-specific transcription factor, MORE ROOT (TaMOR). Overexpression wheat TaMOR gene in rice (Oryza sativa) results in larger root system and higher grain yield27. Another study showed that overexpression of a wheat NAC transcription factor gene can promote root growth and increase plant drought tolerance28. However, despite these advances, the molecular mechanisms controlling wheat root growth and development remain unclear.
Previously we developed a “XY54 × J411” recombinant inbred line (RIL) population. XY54 was the female parent of the RIL population and had significantly longer PRL (Primary root length) and MRL (Maximum root length) than the male parent J41124,29. In this study, fifteen long-root lines (Long root group, LR group) and another fifteen short-root lines (Short root group, SR group) were selected based on previous studies from the RIL population24. To minimize differences in genetic background, the SRM (Short Root Mixture, a root mixture of the SR group) and LRM (Long Root Mixture, a root mixture of the LR group), and the roots of XY54 and J411 were used as materials for comparative proteomic analysis to identify candidate regulatory proteins of wheat root growth. Differentially expressed proteins and significantly enriched pathways involved in the regulation of wheat primary root growth were identified. Further molecular and physiological analyses suggested that the regulation of wheat primary root growth is closely related to BR biosynthesis pathway and BR-mediated ROS distribution.
Results
Phenotype analysis of the LR and SR groups
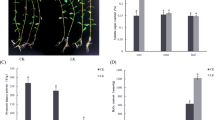
In this study, XY54 had significantly longer PRL and MRL than J411, which is consistent with our previous studies24,29 (Fig. 1A,B). Similarly, the PRL and MRL of the lines in the LR group were significantly greater than those of the lines in the SR group (Fig. 1C,D). The MRL of lines in the LR group varied from 13.6 cm to 19.4 cm, while the MRL of lines in the SR group varied from 7.2 cm to 9.4 cm. On average, the MRL of the LR group was 81.2% longer than that of the SR group (Fig. 1E).

Root phenotype of wheat varieties XY54, J411, the LR group (Long root group) and the SR group (Short root group). (A, B) Roots (A) and root morphologies (B) of XY54 and J411; (C, D) root morphologies (C) and roots (D) of eight randomly selected lines from the LR group and SR group, respectively; (E) average values of MRL (maximum root length) of XY54, J411, LR group and SR group. The asterisks above the columns indicate significant differences at p < 0.05 (n = 15). MRL, maximum root length. Bar = 5 cm.
Identification and quantitative results of differential proteins
To identify proteins regulating wheat primary root growth, root protein profiles of XY54, J411, LRM and SRM were obtained. In total, we identified 6294 reliable quantified proteins for the following DEPs analysis (False discovery rate, FDR ≤1%; detected in at least two biological replicates; Supplementary Table S1). We identified 723 proteins as DEPs in one to one comparison between XY54 and J411 based on their respective abundance in roots (Supplementary Table S2). Among the DEPs, 373 proteins were up-regulated and 350 proteins were down-regulated in the roots of XY54. We also identified 452 proteins as DEPs in the LRM-SRM comparison. Among them, 235 DEPs exhibited up-regulation and 217 DEPs showed down-regulation in the roots of LRM (Supplementary Table S3).
Among the DEPs identified above, 96 existed in both the XY54-J411 comparison and LRM-SRM comparison (Fig. 2A). Of these 96 DEPs, 16 DEPs exhibited opposite expression patterns in the two subsets (up-regulated in one subset and down-regulated in the other), while the remaining 80 DEPs exhibited similar expression patterns (Supplementary Table S4). As described above, XY54 and lines of the LR group exhibited longer primary roots than J411 and lines of the SR group. Therefore, there is reason to believe that these 80 DEPs may be associated with PRL and MRL, because most of the differences in genetic background between the parents had been eliminated. The 80 DEPs that existed in both subsets are listed in Supplementary Table S4. Among them, 32 proteins were up-regulated and 48 proteins were down-regulated in the roots of XY54 and LRM, compared with J411 and SRM (Supplementary Table S4). Hierarchical cluster analysis of the 80 proteins showed that a distinct difference exists in abundance of these proteins between the long root genotype samples (XY54 and LRM) and the short root genotype samples (J411 and SRM). As expected, the 80 DEPs could be divided into two major distinct sub-clusters containing proteins with different levels of abundance (Fig. 3). According to the classification of cell component, molecular function and biological process, 80 DEPs were classified by GO (gene ontology) terminology. All the quantified protein annotations were taken as a background dataset, significantly enriched GO terms are shown in Fig. 2B (p < 0.05). Results showed that proteins involved in biological processes, such as the categories of metabolic process, cellular process, biological process and response to stimuli, were most common; while proteins associated with molecular functions were predicted to be involved in catalytic activity, antioxidant activity, binding, structural molecules, transport activity, molecular function regulation, nutrient reservoir activity and protein tags. Among all cellular components, ‘cell’ and ‘cell part’ were predicted to have the largest number of DEPs (Fig. 2B).

Results of Venn diagram analysis, and GO and pathway enrichment analysis of the DEPs. (A) Result of Venn diagram analysis; (B) GO enrichment analysis; (C) significantly enriched pathways involving the up-regulated DEPs. The values on the vertical ordinate represent the percentage of DEPs to the background proteins in the pathway. DEPs, differentially expressed proteins.

