
Abstract
The erythroid growth factor erythropoietin (Epo) is produced by renal interstitial fibroblasts, called REP (renal Epo-producing) cells, in a hypoxia-inducible manner. In chronic kidney disease (CKD), REP cells lose their Epo-production ability, leading to renal anaemia. Concurrently, REP cells are suggested to be transformed into myofibroblasts, which are the major player of renal fibrosis. Although establishment of cultured cell lines derived from REP cells has been a long-term challenge, we here successfully established a REP-cell-derived immortalized and cultivable cell line (Replic cells) by using a genetically modified mouse line. Replic cells exhibited myofibroblastic phenotypes and lost their Epo-production ability, reflecting the situation in renal fibrosis. Additionally, we found that cell-autonomous TGFβ signalling contributes to maintenance of the myofibroblastic features of Replic cells. Furthermore, the promoters of genes for Epo and HIF2α, a major activator of Epo gene expression, were highly methylated in Replic cells. Thus, these results strongly support our contention that REP cells are the origin of myofibroblasts in fibrotic kidneys and demonstrate that cell-autonomous TGFβ signalling and epigenetic silencing are involved in renal fibrosis and renal anaemia, respectively, in CKD. The Replic cell line is a useful tool to further investigate the molecular mechanisms underlying renal fibrosis.
Similar content being viewed by others

Introduction
Because kidneys are the major organ producing the erythroid growth factor erythropoietin (Epo), Epo-deficiency anaemia often arises as a complication of chronic kidney disease (CKD)1. In addition to renal anaemia, renal fibrosis, in which myofibroblasts producing extracellular matrix emerge in peritubular interstitia, is commonly observed among a large number of CKD patients2. Renal myofibroblasts are considered to originate in diverse ways, for example, through transformation of tubular epithelial cells3, capillary endothelial cells4, and interstitial fibroblasts5,6,7 in kidneys as well as the migration of bone marrow cells8,9. Of the multiple myofibroblast origins, interstitial fibroblasts are thought to mainly contribute to renal fibrosis in response to inflammation signalling activated by injury to nephrons6,7,8,9. Importantly, interstitial fibroblasts distributed in renal cortices and corticomedullary boundaries are the site of Epo production10,11,12,13, and the Epo-production ability of fibroblasts (renal Epo-producing [REP] cells) is inactivated by myofibroblastic transformation6,7. Thus, renal anaemia and fibrosis are tightly related to each other in REP cells during CKD progression.
REP cells express the Epo gene in a hypoxia-inducible manner to maintain a systemic oxygen supply via erythrocytes14. For hypoxic induction of the Epo gene, hypoxia-inducible transcription factor 2α (HIF2α) is essential15. The α subunits of hypoxia-inducible factors (HIFs), including HIF2α, are degraded and inactivated under normal oxygen conditions (normoxia) through hydroxylation of their specific proline residues followed by ubiquitin-proteasome degradation, whereas hypoxia stabilizes and activates HIFs by blocking hydroxylation16,17,18. Prolyl hydroxylase domain enzymes (PHDs) are hypoxia-sensing molecules that catalyse the hydroxylation of HIFs in an oxygen-dependent manner19. We previously reported that HIF2α is inactivated in myofibroblast-transformed REP (MF-REP) cells in damaged mouse kidneys regardless of their hypoxic milieu15. Because forced activation of HIF2α by deletion of genes for PHDs restores the Epo-production ability in MF-REP cells, inactivation of HIF2α in MF-REP cells is considered to be due to abnormal activation of PHDs in hypoxic myofibroblasts15. HIF2α is one of the most important molecules in renal anaemia development. In fact, chemicals that are inhibitory to PHDs (PHDi) are undergoing clinical trials for renal anaemia treatment20.
The myofibroblastic transformation of REP cells is reversed after the restoration of kidney injuries at least partially, and inflammatory signalling such as transforming growth factor β (TGFβ) and tumour necrosis factor α signalling, are involved in this transformation7. Although understanding the mechanisms of renal fibrosis is critical for elucidating renal anaemia and CKD progression, the molecular characterization of REP cells has not been investigated due to the lack of appropriate culture cell models for REP cells. In this study, we isolated REP cells from mouse kidneys and immortalized them by the exogenous expression of oncogenic H-RAS. Consequently, one cell line referred to as “Replic (REP-cell lineage cells immortalized and cultivable) cells” was successfully established. Replic cells exhibited myofibroblastic features with high-level TGFβ expression, and inhibition of TGFβ signalling attenuated the myofibroblast-related gene expression pattern in the cells. Additionally, the cells lost their Epo-production ability by epigenetic suppression of HIF2α expression. These results directly indicate that renal myofibroblasts emerge from transformation of REP cells in injured kidneys and that the cell-autonomous TGFβ signal is involved in REP cell transformation to myofibroblasts. Furthermore, it is demonstrated that epigenetic silencing of genes for Epo and/or HIF2α is one of the major causes for loss of the Epo-production ability in MF-REP cells. We also propose that Replic cells are a valuable tool to understand the mechanisms underlying renal fibrosis and renal anaemia, both of which are significant complications of CKD associated with disease progression21,22.
Results
Cultivation and immortalization of REP cells isolated from ISAM-REC mice
We previously established a gene-modified mouse line in which REP cells are efficiently labelled with tdTomato red fluorescent protein expression13. In this mouse line, referred to as ISAM-REC mice (EpoGFP/GFP:Rosa26CAG-LSL-tdTomato/WT:Tg3.3K-Epo:TgEpoCre genotype)14, the expression of transgenic Cre recombinase under the control of the Epo gene regulatory region is highly induced by severe anaemic conditions due to Epo deficiency, and most REP cells permanently express tdTomato as a marker for Cre-mediated recombination without any treatment14,23. Thus, tdTomato-positive cells from ISAM-REC kidneys were applied for ex vivo cultivation and immortalization to generate cell lines derived from REP cells.
First, tdTomato-positive cells were isolated from ISAM-REC kidney cell suspensions by using a cell sorter, and the cells were incubated with mesenchymal stem cell growth medium (MSCM). However, no cells survived after the 10-day ex vivo cultivation, suggesting that cell-cell communications and/or soluble factors secreted by kidney cells other than REP cells are required for REP cell expansion and maintenance. Therefore, the cell suspensions were directly incubated without cell sorting. After a week of cultivation, we observed that tdTomato-positive cells grew with tdTomato-negative cells, attaching to the bottoms of culture dishes (Fig. 1a).