Hierarchical clustering of DEPs involved in the regulation of primary root growth. The four columns represent the protein expression levels in XY54, the long root mixture (LRM), J411 and the short root mixture (SRM). Red and blue indicate the higher and lower expression levels, respectively. DEPs, differentially expressed proteins.
To gain insight into the DEPs involved in the regulation of PRL, we used the web-based software KOBAS 3.0 and identified nine significantly enriched pathways in the up-regulated DEPs in long rooted plants based on hypergeometric distributions (p < 0.05 after the FDR correction; Fig. 2C; Supplementary Table S5). Interestingly, plant steroid biosynthesis pathway was significantly enriched. There were two DEPs (one obtusifoliol 14-alpha demethylase and one delta (24)-sterol reductase) involved in plant steroid biosynthesis pathway (Fig. 2C). Obtusifoliol 14-alpha demethylase, encoded by the CYP51 gene, catalyses the demethylation of obtusifoliol30,31. Delta (24)-sterol reductase, encoded by the Dwarf1 (DWF1) gene, is involved in the formation of campesterol (CR), an early precursor of BRs32. Both CYP51 and DWF1 are essential for the biosynthesis of plant steroids. The expression of these two proteins was up-regulated in the roots of the LR group and long-root parent, XY54, compared with the SR group and short-root parent J411 (Fig. 3; Supplementary Table S4). Most DEPs in the phenylpropanoid biosynthesis pathway are peroxidases. In addition, the pathways for fatty acid biosynthesis and metabolism, phenylalanine metabolism and some carbon metabolism pathways (i.e. citrate cycle, glyoxylate and dicarboxylate metabolism) were also elevated in XY54 and LRM (Fig. 2C; Supplementary Table S5).
Real-time PCR verification
To evaluate the correlation of protein expression with their corresponding mRNA level, all nine differentially expressed peroxidases and another nine randomly selected DEPs were chosen for RNA level examination using real-time PCR (Fig. 4). Of the nine peroxidases genes, six genes closely matched their mRNA levels with their translation products and the other three genes (A0A1B6QGB6, I1GR63 and W5ERP5) had poor correlations between mRNA and protein expression levels (Fig. 4A). Of the nine randomly selected genes, five genes had closely matched mRNA levels with their translation products while the expression levels of the other four genes (A0A1D5VID7, A0A1D6CR70, A0A1D5TH00 and A0A1D5XW89) exhibited poor correlations with their changes in protein abundance (Fig. 4B).

Relative mRNA expression analysis of DEPs using real-time PCR. (A) Relative mRNA expression of nine differentially expressed peroxidases; (B) relative mRNA expression of nine randomly selected DEPs. For the expression level of a protein, the lowest of the four samples was defined as a reference and its expression value was set as 1.0. Columns marked with different lowercase letters indicate that the difference between them (in gene expression levels) is significant according to Duncan’s multiple range test (n = 3, p < 0.05).
Real-time PCR analysis of BR biosynthesis related genes
CYP51 and DWF1 are two components involved in the biosynthesis of plant steroids. As both were significantly up-regulated in roots of the LR group and long-root parent XY54, compared with the SR group and short-root parent J411, we further checked the relative expression of TaCYP51 and TaDWF1, and four other genes (TaDWF4, TaDWF6, TaDWF7 and TaCYP90D1) involved in the BR biosynthesis pathway at an RNA level. Results showed that all of them were significantly up-regulated in XY54 and the LR group compared with J411 and the SR group (Fig. 5).

Relative mRNA expression analysis of genes involved in brassinosteroid biosynthesis using real-time PCR. The sample with the lowest expression level was set as 1.0. Columns marked with different lowercase letters indicate that the difference between them (in gene expression levels) is significant according to Duncan’s multiple range test (n = 3, p < 0.05).
In Situ O 2.- and H2O2 detection
As mentioned above, a significant number (nine) of peroxidases were present in the DEPs and most of them also exhibited differential expression at an RNA level (Fig. 4A). As peroxidases regulate the balance of ROS between the meristematic and elongation zones33, we further analysed the distribution of ROS (O2.- and H2O2) in the root tips by staining roots with nitroblue tetrazolium (NBT) and 3,3′-diaminobenzidine (DAB), respectively; these reagents are widely used as indicators of O2.- and H2O2 levels34,35. As shown in Fig. 6, for XY54 and LR group lines, apparent staining signals were observed in both the root meristematic zone and elongation zone, however, apparent signals of NBT staining were observed only in the root meristematic zone of J411 and lines in the SR group (Fig. 6A). These results suggested that O2.- was distributed abundantly both in the meristematic and elongation zones in XY54 and LR group lines but only in the meristematic zone of J411 and SR group lines. The results of DAB staining for H2O2 were similar to those of NBT staining (Fig. 6B).

Results of in situ detection of root O2.- and H2O2 in XY54, J411 and eight randomly selected lines from the long root (LR) group and short root (SR) group, respectively. NBT (Nitroblue tetrazolium) and DAB (3,3′-diaminobenzidine) were used to stain O2.- and H2O2, respectively. (A) Results of NBT staining; (B) results of DAB staining.
Real-time PCR analysis of peroxidase genes in different zones of root tip
We totally identified nine peroxidases in the DEPs. Compared with the SR group, six of them were up-regulated and three were down-regulated in the LR group (Supplementary Table S4). As O2.- and H2O2 were differentially distributed in the meristematic and elongation zones between the long root genotype and short root genotype (Fig. 6), and peroxidases can regulate ROS production33, we further analysed the expression levels of these peroxidase genes in the meristematic, elongation and maturation zones of XY54 and J411 root tips (Fig. 7A). Results showed that the expression levels of different peroxidase genes varied greatly in different root apical zones and different varieties (Fig. 7B). Among the nine peroxidase genes, five of them had significantly higher expression levels in the elongation zone than in the meristematic and maturation zones, while two of them had significantly higher expression levels in the meristematic zone (A0A1D5Z2R6, A0A1D5SWP7) than in the elongation and maturation zones (Fig. 7B). In addition, the other two peroxidase (A0A1D5T6G6 and I1GR63) genes had the highest expression levels in the maturation zone in the long root plants XY54, but not in the short root plants J411. In XY54, the expression levels of four peroxidase (A0A1D5SWP7, M7Y9R8, A0A1D6AFC1 and A0A1D5Z2R6) genes in both the meristematic and elongation zones were significantly higher than those in J411 (Fig. 7B). While the expression levels of another four peroxidase genes (A0A1D6D586, A0A1B6QGB6, I1GR63 and W5ERP5) in both the meristematic zone and elongation zone of J411 were significantly higher than those of XY54 (Fig. 7B).

Relative mRNA expression analysis of differentially expressed peroxidase genes in the meristematic, elongation and maturation zones of root tips using RT-qPCR. ‘Me’ represent meristematic zone; ‘En’ represent the elongation; ‘Ma’ represent maturation zone. The lowest expression level in the samples was set as 1.0. Columns marked with different lowercase letters indicate that the difference between them (in gene expression levels) is significant according to Duncan’s multiple range test (n = 3, p < 0.05).
Discussion
Deeper roots contribute to plant production and survival under water and nitrogen limiting conditions36,37,38. However, the molecular mechanisms controlling root growth in wheat remain unclear. Here, an integrated comparative proteome study was performed to explore this question. Eighteen DEPs including the nine differentially expressed peroxidases were selected to evaluate the correlation of protein expression with their corresponding mRNA level using real-time PCR. Eleven genes had closely matched mRNA levels with their translation products while the expression levels of the other seven genes exhibited poor correlations with their changes in protein abundance (Fig. 4B). One possible reason is that post-transcriptional, translational and post-translational regulation may occur in the expression of those poorly correlated genes39,40,41. Another possible reason is that the peroxidase genes belong to a huge gene family in hexaploid wheat, and some peroxidase members have a high degree of homology. Therefore, the poor correlation may be due to the lack of specificity for the genes encoding the identified proteins.
In this work, 80 DEPs associated with the regulation of primary root growth were identified, including two plant steroid biosynthesis related proteins (CYP51 and DWF1) (Fig. 3; Supplementary Table S4). Plant steroid biosynthesis pathway was significantly enriched in the up-regulated DEPs (Fig. 2C). Coincidentally, TaTRIP1, a negative regulator in BR signalling, has been shown to be up-regulated in short-root wheat varieties28. Both CYP51 and DWF1 are essential for the biosynthesis of plant steroids. DWF1 catalyses the formation of CR, while CR is the first molecule specifically entering BR biosynthetic pathways42. Moreover, the expression levels of TaCYP51 and TaDWF1, and four other genes (TaDWF4, TaDWF6, TaDWF7 and TaCYP90D1) involved in BR biosynthesis pathway also exhibited significantly up-regulated pattern in XY54 and the LR group compared with J411 and the SR group (Fig. 5). These results indicated that the primary root growth was possibly promoted by elevated BR biosynthesis. Brassinolide (BL) is reported as a potent BR in plants43,44, so we measured the content of endogenous BL in LRM and SRM using high performance liquid chromatography (HPLC)-MS/MS. A small amount of BL was detected in the LRM, but not in the SRM (below the detection limit; data not shown). We repeated the experiment and obtained a similar result. As a matter of fact, similar results have also been reported in another gramineous plant, rice45,46. Therefore, it is likely that some other sterols (e.g. castasterone) other than BL act as the main active BRs in wheat. However, further studies are needed to verify this point.
Noticeably, among the 80 DEPs identified in this study, nine of them were annotated as class III peroxidases of which six were up-regulated and three were down-regulated (Fig. 3; Supplementary Table S4). Class III peroxidases belong to a large multigene family in plants. 73 and 138 genes encoding class III peroxidase genes have been identified in Arabidopsis and rice, respectively47,48,49. Class III peroxidases can catalyse the reduction of H2O2 by taking electrons to various donor molecules (such as lignin precursors or secondary metabolites) in the regular peroxidative cycle49,50. Real-time PCR analysis of the peroxidase genes in different zones of root tips showed that some peroxidase genes had higher expression in the meristematic zone, while some others in the elongation zone. The results suggest that different members of the peroxidase gene family may play roles in different zones of root tips. Furthermore, the expression levels of different peroxidase genes also varied greatly in the root tips of XY54 and J411 (Fig. 7B). It is not clear why some differentially expressed peroxidases were highly expressed in the long root genotype, while others were expressed in the short root genotype. One possible reason is that different members of this large gene family may have functional differentiation. Actually, previous studies have shown that peroxidases can not only catalyze the cleavage of H2O2, but also has oxidase activity, which can also catalyze the formation of O2.- and H2O2 in the presence of NAD(P)H51,52,53. Thus, the regulation of peroxidases on reactive oxygen species (ROS) is very complicated. As ROS balance has an important role in maintaining root meristem activity54, different peroxidases may play different roles in mediating the balance of ROS between the root meristematic zone and elongation zone. In our study, ROS (O2.- and H2O2) accumulated both in the root meristematic zone and elongation zone of XY54 and LR, while accumulation was observed only in the root meristematic zone of J411 and the SR group (Fig. 6). The differential accumulation of ROS between the LR and SR groups may lead to different meristem activity and thus different PRL and MRL. BRs are important regulators of many important biological processes55. Ca2+ and ROS can act as second messengers to function in BR-regulated biological processes56,57,58,59. In addition, MAPK cascades also control ROS production in BR-mediated responses60,61. Such evidence suggests that ROS may be an integration node for BR signalling pathway with root developmental pathways.