Establishment of a Replic cell line. (a) A representative image of cells cultured in MSCM for one week after isolation from ISAM-REC kidneys. The mixed cell culture contains cells positive (red) and negative (asterisk) for tdTomato fluorescence. (b) A colony of tdTomato-positive cells was generated after immortalization and isolation of tdTomato-positive REP cell lineage cells from the mixed renal cell culture. (c) Phase-contrast (left) and tdTomato fluorescent (right) images of Replic cells cultured with MSCM (upper) or DMEM (lower). (d) Growth curves of Replic cells cultured with DMEM (blue) or MSCM (red). On Day 0, 1.0 × 104 cells were seeded onto 3.5-cm dishes with 2.5 mL medium, and cell numbers were counted daily. The data are shown as the means ± standard errors. n = 3 for each group. **p < 0.01 by two-tailed and unpaired Student’s t tests. (e) Genomic PCR of Replic cells compared with that of organs from wild-type C57BL/6 (WT) and ISAM-REC mice. N, a non-template negative control.
Because it was considered that the 1-week cultivation let the kidney cells adapt to the culture conditions, the cells were integrated with a lentivirus vector expressing the human HRAS gene bearing an oncogenic mutation in order to immortalize the cells. After expansion of the infected cells for 2 weeks, tdTomato-positive REP cells in the dishes were sorted and subjected to pure cultivation. Finally, a single colony of tdTomato-positive cells was developed (Fig. 1b). It was possible for the colony-derived cells in culture dishes to grow with MSCM for more than 1 year, to recover from freeze-thawing, and to be passaged more than 10 times. We thus concluded that a REP-cell-derived cell line was established and named the cells Replic (REP cell-lineage immortalized and cultivable) cells.
Growth of Replic cells
Replic cells and natural REP cells in ISAM-REC mice shared a fibroblast-like shape that exhibited bipolar stretching with bright fluorescence of tdTomato (Fig. 1c)12. The cell shape was narrow and stretched with many processes when the cells were cultured with MSCM rather than with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS) (Fig. 1c). This observation suggests that MSCM is a more appropriate culture medium than DMEM for maintaining the features of in vivo REP cells under ex vivo culture conditions because the REP cells show a sharp and extended morphology with many processes in the renal interstitia11,12. Therefore, MSCM was used for characterization of Replic cells in this study. Replic cells proliferated significantly in MSCM rather than in DMEM and doubled daily in the logarithmic growth phase on normal polystyrene dishes without any coating (Fig. 1d). Although the contents of MSCM are veiled by the manufacturer, the cell-growth-optimized MSCM likely promotes proliferation of Replic cells compared with the basic medium.
Genotyping PCR confirmed that the immortality and proliferative activity of Replic cells were supported by integration of the oncogenic mutant HRAS gene (Fig. 1e). The ISAM-REC kidney origin of Replic cells was confirmed by PCR data demonstrating that Replic cells and ISAM-REC mice commonly harboured the Cre, tdTomato, and GFP genes but lacked the Epo gene (Fig. 1e). From these results, we concluded that the first REP-cell-derived cell line has been established and that the cells vigorously proliferate, maintaining their fibroblastic features under culture conditions optimized for mesenchymal stem cells.
Fibroblastic phenotype of Replic cells
Flow cytometry of Replic cells detected high-level tdTomato fluorescence in every Replic cell (Fig. 2a), showing the clonality of the cell line. The data also revealed that compared to wild-type mouse embryonic fibroblasts (MEFs), Replic cells strongly expressed CD73 (ecto-5′-nucleotidase), a fibroblastic surface marker of REP cells in mouse kidneys12,24,25. Consistently, gene expression analyses showed that Replic cells expressed the gene for CD73 (Nt5e) at higher levels than MEFs did (Fig. 2b).

Fibroblastic phenotype of Replic cells. (a) Flow cytometry of Replic cells (upper) and MEFs (lower) cultured with MSCM. Every Replic cell expressed high-level tdTomato fluorescence. The mean fluorescent intensity of CD73 in Replic cells was higher than that in MEFs (dotted red lines). (b,c) The mRNA expression levels of marker genes for fibroblasts (b) and non-fibroblastic renal cells (c) in Replic cells were compared to those in wild-type (WT) kidneys and MEFs. Arrows indicate undetectable levels. Cell culture was conducted with MSCM. Ten biologically independent samples for each cell line were analysed. The average expression level of WT kidneys (n = 3) was set at 1.0, and error bars indicate standard errors. **p < 0.01 by multiple comparisons using one-way ANOVA with Tukey-Kramer tests.
Replic cells did not express the Kdr, Cdh1, and Nphs1 genes, which are lineage markers for vascular endothelial cells, tubular epithelial cells, and podocytes, respectively (Fig. 2c). Taken together, the findings revealed that the cell line originated from tdTomato+CD73+ REP cells of the ISAM-REC kidney but not from other lineage cells of kidneys. However, mRNA expression of the Pdgfrb gene encoding PDGFRβ (platelet-derived growth factor receptor β), another fibroblastic marker of REP cells6,12,26, was undetectable in Replic cells (Fig. 2b). These data suggested that the cell phenotype was altered during isolation, cultivation, and/or immortalization.
Epo gene activity and hypoxic response of Replic cells
Because the Epo gene of ISAM-REC mice was homozygously replaced with the gene for green fluorescent protein (GFP), the Epo gene activity of Replic cells derived from an ISAM-REC mouse was able to be evaluated by measuring the amounts of mRNA transcribed from the EpoGFP recombinant gene13,27. In REP cells, Epo gene expression is induced in a hypoxia-inducible manner via stabilization and activation of HIF2α15. Therefore, we investigated EpoGFP mRNA levels in Replic cells incubated under hypoxic (1% O2) conditions.
In ISAM-REC kidneys, EpoGFP mRNA was highly expressed by severe anaemia, in agreement with our previous report (Fig. 3a,b)27. However, EpoGFP mRNA expression was undetectable in Replic cells regardless of the presence of hypoxic stimuli, while the 24-hour hypoxic exposure induced expression of the Vegfa and Slc2a1 genes (encoding vascular endothelial cell growth factor A and glucose transporter 1, respectively), both of which are well known as HIF target genes (Fig. 3a)28,29. Additionally, expression of the gene for HIF2α (Epas1) was undetectable in both normoxic and hypoxic Replic cells, whereas Hif1a mRNA was consistently expressed under both conditions (Fig. 3a).

Hypoxic response of Replic cells. (a,b) mRNA expression levels of genes related to the hypoxic response in Replic cells exposed to hypoxia (a) or GSK360A (b) for 24 hours were compared to those in ISAM-REC kidneys. Twelve biologically independent samples for each group of Replic cells were analysed. The average expression level of ISAM-REC kidneys (n = 3) was set at 1.0, and error bars indicate standard errors. Arrows indicate undetectable levels. *p < 0.05 and **p < 0.01 by multiple comparisons using one-way ANOVA with Tukey-Kramer tests. (c) Immunoblots for HIF1α and HIF2α in Replic cells cultured under normoxic (N) or hypoxic (H, 1% oxygen) conditions for 24 hours (upper) and in Replic cells cultured with or without GSK360A (G and V, respectively) for 24 hours (lower). The HIF1α- and HIF2α-derived band intensities normalized with βTubulin-derived band intensities were quantified and are shown in the right panels. Hep3B cells was used as positive controls for the hypoxia-inducible accumulation of HIF1α and HIF2α. βTubulin was used as an internal control. Cell culture was conducted with MSCM (a–c).
We then tested effects of the PHDi GSK360A27 on EpoGFP mRNA expression of Replic cells. The results showed that, similar to hypoxic exposure, GSK360A treatment induced neither EpoGFP nor Epas1 expression (Fig. 3b). Because GSK360A enhanced the expression of the Vegfa and Slc2a1 genes (Fig. 3b), it was demonstrated that GSK360A activated HIF signalling in Replic cells. Indeed, HIF1α protein accumulated in Replic cells after GSK360A treatment, and hypoxic exposure also induced HIF1α accumulation (Fig. 3c). Additionally, GSK360A treatment resulted in the accumulation of both HIF1α and HIF2α in a Hep3B human hepatoma cell line (Fig. 3c). However, in accordance with the data showing the absence of Epas1 mRNA in Replic cells, HIF2α protein was undetectable in the cells even in the presence of GSK 360A or a hypoxic stimulus (Fig. 3c).
DNA methylation in the promoters of genes for Epo and HIF2α
We surmised that epigenetic silencing blunted Epo and Epas1 gene expression in Replic cells. It has been reported that DNA methylation in the Epo gene promoter region is involved in the loss of Epo production in fibrotic kidneys30. Bisulfite genomic sequence analyses of the Epo and Epas1 gene promoters in Replic cells were conducted by a comparison with ISAM-REC livers, in which the Epo gene promoter is active in the majority of cells13.
The results revealed that the proximal promoter regions of both genes were significantly methylated in Replic cells, whereas a few CpG methylations were detected in the promoters of the ISAM-REC livers (Fig. 4a). These data suggest that the expression of Epo and HIF2α in Replic cells was silenced by DNA methylation in their gene promoters. Additionally, it was thought that Replic cells may undergo transformation to myofibroblasts, in which the Epo gene is methylated and inactivated30, maintaining their fibroblastic features.

Epigenetic suppression of the Epo-production ability in Replic cells. (a) Summary of results from bisulfite sequencing in the Epo (upper) and Epas1 (lower) gene promoters of Replic cells and ISAM-REC livers. Genomic regions between red arrows were sequenced after cloning 8 clones for each promoter from bisulfite-converted genomic DNAs. Each row represents a single clone. Vertical bars indicate CpG sites in the tested regions, and white and black dots represent unmethylated and methylated CpG sites, respectively. TSS, transcription start site. *p < 0.05 and **p < 0.01 between the methylated ratio of each CpG site in Replic cells and ISAM-REC livers, using χ2 tests. (b) Immunoblots for HIF2α in Replic cells transiently transfected with plasmids expressing GFP or constitutively active HIF2α. βTubulin was used as an internal control for immunoblots. (c) EpoGFP mRNA expression levels in Replic cells transiently transfected with plasmids expressing GFP or constitutively active HIF2α. ISAM-REC kidneys were used as positive controls for EpoGFP mRNA expression. Six biologically independent samples for each group of Replic cells were analysed. The average expression level of ISAM-REC kidneys (n = 3) was set at 1.0, and error bars indicate standard errors. Arrows indicate undetectable levels. **p < 0.01 by multiple comparisons using one-way ANOVA with Tukey-Kramer tests. Cell culture was conducted with MSCM (a–c).
A 1-week incubation with 5-aza-2′-deoxycytidine (5-Aza), an inhibitor of DNA methyltransferase 1 (DNMT1), which is essential for maintenance of DNA methylation patterns beyond mitosis31, did not alter DNA methylation of the Epo gene promoter region, whereas the upstream enhancer region (the -8kHRE-flanking region) of the Epo gene32 was significantly demethylated by the inhibitor in Replic cells (Supplementary Fig. 1). Therefore, the contribution of continuous de novo DNA methylation to the hyper-methylation of Epo and Epas1 promoters in Replic cells was assumed33. We then tested whether exogenous HIF2α overexpression could activate EpoGFP gene expression in Replic cells. Constitutively active HIF2α, in which PHD-target prolyl residues were replaced with alanine residues34, was transiently overexpressed in Replic cells (Fig. 4b). However, EpoGFP expression was not induced (Fig. 4c), indicating that exogenous HIF2α overexpression was insufficient to reactivate the Epo-production ability in Replic cells. Thus, we propose that Replic cells lose their Epo-production ability due to continuous DNA methylation in the Epo gene promoter, which blocks the transcriptional activation signal of HIF2α.
Myofibroblastic property of Replic cells
Because REP cells transform to myofibroblasts in injured kidneys with loss of the Epo-production ability and DNA hyper-methylation of the Epo gene promoter6,7,30, the myofibroblastic property of Replic cells was investigated by a comparison to fibrotic kidneys of normal mice subjected to unilateral ureteral obstruction (UUO) for 14 days. Ureteral obstruction is an established model of renal fibrosis7,35 and Masson’s trichrome staining of kidney sections confirmed that our ureteral obstruction surgery induced renal fibrosis (Supplementary Fig. 2a)15. Although MEFs were used in this experiment as the normal fibroblast control, strong expression of myofibroblastic genes was reported in MEFs36. Gene expression analyses of Replic cells revealed that the cells highly expressed the Acta2 and Fn1 genes, both of which are known to be expressed in myofibroblasts37,38, compared to the expression levels in MEFs or injured kidneys (Fig. 5a). The data clearly indicated that Replic cells exhibit myofibroblastic properties.

Myofibroblastic property of Replic cells. (a) mRNA expression levels of genes related to myofibroblasts in Replic cells cultured with MSCM. In addition to MEFs cultured with DMEM, injured and contralateral kidneys (n = 3 for each) of UUO-treated normal mice were compared to Replic cells with respect to mRNA expression. Twelve biologically independent samples for each cell line were analysed. (b) mRNA expression levels of genes for TGFβ superfamily receptors in Replic cells cultured with MSCM were compared to those in mouse kidneys (n = 3 for ISAM-REC kidneys and WT kidneys) and MEFs. Six biologically independent samples for each cell line were analysed. (c) Flow cytometry of TGFβR2 expression on the cell surface of Replic cells and MEFs cultured with DMEM. Negative controls (NC) were stained only with APC-conjugated streptavidin. The mean fluorescent intensities are shown (red dotted lines). (d) mRNA expression levels of genes related to myofibroblasts in Replic cells cultured with DMEM or MSCM. Six biologically independent samples for each cell line were analysed. The average expression levels of injured kidneys (a), ISAM-REC kidneys (b), or Replic cells cultured with DMEM (d) were set at 1.0. Error bars are indicated standard errors, and arrows indicate undetectable levels (a,b). *p < 0.05 and **p < 0.01 by multiple comparisons using one-way ANOVA with Tukey-Kramer tests (a,b) or by two-tailed, unpaired Student’s t-tests (d).