BRs have been reported to regulate RAM size by promoting cell cycle progression in Arabidopsis62,63. Overexpression of a negative regulatory gene of wheat BR signalling, TaTRIP1, in Arabidopsis, reduces Arabidopsis root meristem size, and hence PRL28. Plants treated with BR inhibitor have reduced ROS levels, whereas exogenous BRs induce moderate ROS accumulation64. Moreover, BR biosynthesis pathway was elevated in plants of the LR group compared with the SR group (Figs 2C and 5). Taken together, the regulation of wheat primary root growth is closely related to BR biosynthesis pathway and BR-mediated ROS distribution. Based on our experimental evidences and previous reports, we proposed a possible regulatory mechanism: the BR-mediated ROS accumulation and distribution in root tips determined root meristem size, which determines the growth rate of primary roots. And the differential accumulation and distribution of ROS in the root tips of different genotypes may be due to the differential expression of a number of peroxidases.
Materials and Methods
Plant materials and evaluation of root morphology
The LR group and SR group were selected based on previous studies from a RIL population25,29. The plants were grown in a greenhouse with six replications each. Methods for seed sterilization, germination and the growth conditions of wheat plants were as previously described by Ren et al.25. 12 d after transfer, the root phenotype of each line was investigated. Roots of each line from the LR and SR groups were mixed, respectively (LRM and SRM). The reason for choosing this stage is that the primary root keeps elongate in the long root genotype, but the elongation of the short root genotype almost stops after two weeks. The root samples of LRM, SRM and their parents (XY54 and J411) were quick-frozen in liquid nitrogen and stored in a −80 °C freezer for subsequent protein and RNA extraction. Three biological replicates were analysed to minimize experimental error.
Protein extraction, digestion and iTRAQ labeling
Protein extraction was conducted according to the method described by Thiellement et al.65. Protein digestion was conducted using the FASP procedure66. Protein samples were digested overnight in 50 μL trypsin solution at 37 °C, and then labelled with iTRAQ reagents based on the manufacturer’s instructions (Applied Biosystems, USA). Three biological replicates were performed. Four eight-plex iTRAQ sets was used to label the samples. XY54-1, XY54-2 and XY54-3 were labeled with 113 of set 1, set 2 and set 3, respectively; J411-1, J411-2 and J411-3 were labeled with 114 of set 1, set 2 and set 3, respectively; LRM-1, LRM-2 and LRM-3 were labeled with 113, 114 and 115 of set 4, respectively; SRM-1, SRM-2 and SRM-3 were labeled with 116, 117 and 118 of set 4, respectively. The remaining labels of each set together with the 113 and 114 of the first three sets were used for another study.
SCX fractionation and LC-ESI-MS/MS analysis
AKTA Purifier system (GE Healthcare, USA) was used for the SCX (strong cationic exchange) chromatography to fractionate the iTRAQ labelled peptides as described previously with minor modifications67,68,69. The elution was surveyed using the absorbance at 214 nm. In total, 30 fractions were collected (1 fraction/min) and merged into 10 parts, and each part were then desalted and reconstituted in 40 µl 0.1% (v/v) trifluoroacetic acid. A Q Exactive mass spectrometer coupled to an Easy-nLC (Proxeon Biosystems) was used for the LC ESI-MS/MS (liquid chromatography-electrospray ionization tandem MS) analysis according to previous reports67,68,69.
Protein identification and quantification
The LC-MS/MS analysis were performed in positive ion mode. The most abundant precursor ions were chosen from the survey scan (300–1800 m/z) for HCD fragmentation using the data-dependent TOP10 method; the peptide recognition mode was run in the instrument69. Raw data were collected by Proteome Discoverer 1.4 software (Thermo Fisher Scientific), and fragmentation spectra were identified using the MASCOT 2.2 search engine running against the uniprot_Poaceae database (http://www.uniprot.org). The parameters for protein identification were set as follows: missed cleavages ≤2, the mass tolerance was 20 ppm for precursor ions and 0.1 Da for fragmented ions, FDR ≤1%. For the quantitative analysis of the identified proteins, all peptide ratios were normalized by the median protein ratio using Proteome Discoverer 1.4. After the normalization, the median protein ratio is 1.0. The relative expression values of each protein were normalized using the median ratio in Mascot69,70,71. Only those proteins detected in at least two replicates were used for the following DEP analysis. Protein species with a ratio fold change greater than or equal to 1.2 (p < 0.05) were defined as DEPs40,72. Here, we selected a relatively lower ratio (1.2-fold change) because the DEPs involved in the regulation of wheat primary root growth were defined by two steps to reduce the possible interference of the genetic background of XY54 and J411. Only those DEPs that coexist in XY54-J411 and LRM-SRM comparisons were considered to be the final DEPs.