TGFβ is known to be secreted by fibrotic kidneys and to promote organ fibrosis7,39,40,41. In fact, our analyses clearly demonstrated that mRNA expression of the gene for TGFβ (Tgfb1) was dramatically induced by kidney injury (Fig. 5a). Notably, Replic cells expressed the Tgfb1 gene at a comparable level to that in injured kidneys (Fig. 5a). The expression levels of genes for the major TGFβ receptors TGFβR1 (Tgfbr1) and TGFβR2 (Tgfbr2) were also detected in Replic cells, while other TGFβ superfamily receptor genes (Acvrl1 and Bmpr1b) were not expressed (Fig. 5b). Flow cytometry confirmed that TGFβR2 is apparently expressed on the cell surface of the majority of Replic cells (Fig. 5c). Additionally, the gene expression levels of these receptors were significantly higher in injured kidneys than in contralateral kidneys of UUO-subjected mice (Supplementary Fig. 2b), supporting the idea that TGFβ signalling is involved in progression of renal fibrosis42. These data led us to assume that cell-autonomous TGFβ signalling is involved in the myofibroblastic property of Replic cells.
Based on the cell shape described above, we proposed that Replic cells seemed to differentiate into myofibroblasts by changing the culture medium from MSCM to DMEM (see Fig. 1c). This proposal was supported by the gene expression data showing that expression levels of the myofibroblastic marker genes Acta2 and Fn1 were significantly higher in Replic cells cultured with DMEM than in Replic cells cultured with MSCM (Fig. 5d). In contrast, the expression levels of genes for TGFβ and TGFβ receptors were not responsive to changes in the medium (Fig. 5d). Because TGFβ signalling activates expression of the Acta2 and Fn1 genes but not the Tgfb1, Tgfbr1, and Tgfbr2 genes38,43, MSCM likely contains factors that inhibit TGFβ signalling, selectively suppressing Acta2 and Fn1 gene expression in Replic cells.
Activation of cell-autonomous TGFβ signalling in Replic cells
In the TGFβ signal, TGFβ binding to a TGFβR1/TGFβR2 heterodimeric receptor results in phosphorylation of Smad2 and/or Smad3 transcription factors followed by induction of Acta2 and Fn1 gene expression44. Consistent with the myofibroblastic gene expression profiles (Fig. 5a,d), Smad2 and/or Smad3 (Smad2/3) were highly phosphorylated in Replic cells compared to MEFs when cells were cultured with DMEM, and the phosphorylation was reduced by cultivation with MSCM (Fig. 6a). Consequently, cell-autonomous TGFβ signalling through TGFβR1/TGFβR2-Smad2/3 was considered one of the major pathways promoting myofibroblastic transformation of Replic cells.

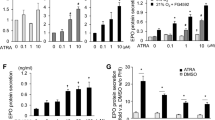
Upregulation of profibrotic TGFβ-signalling in Replic cells. (a) Immunoblots for p-Smad2/3 and total Smad2/3 in Replic cells cultured with DMEM were compared to those in MEFs cultured with DMEM (left) or Replic cells cultured with MSCM (right). βTubulin was used as an internal control. (b) TGFβ concentrations in serum-free DMEM incubated with Replic cells or MEFs for 24 hours. Three biologically independent samples for each cell line were analysed. (c) Relative amounts of the listed factors in culture supernatants of Replic cells and MEFs, both of which were cultured with serum-free DMEM for 24 hours, were determined by the antibody array based on a double antibody sandwich assay. The data indicate the chemiluminescent intensities of each spot (duplicate for each antibody, A and B). Undetectable cytokines, both spots of which showed signal intensities lower than 50, are listed in the right column. The scanned image of the antibody array is shown in Supplementary Fig. 3.
To confirm TGFβ production by Replic cells, TGFβ concentrations in the supernatant incubated with Replic cells for 24 hours were measured by enzyme-linked immunosorbent assay (ELISA). The data demonstrated that Replic cells produce significantly higher amounts of TGFβ than MEFs (Fig. 6b), although these cell lines expressed comparable levels of Tgfb1 mRNA (Fig. 5a). ELISA data from mouse plasma showed that plasma TGFβ concentrations are elevated in mice suffering from renal fibrosis (Supplementary Fig. 2c). These data suggest that TGFβ is secreted by fibrotic kidney cells including myofibroblast-transformed REP cells.
We further performed an antibody-based cytokine array on the culture supernatants. In agreement with the ELISA data, intense signals were detected in samples from Replic cells compared to those from MEFs (Fig. 6c, Supplementary Fig. 3a,b). The data also showed that Replic cells produced proinflammatory cytokines such as MCP1 (monocyte chemotactic protein 1, also known as CCL2) and CXCL5 (C-X-C chemokine ligand 5) at higher levels than MEFs (Fig. 6c, Supplementary Fig. 3a,b). These results suggest that Replic cells acquire myofibroblastic properties related to cell-autonomous TGFβ signalling during isolation, cultivation, and/or immortalization of fibroblastic REP cells.
TGFβ-dependent myofibroblastic property of Replic cells
To investigate the necessity of cell-autonomous TGFβ signalling for the myofibroblastic property of Replic cells, Replic cells were incubated with SB431542, a kinase inhibitor specific for TGFβR1, ALK4 (activin receptor-like kinase 4), and ALK745. The mRNA expression levels of the Acta2 and Fn1 genes were markedly decreased by SB431542, while Tgfb1 mRNA expression was unaltered (Fig. 7a). In addition to SB431542, TGFβ neutralizing antibody (TGFβ N-Ab) reduced Acta2 gene expression in Replic cells (Fig. 7b). Although it was predicted that the original fibroblastic property of REP cells was, at least in part, restored by inhibition of TGFβ signalling in Replic cells, EpoGFP mRNA expression was still undetectable in Replic cells cultured with SB431542 for 24 hours (Fig. 7a). Inhibition of TGFβ signalling by SB431542 was confirmed by immunoblotting data showing a decrease in phosphorylated Smad2/3 (p-Smad2/3, Fig. 7c).

TGFβ-dependent myofibroblastic property of Replic cells. (a) mRNA expression levels of genes related to myofibroblasts in Replic cells incubated with or without SB431542 in DMEM for 24 hours. EpoGFP mRNA levels were also examined. ISAM-REC kidneys (n = 3) were used as controls. Ten biologically independent samples for each treatment of Replic cells were analysed. (b) The Acta2 mRNA expression levels in Replic cells incubated with vehicle or neutralizing antibody against TGFβ (N-Ab) in DMEM for 24 hours. Six biologically independent samples for each treatment of Replic cells were analysed. (c) Immunoblots for p-Smad2/3 and total Smad2/3 in Replic cells cultured with or without SB431542 in DMEM for 1 hour. βTubulin was used as an internal control. (d) Relative mRNA expression levels of genes related to hypoxic response were measured in Replic cells cultured with DMEM in the presence or absence (Vehicle) of the inhibitor cocktail. ISAM-REC kidneys (n = 3) were used as controls. As the inhibitor cocktail, SB431542, 5-Aza, and GSK360A were added to the cell culture at 24 hours, 5 days, and 24 hours before sampling, respectively. Five biologically independent samples for each treatment of Replic cells were analysed. The average expression level of ISAM-REC kidneys (a,d) or vehicle-treated Replic cells (b) was set at 1.0, and the error bars represent standard errors for each experimental group. (a,b,d) Arrows indicate undetectable levels. *p < 0.05 and **p < 0.01 by multiple comparisons using one-way ANOVA with Tukey-Kramer tests (a,d) or by two-tailed, unpaired Student’s t-tests (b).
Finally, we tested whether the Epo-production ability could be restored by simultaneous supplementation with SB431542, GSK360A, and 5-Aza. As the result, the inhibitor cocktail did not activate EpoGFP mRNA expression, while the expression of other HIF target genes was significantly induced (Serpine1 and Slc2a1, Fig. 7d)29,46. The Epas1 gene remained silent under the cocktail presence, although Hif1a mRNA was constantly expressed in Replic cells with or without the cocktail (Fig. 7d). Therefore, expression of the Serpine1 and Slc2a1 genes in Replic cells is thought to be driven through HIF1α activation by the inhibitor cocktail, which includes a PHDi. Consequently, we propose that Replic cells would be useful for elucidating the mechanisms of the myofibroblastic transformation of REP cells even though the cells may irreversibly lose their Epo-production ability.
Discussion
Because REP cells play a central role in renal anaemia and renal fibrosis in CKD patients, cell lines derived from REP cells have long been waited for use in studies elucidating the molecular pathophysiology of REP cells. In this study, we successfully generated a REP-cell-derived cell line “Replic cells” for the first time. Because Replic cells exhibit myofibroblastic features and defects in Epo production, we are certain that Replic cells will allow us to understand the molecular mechanisms underlying the development of renal anaemia and fibrosis during kidney disease progression.
We previously reported that REP cells are inactive in proliferation and energy metabolism in healthy kidneys, whereas kidney injuries stimulate the proliferation of REP cells with Ki67 expression7,15. In this study, we observed that REP cells isolated from single-cell suspensions of mouse kidneys failed to survive for more than 1 week under any culture conditions tested here. Then, we tested mixed cell cultures of renal cells and found that REP cells require intercellular communication with renal cells for survival and growth in vitro. During the mixed culture, REP cells gained proliferative activity. Although our preliminary experiments showed that the primary REP cells in the mixed culture for 3 days expressed the EpoGFP gene in response to the PHDi, the cells lost their Epo-production ability within 1 week. Thus, it is proposed that the renal microenvironment, including tubulointerstitial and/or vasculointerstitial interactions, keeps REP cells quiescent in healthy kidneys and that damage to tubules and/or vessels in injured kidneys or in culture dishes induces the transformation of REP cells through activating proliferation and inactivating Epo production. Additionally, quiescence may be required for the Epo-producing ability of REP cells.
Replic cells preferentially grow and maintain their sharp configuration in a special culture medium optimized for mesenchymal stem cell (MSC) growth rather than in a general medium. In addition, Replic cells and REP cells commonly express CD73 at high levels12,24,25; CD73 is an MSC marker47. Thus, it is reasonable to conclude that REP cells include MSCs. Indeed, there are reports demonstrating that MSCs play essential roles in kidney development by secreting growth factors48, that renal interstitial fibroblasts in healthy kidneys and renal myofibroblasts in injured kidneys are derivatives of MSCs5, and that cells bearing MSC properties exist in kidneys for tissue repair25,49,50. Importantly, a subset of MSCs isolated from adult mouse kidneys expresses Epo25.
A variety of cell types have been reported as origins of myofibroblasts emerging in kidney diseases. For instance, interstitial fibroblasts such as REP cells5,6,7, bone marrow-derived cells8,9, tubular epithelial cells3 and vascular endothelial cells4. The myofibroblastic features of Replic cells clearly demonstrate that REP cells are one of the sources of renal myofibroblasts. This conclusion coincides with our previous articles reporting that the majority of renal myofibroblasts in injured mouse kidneys originate from REP cells6,7.
TGFβ is known as a master regulator of fibrosis2,51, but its source is controversial in renal fibrosis. Injured tubular epithelial cells, endothelial cells, and macrophages are major TGFβ-producing cell candidates in fibrotic kidneys52,53,54. On the basis of data from Replic cells, we further argue that cell-autonomous TGFβ signalling promotes the myofibroblastic transformation of REP cells. We and others previously demonstrated that activation of TGFβ signalling, which is defined by the phosphorylation of Smad2/3, is detected in MF-REP cells in injured kidneys7,30. However, further studies using Replic cells are required to identify triggers of cell-autonomous TGFβ induction in myofibroblasts of injured kidneys. In addition to TGFβ, MCP1 and CXCL5 are secreted at high levels from Replic cells, and both chemokines have been suspected to be secreted from injured kidneys and to contribute to inflammation and fibrosis by recruiting myeloid cells from the bone marrow55,56. Analyses of Replic cells will allow us to deeply understand the molecular mechanism of organ fibrosis.
Hypoxia-inducible Epo production in REP cells is regulated at the gene transcription level23. In Replic cells exhibiting features of MF-REP cells, the promoter region of the Epo gene is highly methylated. Because DNA methylation in gene promoters blocks the association of basic transcription factors and RNA polymerases57, it is proposed that REP cells lose their Epo-production ability due to Epo gene silencing by DNA methylation during the transformation to myofibroblasts in injured kidneys. Similarly, a previous report demonstrated that deficient Epo production is caused by hyper-methylation of the Epo gene promoter in renal myofibroblasts30 although this study could not exclude the possibility that H-RAS overexpression but not myofibroblastic transformation directly mediated the DNA methylation. This content is confirmed by our experiment showing that the Epo-production ability of Replic cells was not induced by overexpression of HIF2α, a major activator of Epo gene transcription in REP cells15. In this study, we revealed that the expression of HIF2α at the mRNA and protein levels is blunted due to hyper-methylation of the Epas1 promoter region in Replic cells. Therefore, epigenetic silencing in the Epo and Epas1 loci is involved in the development of renal anaemia.