Bioinformatics analysis
NCBI BLAST + client (ncbi-blast-2.2.28 + -win32.exe) and BLAST2GO were used for GO annotation73. GO enrichment analysis was applied based on Fisher’s exact test using all the quantified protein annotations as a background dataset (p < 0.05). The protein pathway analysis was performed using KOBAS 3.0 (http://kobas.cbi.Pku.edu.cn./anno_iden.php). Functional categories and pathways with a corrected p-value < 0.05 were considered as significant. Cluster3.0 (http://bonsai. hgc.jp/~mdehoon/software/cluster/software.htm) together with the Java Treeview software (http://jtreeview.sourceforge.net) were used to perform hierarchical clustering analysis.
Quantitative Real-time PCR
The meristematic, elongation and maturation zones of root tips were dissected by a shaving blade under a microscope (OLYMPUS BX-53, Japan). The whole root samples and different root apical zone samples were quick-frozen in liquid nitrogen and stored in a −80 °C freezer for RNA extraction. Total RNA of each sample was isolated using TRIzol Reagent (Invitrogen, USA) with genomic DNA removed using DNase I (Promega, USA). Each total RNA sample was used to synthesize the first strand of cDNA using the GoScript Reverse Transcription System (Promega, USA) following the manufacturer’s instructions. Primer 5.0 software (Premier, Canada) was used to design real-time PCR primers (Supplementary Table S6). SYBR Green PCR Real Master Mix (Tiangen, Beijing, China) was used for real-time PCR analysis on a Thermal Cycler CFX96 Real-Time System (Bio-Rad, USA) according to the instructions. The relative expression levels of target genes were computed using the 2−ΔΔCT method, with TaActin as the reference gene.
DAB and NBT staining
In situ H2O2 and O2.- was stained using DAB (3,3′-diaminobenzidine) and NBT (nitroblue tetrazolium) respectively according to previous descriptions35,74. The results of NBT and DAB staining were photographed using a camera (Canon EOS 70D, Japan) mounted on a microscope (OLYMPUS BX-53, Japan).
Statistical analysis
The mean values and standard errors were analysed using Excel 2017 and IBM SPSS statistics 21 software. The statistical significance of differences was analysed using Duncan’s multiple range test for all traits evaluated and gene expression levels, with a p-value < 0.05 considered significant.
Data Availability
MS data in this study has been deposited in the iProX repository (ww.iprox.org) with identifier PX0001149000/PXD008793.
References
Waines, J. G. & Ehdaie, B. Domestication and crop physiology: roots of Green-Revolution wheat. Ann. Bot. 100, 991–998, https://doi.org/10.1093/aob/mcm180 (2007).
Den Herder, G., Van Isterdael, G., Beeckman, T. & De Smet, I. The roots of a new green revolution. Trends Plant Sci. 15, 600–607, https://doi.org/10.1016/j.tplants.2010.08.009 (2010).
Hochholdinger, F. Untapping root system architecture for crop improvement. J. Exp. Bot. 67, 4431–4433, https://doi.org/10.1093/jxb/erw262 (2016).
Wachsman, G., Sparks, E. E. & Benfey, P. N. Genes and networks regulating root anatomy and architecture. New Phytol. 208, 26–38, https://doi.org/10.1111/nph.13469 (2015).
Rosegrant, M. W., Ringler, C. & Zhu, T. Water for agriculture: maintaining food security under growing scarcity. Annu. Rev. Environ. Resour. 34, 205–222, https://doi.org/10.1146/annurev.environ.030308.090351 (2009).
Good, A. G. & Beatty, P. H. Fertilizing nature: a tragedy of excess in the commons. Plos Biol. 9, e1001124, https://doi.org/10.1371/journal.pbio.1001124 (2011).
White, P. J. et al. Matching roots to their environment. Ann. Bot. 112, 207–222, https://doi.org/10.1093/aob/mct123 (2013).
Jiao, X. et al. Grain production versus resource and environmental costs: towards increasing sustainability of nutrient use in China. J. Exp. Bot. 67, 4935–4949, https://doi.org/10.1093/jxb/erw282 (2016).
de Dorlodot, S. et al. Root system architecture: opportunities and constraints for genetic improvement of crops. Trends Plant Sci. 12, 474–481, https://doi.org/10.1016/j.tplants.2007.08.012 (2007).
Lynch, J. Roots of the second green revolution. Aust. J. Bot. 55, 493–512, https://doi.org/10.1071/BT06118 (2007).
Yu, P., Li, X. X., White, P. J. & Li, C. J. A large and deep root system underlies high nitrogen-use efficiency in maize production. Plos one 10, e0126293, https://doi.org/10.1371/journal.pone.0126293 (2015).
Pacifici, E., Polverari, L. & Sabatini, S. Plant hormone crosstalk: the pivot of root growth. J. Exp. Bot. 66, 1113–1121, https://doi.org/10.1093/jxb/eru534 (2015).