We previously reported that the Epo-production ability is retained beyond myofibroblastic transformation by the constitutive activation of HIF2α in REP cells before injury15. During Replic cell establishment under normal air conditions, HIF2α was probably inactivated, and the Epo and Epas1 promoter regions were methylated. We then propose that active HIF2α or active Epo gene transcription may protect the Epo gene promoter from hyper-methylation in renal myofibroblasts. Overexpression of the constitutively active H-RAS may also contribute to the DNA-methylation-mediated gene silencing, because RAS signalling has been reported to induce DNA methylation primarily through the MAPK pathway58,59. Although the molecular mechanism of gene-specific methylation in renal myofibroblasts has not been elucidated, TGFβ signalling is supposed to induce DNA methylation through enhancing the expression of DNA methyltransferases in renal myofibroblasts15,30. This study proposes that de novo methylation of specific genomic regions, including the Epo and Epas1 gene promoters, is constitutively induced by cell-autonomous TGFβ production in renal myofibroblasts.
Using mouse models, we previously demonstrated that MF-REP cells restore REP cell properties by ameliorating kidney injury7. Although this study failed to recover the Epo-production ability in Replic cells by TGFβ signal blockage, the inhibition of TGFβ signalling partially debilitated the myofibroblastic phenotype of Replic cells in the gene expression profile. Further attempts to restore REP-cell properties in Replic cells are directly linked to the identification of therapeutic targets for both renal fibrosis and anaemia in CKD. To treat renal anaemia, PHDi is effective for inducing renal Epo production in CKD patients, including end-stage renal disease patients20. However, the Epo gene in Replic cells is not responsive to PHDi. Our preliminary data show that the Epo-induction effect of PHDi is gradually weakened with the progression of kidney disease in mice. These observations suggest that REP cells may transform into myofibroblasts in a stepwise manner during kidney disease progression and that Replic cells may correspond to severely myofibroblast-transformed REP cells, in which Epo and HIF2α expressions are epigenetically silenced. Finally, we conclude that Replic cells will provide significant information on the molecular pathology of CKD, from which more than 10% of people suffer worldwide and for which no plausible treatments have been established due to a poor understanding of the mechanisms of CKD pathogenesis.
Methods
Ethics statement
All animal experiments were conducted in accordance with “Regulations for Animal Experiments and Related Activities at Tohoku University” by “Institutional Animal Care and Use Committee of Tohoku University (http://www.clar.med.tohoku.ac.jp/en.html; Approval No. is 2017-MdA-090)”.
Mice
All animals, including ISAM-REC mice13,14, were maintained under specific pathogen-free conditions. Male mice at 11–14 weeks of age were used in all experiments. For UUO surgery, the left ureter was cut between 2 ligatures made by 4–0 Polysorb (Syneture) at the level of the lower pole of the kidney7. At 10 days after UUO surgery, both kidneys and plasma from the buccal vein were collected. Kidneys were fixed with 10% formalin (Nakalai Tesque) for 24 hours, and sections (5-µm thickness) of paraffin-embedded kidneys were stained with Masson’s trichrome stain (Polysciences). Kidney samples for RT-qPCR analyses were obtained 14 days after UUO surgery.
Isolation and immortalization of REP cells
Kidneys of ISAM-REC mice were chopped and digested with 1.5 mg mL−1 of collagenase type II (Worthington) for 60 minutes at 37 °C followed by dissociation using a gentleMACS dissociator (Miltenyi Biotec). The cell suspension was incubated for one week, and adherent cells were infected with a lentivirus vector expressing the G12V oncogenic mutant of human H-RAS (abm). From the immortalized cells, REP-cell-derived cells positive for tdTomato expression were isolated using a FACS Jazz cell sorter with FACS Diva software (Becton Dickinson). In the isolated cell culture, a single colony was developed and expanded as the Replic cell line.
Cell culture
MEFs were established from embryos of wild-type C57BL/6 mice (CREA Japan) at embryonic day 13.5. Replic cells, MEFs, and Hep3B cells (ATCC) were maintained in DMEM (Nakalai Tesque) containing 10% foetal bovine serum (FBS, Biosera) or MSCM (PromoCell) in a humidified atmosphere containing 5% CO2 at 37 °C. Unless otherwise noted, cells were seeded onto 6- or 12-well polystyrene culture plates (Merck) within 10 passages and harvested when the cells grew to 80–90% confluency. Cell images were taken using a BZ9000 fluorescence microscope (Keyence). The growth ratio of cells was examined daily using a Tali image-based cytometer (Thermo Fisher Scientific) after 1.0 × 104 cells were seeded onto 3.5-cm dishes. For exposure to hypoxic air (consisting of 1% O2, 5% CO2, and 94% N2), cells were incubated in a Sci-tive hypoxia workstation (Ruskinn) for 24 hours.
Transient transfection
A pCMX-HIF2αCA plasmid expressing mouse HIF2α, of which 2 specific prolyl residues were replaced with alanine residues to become resistant to PHD-mediated hydroxylation and constitutively active in mammalian cells34, and pEGFP-N1 (TaKaRa) expressing enhanced GFP were used for transient transfection assays. Cells with 80% confluency in a well of 6-well plates were transfected with a total of 600 ng of pCMX-HIF2αCA and/or pEGFP-N1 using Lipofectamine 2000 reagent (Thermo Fisher Scientific) and harvested 48 hours after transfection. Transfection efficiency was estimated by measuring the ratio of GFP-expressing cells in total cells using Tali.
Compounds
SB431542 (Adooq Bioscience), GSK360A (Toronto Research Chemicals), and 5-Aza (Sigma-Aldrich) were resolved in dimethyl sulfoxide (DMSO, Nacalai Tesque) at 10 mM, 50 mM, and 5 mM concentrations, respectively. TGFβ N-Ab (1D11; Novus Biologicals) was dissolved in culture medium at a 2 mg mL−1 concentration just before administration. The resolved compounds were administered to the cells at a 1:1000 dilution. DMSO or culture medium was used as the vehicle control.
Genomic PCR
Genomic DNA was purified with phenol/chloroform/isoamyl alcohol (Nakalai Tesque) from cells or mouse organs. Genotype PCR was performed with primers listed in Supplementary Table 1 using Ex-Taq polymerase (TaKaRa). Amplified DNA fragments were electrophoresed on 2% agarose gels containing ethidium bromide (Nakalai Tesque) and detected using an E-BOX imaging system with ultra-violet light (VILVER).
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted and purified using ISOGEN (Nippon-Gene) immediately after cells were removed from incubators. For control samples, total RNA was purified from whole mouse kidneys using a Precellys 24-bead homogenizer (Bertin) and ISOGEN (Nippon-Gene). cDNA was synthesized using the reverse transcriptase from a Superscript III kit (Thermo Fisher Scientific). Quantitative PCR was performed with primers and probes listed in Supplementary Table 1 using FastStart SYBR green master mix (Roche) or TaqMan master mix (Roche) with a LightCycler 96 system (Roche). The expression levels of housekeeping Hprt (hypoxanthine phosphoribosyl transferase) mRNA were used as internal standards.