Aida, M. et al. The PLETHORA genes mediate patterning of the Arabidopsis root stem cell niche. Cell 119, 109–120, https://doi.org/10.1016/j.cell.2004.09.018 (2004).
Galinha, C. et al. PLETHORA proteins as dose-dependent master regulators of Arabidopsis root development. Nature 449, 1053–1057, https://doi.org/10.1038/nature06206 (2007).
Bishopp, A., Ursache, R. & Helariutta, Y. Plant development: how long is a root? Curr. Biol. 22, 919–921, https://doi.org/10.1016/j.cub.2012.09.048 (2012).
Dello Ioio, R. et al. A genetic frame work for the control of cell division and differentiation in the root meristem. Science 322, 1380–1384, https://doi.org/10.1126/science.1164147 (2008).
Vragovic, K. et al. Translatome analyses capture of opposing tissue-specific brassinosteroid signals orchestrating root meristem differentiation. Proc Natl Acad Sci USA 112, 923–928, https://doi.org/10.1073/pnas.1417947112 (2015).
Li, J., Nagpal, P., Vitart, V., McMorris, T. C. & Chory, J. A role for brassinosteroids in light-dependent development of Arabidopsis. Science 272, 398–401, https://doi.org/10.1126/science.272.5260.398 (1996).
Clouse, S. D., Langford, M. & McMorris, T. C. A brassinosteroid-insensitive mutant in Arabidopsis thaliana exhibits multiple defects in growth and development. Plant Physiol. 111, 671–678, https://doi.org/10.1104/pp.111.3.671 (1996).
Laperche, A., Devienne-Barret, F., Maury, O., Le Gouis, J. & Ney, B. A simplified conceptual model of carbon/nitrogen functioning for QTL analysis of winter wheat adaptation to nitrogen deficiency. Theor. Appl. Genet. 113, 1131–1146, https://doi.org/10.1007/s00122-006-0373-4 (2006).
Sanguineti, M. C. et al. Genetic dissection of seminal root architecture in elite durum wheat germplasm. Ann. Appl. Biol. 151, 291–305, https://doi.org/10.1111/j.1744-7348.2007.00198.x (2007).
Canè, M. A. et al. Association mapping for root architectural traits in durum wheat seedlings as related toagronomic performance. Mol. Breeding 34, 1629–1645, https://doi.org/10.1007/s11032-014-0177-1 (2014).
Maccaferri, M. et al. Prioritizing quantitative trait loci for root system architecture in tetraploid wheat. J. Exp. Bot. 67, 1161–1178, https://doi.org/10.1093/jxb/erw039 (2016).
Iannucci, A. et al. Mapping QTL for root and shoot morphological traits in a Durum Wheat×T. dicoccum segregating population at seedling stage. Int. J. Genomics 3, 1–17, https://doi.org/10.1155/2017/6876393 (2017).
Ren, Y. Z. et al. Major quantitative trait loci for seminal root morphology of wheat seedlings. Mol. Breeding 30, 139–148, https://doi.org/10.1007/s11032-011-9605-7 (2012).
He, X. et al. A genotypic difference in primary root length is associated with the inhibitory role of transforming growth f-actor-beta receptor-interacting protein-1 on root meristem size in wheat. Plant J. 77, 931–943, https://doi.org/10.1111/tpj.12449 (2014).
Li, B. et al. Overexpression of wheat gene TaMOR improves root system architecture and grain yield in Oryza sativa. J Exp Bot. 67, 4155–4167, https://doi.org/10.1093/jxb/erw193 (2016).
Chen, D., Chai, S., McIntyre, C. L. & Xue, G. P. Overexpression of a predominantly root-expressed NAC transcription factor in wheat roots enhances root length, biomass and drought tolerance. Plant Cell Rep. 37, 225–237, https://doi.org/10.1007/s00299-017-2224-y (2018).
Ren, Y. Z. et al. Characterization of QTLs for root traits of wheat grown under different nitrogen and phosphorus supply levels. Front. Plant Sci. 8, 2096, https://doi.org/10.3389/fpls.2017.02096 (2017).
Aoyama, Y. & Yoshida, Y. The 4 beta-methyl group of substrate does not affect the activity of lanosterol 14 alpha-demethylase (P-450(14)DM) of yeast: difference between the substrate recognition by yeast and plant sterol 14 alpha-demethylases. Biochem. Biophys. Res. Commun. 183, 1266–1272, https://doi.org/10.1016/S0006-291X(05)80327-4 (1992).
Bak, S., Kahn, R. A., Olsen, C. E. & Halkier, B. A. Cloning and expression in Escherichia coli of the obtusifoliol 14 alpha-demethylase of Sorghum bicolor (L.) Moench, a cytochrome P450 orthologous to the sterol 14 alpha-demethylases (CYP51) from fungi and mammals. Plant J. 11, 191–201, https://doi.org/10.1046/j.1365-313X.1997.11020191.x (1997).
Klahre, U. et al. The Arabidopsis DIMINUTO/DWARF1 gene encodes a protein involved in steroid synthesis. Plant Cell 10, 1677–1690, https://doi.org/10.1105/tpc.10.10.1677 (1998).
Tsukagoshi, H., Busch, W. & Benfey, P. N. Transcriptional regulation of ROS controls transition from proliferation to differentiation in the root. Cell 143, 606–616, https://doi.org/10.1016/j.cell.2010.10.020 (2010).