Immunoblotting
Whole cell lysates were prepared using lysis buffer (50 mM Tris-Cl, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, and 1.0% NP-40) supplemented with MG132 (LifeSensors), Protease Inhibitor Cocktail (Nacalai Tesque), and Phosphatase Inhibitor Cocktail (Nacalai Tesque). The lysates were transferred to nitrocellulose membranes (Bio-Rad) after sodium dodecyl sulphate polyacrylamide (8.0%) gel electrophoresis (SDS-PAGE). The membranes were immunoblotted with primary antibodies against HIF1α (PAB12138; Abnova), HIF2α (C150132; LSBio), p-Smad2/3 (8828 S; Cell Signaling Technology), total Smad2/3 (8685 S; Cell Signaling Technology), or βTubulin (PA1-21153; Thermo Fisher Scientific). Subsequently, horseradish peroxidase (HRP)-conjugated secondary antibodies against rabbit (P0448; Dako or ab6802; Abcam), goat (205-035-108; Jackson ImmunoResearch Laboratories), or mouse (01803-44; Nacalai Tesque) immunoglobulins were used with chemiluminescence staining reagents (GE Healthcare) followed by detection and quantification of the signals using a C-DiGit blot scanner with an Image Studio software (LI-COR).
Flow cytometry
Cells in culture plates were dissociated with accutase (Nakalai Tesque). Single-cell suspensions were stained with biotin-conjugated CD73 antibody (Thermo Fisher Scientific) or biotin-conjugated TGFβR2 antibody (BAF532; R&D Systems) for 30 minutes on ice and then washed and stained with streptavidin-APC for 30 minutes. After washing, flow cytometry was performed using FACS Jazz.
Bisulfite sequencing
Genomic DNA was purified from Replic cells and chopped mouse organs incubated with proteinase K (Nakalai Tesque) overnight, and bisulfite conversion of genomic DNA was performed using an EpiTect Bisulfite Kit (Qiagen). Bisulfite-converted DNA of the Epo and Epas1 gene promoters was amplified by PCR using primers listed in Supplementary Table 1. Subsequently, TA cloning of the amplified genomic DNA fragments into the pT7Blue T-vector (Novagen) was conducted. Eight clones for each promoter were bilaterally sequenced by using primers (Supplementary Table 1) annealing to the cloning site of the pT7Blue T-vector.
Cytokine array
Subconfluent Replic cells and MEFs in 3.5-cm dishes were incubated with 2 mL of serum-free DMEM for 24 hours, and 1 mL of the culture supernatants were incubated with the membranes (Mouse Neuro Antibody Array III, Abcam), on which capture antibodies against 23 cytokines (including TGFβ) were printed as shown in Supplementary Fig. 3a. After a 2-hour incubation, the membranes were washed and then incubated with a cocktail of biotin-conjugated detector antibodies against each cytokine for 2 hours. Chemiluminescent signals were detected and quantified using C-DiGit after incubation of the membranes with HRP-conjugated streptavidin for 2 hours. All procedures were performed at room temperature.
ELISA
Subconfluent Replic cells (4.43 ± 0.25 [×104 cells]) or MEFs (4.00 ± 0.38 [×104 cells]) in a well of 12-well plates were incubated with 1 mL of serum-free DMEM for 24 hours. Total TGFβ concentrations in these samples were measured using a Total TGFβ1 Pre-coated ELISA kit (BioLegend) according to the manufacturer’s instructions. Briefly, samples were appropriately diluted and incubated in a 96-well plate coated with anti-TGFβ1 capture antibody. After a 2-hour incubation, the cells were washed and then incubated with biotin-conjugated TGFβ detection antibody for 1 hour. Developed colour intensity was measured using a FilterMax F5 microplate reader (Molecular Devices) after incubation of the cells with HRP-conjugated streptavidin and fluorescent substrate.
Statistics
The data are shown as the mean ± standard error (SE). Comparisons between groups were conducted using one-way ANOVA with the Tukey-Kramer test or two-tailed, unpaired Student’s t-test or χ2 test as appropriate. The data were considered statistically significant at P < 0.05.
Data Availability
All data supporting the results of this study are available in the article and supplementary information files or are available from the authors upon request.
References
Nangaku, M. & Eckardt, K. U. Pathogenesis of renal anemia. Semin Nephrol. 26, 261–268 (2006).
Friedman, S. L., Sheppard, D., Duffled, J. S. & Violette, S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 5, 167sr1 (2013).
Iwano, M. et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 110, 341–350 (2002).
Zeisberg, E. M., Potenta, S. E., Sugimoto, H., Zeisberg, M. & Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 19, 2282–2287 (2008).
Humphreys, B. D. et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 176, 85–97 (2010).
Asada, N. et al. Dysfunction of fibroblasts of external origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 121, 3981–3990 (2011).
Souma, T. et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. J Am Soc Nephrol. 24, 1599–1616 (2013).
LeBleu, V. S. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 19, 1047–1053 (2013).
Buchtler, S. et al. Cellular origin and functional relevance of collagen I production in the kidney. J Am Soc Nephol. 29, 1859–1873 (2018).
Koury, S. T., Bondurant, M. C. & Koury, M. J. Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood. 71, 524–527 (1988).
Obara, N. et al. Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood. 111, 5223–5232 (2008).
Pan, X. et al. Isolation and characterization of renal erythropoietin-producing cells from genetically produced anemia mice. PloS One. 6, e25839, https://doi.org/10.1371/journal.pone.0025839 (2011).
Yamazaki, S. et al. A mouse model of adult-onset anaemia due to erythropoietin deficiency. Nat Commun. 4, 1950, https://doi.org/10.1038/ncomms2950 (2013).
Suzuki, N. & Yamamoto, M. Roles of renal erythropoietin-producing (REP) cells in the maintenance of systemic oxygen homeostasis. Pflugers Arch. 468, 3–12 (2016).
Souma, T. et al. Erythropoietin synthesis in renal myofibroblasts is restored by activation of hypoxia signaling. J Am Soc Nephrol. 27, 428–438 (2016).
Lendahl, U., Lee, K. L., Yang, H. & Poellinger, L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat Rev Genet. 10, 821–832 (2009).
Haase, V. H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 27, 41–53 (2013).
Suzuki, N., Gradin, K., Poellinger, L. & Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp Cell Res. 356, 182–186 (2017).
Takeda, K. et al. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 111, 3229–3235 (2008).
Martin, E. R., Smith, M. T., Maroni, B. J., Zuraw, Q. C. & deGoma, E. M. Clinical Trial of Vadadustat in Patients with Anemia Secondary to Stage 3 or 4 Chronic Kidney Disease. Am J Nephrol. 45, 380–388 (2017).
Mise, K. et al. Renal prognosis a long time after renal biopsy on patients with diabetic nephropathy. Nephrol Dial Transplant. 29, 109–118 (2014).
Tsubakihara, Y., Akizawa, T., Iwasaki, M. & Shimazaki, R. High hemoglobin levels maintained by an erythropoiesis-stimulating agent improve renal survival in patients with severe renal impairment. Ther Apher Dial. 19, 457–465 (2015).