Bielski, B. H. J., Shiue, G. G. & Bajuk, S. Reduction of nitro blue tetrazolium by CO2 and O2 − radicals. J. Physiol. Chem. 84, 830–833, https://doi.org/10.1002/chin.198030147 (1980).
Thordal-Christensen, H., Zhang, Z., Wei, Y. & Collinge, D. B. Subcellular localization of H2O2 in plants: H2O2 accumulation in papillae and hypersensitive response during the barley-powdery mildew interaction. Plant J. 11, 1187–1194, https://doi.org/10.1046/j.1365-313X.1997.11061187.x (1997).
Uga, Y. et al. Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat. Genet. 45, 1097–1102, https://doi.org/10.1038/ng.2725 (2013).
Comas, L. H., Becker, S. R., Cruz, V. M. V., Byrne, P. F. & Dierig, D. A. Root traits contributing to plant productivity under drought. Front. Plant Sci. 4, 442, https://doi.org/10.3389/fpls.2013.00442 (2013).
Koevoets, I. T., Venema, J. H., Elzenga, J. T. & Testerink, C. Roots withstanding their environment: exploiting root system architecture responses to abiotic stress to improve crop tolerance. Front. Plant Sci. 31, 1335, https://doi.org/10.3389/fpls.2016.01335 (2016).
Fernie, A. R. & Stitt, M. On the discordance of metabolomics with proteomics and transcriptomics: coping with increasing complexity in logic, chemistry, and network interactions scientific correspondence. Plant Physiol. 158, 1139–1145, https://doi.org/10.1104/pp.112.193235 (2012).
Chu, P. et al. iTRAQ-based quantitative proteomics analysis of Brassica napus leaves reveals pathways associated with chlorophyll deficiency. J. Proteome 113, 244–259, https://doi.org/10.1016/j.jprot.2014.10.005 (2015).
Sun, X. C. et al. Unraveling the root proteome changes and its relationship to molecular mechanism underlying salt stress response in Radish (Raphanus sativus L.). Front. Plant Sci. 8, 1192, https://doi.org/10.3389/fpls.2017.01192 (2017).
Wei, Z. & Li, J. Brassinosteroids regulate root growth, development, and symbiosis. Mol. Plant 9, 86–100, https://doi.org/10.1016/j.molp.2015.12.003 (2016).
Grove, M. D. et al. Brassinolide, a plant growth-promoting steroid isolated from Brassica napus pollen. Nature 281, 216–217, https://doi.org/10.1038/281216a0 (1979).
Noguchi, T. et al. Arabidopsis det2 is defective in the conversion of (24R)-24-methylcholest-4-En-3-one to (24R)-24-methyl-5alpha-cholestan-3-one in brassinosteroid biosynthesis. Plant Physiol. 120, 833–840, https://doi.org/10.1104/pp.120.3.833 (1999).
Nakamura, A. et al. The role of OsBRI1 and its homologous genes, OsBRL1 and OsBRL3, in rice. Plant Physiol. 140, 580–590, https://doi.org/10.1104/pp.105.072330 (2006).
Qian, W. L. et al. Novel rice mutants overexpressing the brassinosteroid catabolic gene CYP734A4. Plant Mol Biol. 93, 197–208, https://doi.org/10.1007/s11103-016-0558-4 (2017).
Tognolli, M., Penel, C., Greppin, H. & Simon, P. Analysis and expression of the class III peroxidase large gene family in Arabidopsis thaliana. Gene 288, 129–138, https://doi.org/10.1016/s0378-1119(02)00465-1 (2002).
Duroux, L. & Welinder, K. G. The peroxidase gene family in plants: a phylogenetic overview. J. Mol. Evol. 57, 397–407, https://doi.org/10.1007/s00239-003-2489-3 (2003).
Passardi, F., Longet, D., Penel, C. & Dunand, C. The class III peroxidase multigenic family in rice and its evolution in land plants. Phytochemistry 65, 1879–1893, https://doi.org/10.1016/j.phytochem.2004.06.023 (2004).
Hiraga, S., Sasaki, K., Ito, H., Ohashi, Y. & Matsui, H. A large family of class III plant peroxidases. Plant Cell Physiol. 42, 462–468, https://doi.org/10.1093/pcp/pce061 (2001).
Elstner, E. F. & Heupel, A. Formation of hydrogen peroxide by isolated cell walls from horseradish (Armoracia lapathifolia Gilib.). Planta 130, 175–180, https://doi.org/10.1007/BF00384416 (1976).
Gross, G. G., Janse, C. & Elstner, E. F. Involvement of malate, monophenols, and the superoxide radical in hydrogen peroxide formation by isolated cell walls from horseradish (Armoracia lapathifolia Gilib.). Planta 136, 271–276, https://doi.org/10.1007/BF00385995 (1977).
Chen, S. X. & Schopfer, P. Hydroxyl-radical production in physiological reactions. A novel function of peroxidase. Eur. J. Biochem. 260, 726–735, https://doi.org/10.1046/j.1432-1327.1999.00199.x (1999).
De Tullio, M. C., Jiang, K. & Feldman, L. J. Redox regulation of root apical meristem organization: connecting root development to its environment. Plant Physiol. Biochem. 48, 328–336, https://doi.org/10.1016/j.plaphy.2009.11.005 (2010).