Suzuki, N. Erythropoietin gene expression: developmental-stage specificity, cell-type specificity, and hypoxia inducibility. Tohoku J Exp Med. 235, 233–240 (2015).
Bachmann, S., Le Hir, M. & Eckardt, K. U. Co-localization of erythropoietin mRNA and ecto-5′-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem. 41, 335–341 (1993).
Plotkin, M. D. & Goligorsky, M. S. Mesenchymal cells from adult kidney support angiogenesis and differentiate into multiple interstitial cell types including erythropoietin-producing fibroblasts. Am J Physiol Renal Physiol. 291, F902–F912 (2006).
Armulik, A., Genové, G. & Betsholtz, C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 21, 193–215 (2011).
Suzuki, N. et al. Iron attenuates erythropoietin production by decreasing HIF2alpha concentrations in renal interstitial fibroblasts. Kidney Int. 94, 900–911 (2018).
Forsythe, J. A. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 16, 4604–4613 (1996).
Chen, C., Pore, N., Behrooz, A., Ismail-Beigi, F. & Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 276, 9519–9525 (2001).
Chang, Y. T. et al. DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J Clin Invest. 126, 721–731 (2016).
Jeltsch, A. Molecular enzymology of mammalian DNA methyltransferases. Curr Top Microbiol Immunol. 301, 203–225 (2006).
Hirano, I. et al. Renal Anemia Model Mouse Established by Transgenic Rescue with an Erythropoietin Gene Lacking Kidney-Specific Regulatory Elements. Mol Cell Biol. 37, https://doi.org/10.1128/MCB.00451-16 (2017).
Hsieh, C. L. In vivo activity of murine de novo methyltransferases, Dnmt3a and Dnmt3b. Mol Cell Biol. 19, 8211–8218 (1999).
Tojo, Y. et al. Hypoxia signaling cascade for erythropoietin production in hepatocytes. Mol Cell Biol. 35, 2658–2572 (2015).
Chevalier, R. L., Forbes, M. S. & Thornhill, B. A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 75, 1145–1152 (2009).
Sousa, A. M. et al. Smooth muscle alpha-actin expression and myofibroblast differentiation by TGFbeta are dependent upon MK2. J Cell Biochem. 100, 1581–1592 (2007).
Derby, I., Skalli, O. & Gabbiani, G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. 63, 21–29 (1990).
Ignotz, R. A. & Massagué, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 261, 4337–4345 (1986).
Sato, M., Muragami, Y., Saika, S., Roberts, A. B. & Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 112, 1486–1494 (2003).
Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 15, 1–12 (2004).
Koesters, R. et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 177, 632–643 (2010).
Meng, X. M., Tang, P. M., Li, J. & Lan, H. Y. TGF-beta/Smad signaling in renal fibrosis. Front Physiol. 6, 82 (2015).
Desmoulière, A., Geinoz, A., Gabbiani, F. & Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 122, 103–111 (1993).
Verrecchia, F. & Mauviel, A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 118, 211–215 (2002).
Inman, G. J. et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 62, 65–74 (2002).
Chen, W. et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 539, 112–117 (2016).
Dominici, M. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy. 8, 315–317 (2006).
Bruno, S., Chiabotto, G. & Camussi, G. Concise review: different mesenchymal stromal/stem cell populations reside in the adult kidney. Stem Cells Transl Med. 3, 1451–1455 (2014).
Wang, H. et al. Adult renal mesenchymal stem cell-like cells contribute to juxtaglomerular cell recruitment. J Am Soc Nephrol. 24, 1263–1273 (2013).
Chen, J. et al. Kidney-derived mesenchymal stem cells contribute to vasculogenesis, angiogenesis and endothelial repair. Kidney Int. 74, 879–889 (2008).
Wynn, T. A. & Ramalingam, T. R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 18, 1028–1040 (2012).
Antonelli-Orlidge, A., Saunders, K. B., Smith, S. R. & D’Amore, P. A. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci USA 86, 4544–4548 (1989).
Fan, J. M. et al. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 56, 1455–1467 (1999).
Branton, M. H. & Kopp, J. B. TGF-beta and fibrosis. Microbes Infect. 1, 1349–1365 (1999).
Peng, X., Zhang, J., Xiao, Z., Dong, Y. & Du, J. CX3CL1-CX3CR1 interaction increases the population of Ly6C(−)CX3CR1(hi) macrophages contributing to unilateral ureteral obstruction-induced fibrosis. J Immunol. 195, 2797–2805 (2015).
Disteldorf, E. M. et al. CXCL5 drives neutrophil recruitment in TH17-mediated GN. J Am Soc Nephrol. 26, 55–66 (2015).
Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 13, 484–492 (2012).
MacLeod, A. R., Rouleau, J. & Szyf, M. Regulation of DNA methylation by the Ras signaling pathway. J Biol Chem. 270, 11327–11337 (1995).
Lund, P. et al. Oncogenic HRAS suppresses clusterin expression through promoter hypermethylation. Oncogene. 25, 4890–4903 (2006).
Acknowledgements
We thank Atsuko Konuma, Mizuho Tanno, Yuna Kawamoto, Akinori Ishii (Tohoku University) for technical help and scientific comments. We also thank Unitech Co., Ltd. (http://www.uniqtech.co.jp/english/), for bisulfite sequencing, and the Biomedical Research Core and Centre for Laboratory Animal Research in Tohoku University for technical support. This work was supported in part by Grants-in-Aids from MEXT/JSPS KAKENHI (17K19680 and 15H04691 for N.S.; 26111002 for M.Y.), the Platform Project for Drug Discovery and Life Science Research from MEXT and AMED (M.Y. and N.S.), and Takeda Life Science Foundation (N.S.). The funders had no role in the design of the study, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
K.S. and N.S. designed the study. K.S., I.H., H.S., K.M., T.N., K.K. and N.S. performed the experiments and analysed the data. H.S., K.K. and N.S. provided the gene-modified mouse lines and reagents. K.S. and N.S. constructed the figures and wrote the manuscript with comments and edits from all the authors. S.I., M.Y. and N.S. developed the project.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sato, K., Hirano, I., Sekine, H. et al. An immortalized cell line derived from renal erythropoietin-producing (REP) cells demonstrates their potential to transform into myofibroblasts. Sci Rep 9, 11254 (2019). https://doi.org/10.1038/s41598-019-47766-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47766-5
This article is cited by
-
Role of perivascular cells in kidney homeostasis, inflammation, repair and fibrosis
Nature Reviews Nephrology (2023)
-
Fount, fate, features, and function of renal erythropoietin-producing cells
Pflügers Archiv - European Journal of Physiology (2022)
-
Transforming growth factor-β1 decreases erythropoietin production through repressing hypoxia-inducible factor 2α in erythropoietin-producing cells
Journal of Biomedical Science (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