Haubrick, L. L. & Assmann, S. M. Brassinosteroids and plant function: some clues, more puzzles. Plant Cell Environ. 29, 446–457, https://doi.org/10.1111/j.1365-3040.2005.01481.x (2006).
Zhao, Y., Qi, Z. & Berkowitz, G. A. Teaching an old hormone new tricks: cytosolic Ca2+ elevation involvement in plant brassinosteroid signal transduction cascades. Plant Physiol. 163, 555–565, https://doi.org/10.1104/pp.112.213371 (2013).
Xia, X. J. et al. Interplay between reactive oxygen species and hormones in the control of plant development and stress tolerance. J. Exp. Bot. 66, 2839–2856, https://doi.org/10.1093/jxb/erv089 (2015).
Ogasawara, Y. et al. Synergistic activation of the Arabidopsis NADPH oxidase AtrbohD by Ca2+ and phosphorylation. J. Biol. Chem. 283, 8885–8892, https://doi.org/10.1074/jbc.M708106200 (2008).
Kobayashi, M. et al. Calcium-dependent protein kinases regulate the production of reactive oxygen species by potato NADPH oxidase. Plant Cell 19, 1065–1080, https://doi.org/10.1105/tpc.106.048884 (2007).
Zhang, A. et al. ZmMPK5 is required for the NADPH oxidase-mediated self-propagation of apoplastic H2O2 in brassinosteroid-induced antioxidant defence in leaves of maize. J. Exp. Bot. 61, 4399–4411, https://doi.org/10.1093/jxb/erq243 (2010).
Zhu, Y. et al. MAP65-1a positively regulates H2O2 amplification and enhances brassinosteroid-induced antioxidant defence in maize. J. Exp. Bot. 64, 3787–3802, https://doi.org/10.1093/jxb/ert215 (2013).
González-García, M. P. et al. Brassinosteroids control meristem size by promoting cell cycle progression in Arabidopsis roots. Development 138, 849–859, https://doi.org/10.1242/dev.057331 (2011).
Hacham, Y. et al. Brassinosteroid perception in the epidermis controls root meristem size. Development 138, 839–848, https://doi.org/10.1242/dev.061804 (2011).
Xia, X. J., Chen, Z. & Yu, J. Q. ROS mediate brassinosteroids-induced plant stress responses. Plant Signal Behav. 5, 532–534, https://doi.org/10.4161/psb.10989 (2010).
Thiellement, H., Zivy, M., Damerval, C. & Mechin, V. Plant Proteomics: Methods and Protocols. Methods Mol. Biol. 1, 355, https://doi.org/10.1385/1597452270 (2007).
Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362, https://doi.org/10.1038/nmeth.1322 (2009).
Hu, X. L. et al. Quantitative iTRAQ-based proteomic analysis of phosphoproteins and ABA regulated phosphoproteins in maize leaves under osmotic stress. Sci. Rep. 5, 15626, https://doi.org/10.1038/srep15626 (2015).
Zhao, Y. L. et al. Quantitative proteomic analyses identify ABA-related proteins and signal pathways in maize leaves under drought conditions. Front. Plant Sci. 7, 1827, https://doi.org/10.3389/fpls.2016.01827 (2016).
Xu, Y. et al. Comparative proteomic analysis provides new insights into low Nitrogen-promoted primary root growth in hexaploid wheat. Front Plant Sci. 10, 151, https://doi.org/10.3389/fpls.2019.00151 (2019).
Wu, L. et al. Comparative proteomic analysis of the maize responses to early leaf senescence induced by preventing pollination. J. Proteomics 177, 75–87, https://doi.org/10.1016/j.jprot.2018.02.017 (2018).
Tomizioli, M. et al. Deciphering thylakoid sub-compartments using a mass spectrometry-based approach. Mol. Cell Proteomics 13, 2147–2167, https://doi.org/10.1074/mcp.M114.040923 (2014).
Chen, Y. Y. et al. Proteolysis of chloroplast proteins is responsible for accumulation of free amino acids in dark-treated tea (Camellia sinensis) leaves. J. Proteomics 157, 10–17, https://doi.org/10.1016/j.jprot.2017.01.017 (2017).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435, https://doi.org/10.1093/nar/gkn176 (2008).
Jabs, T., Dietrich, R. A. & Dangl, J. L. Initiation of runaway cell death in an Arabidopsis mutant by extracellular superoxide. Science 27, 1853–1856, https://doi.org/10.1126/science.273.5283.1853 (1996).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31572197), the National Key Research and Development Program of China (2016YFD0300205), the National Natural Science Foundation of China (31201678), and Natural Science Foundation of Henan province (162300410133).
Author information
Authors and Affiliations
Contributions
L.L. and Y.X. performed most experiments; Y.R. and J.L. performed partial work of this research; Z.G. and Y.R. took part in the bioinformatic analysis; T.L. and Y.T. proposed many suggestions during the research; Y.R., T.L. and D.C. conceived and designed the experiments; Y.R. and Y.X. wrote the paper. Y.T. put forward many helpful suggestions on the writing.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Xu, Y., Ren, Y. et al. Comparative Proteomic Analysis Provides Insights into the Regulatory Mechanisms of Wheat Primary Root Growth. Sci Rep 9, 11741 (2019). https://doi.org/10.1038/s41598-019-47926-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47926-7
This article is cited by
-
Wheat root systems as a breeding target for climate resilience
Theoretical and Applied Genetics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
