
Abstract
Spiro-oxindole scaffolds have been studied due to their promising therapeutic potential. In the Amazon rainforest there are two important Uncaria species known as “cat’s claw”, which biosynthesize spirocyclic oxindole alkaloids; Uncaria tomentosa (Willd. ex Schult.) DC. and Uncaria guianensis (Aublet) Gmell. We carried out a precursor-directed biosynthesis approach with U. guianensis and successfully obtained oxindole alkaloid analogues with molecular mass corresponding to the addition of a methyl or fluorine group on the oxindole ring using tryptamine analogue precursors. Two of these novel oxindole alkaloid analogues (3b-7-methyl-isomitraphylline and 3c-6-fluoro-isomitraphylline) were isolated and characterized by NMR spectroscopy and ESI-QTOF-MS/MS. Having established a substrate feeding protocol for these plantlets, the biosynthetic route for mitraphylline (1), rhynchophylline (2), isomitraphylline (3) and isorhynchophylline (4) was also investigated using 13C-precursors (1-13C-D-glucose, 2-13C-tryptophan, 1-13C-DL-glyceraldehyde, and methyl-13C-D-methionine).
Similar content being viewed by others
Introduction
Natural products and their derivatives have been, and continue to be, a source of inspiration in the drug discovery domain. Currently, between 50–70% of all small molecule therapeutics used today are natural product inspired1. In general, modifications to the natural product structure to yield “new-to-nature compounds” often result in either improved biological or pharmacological activity. As an example, fludrocorticoid was the first fluorine drug identified as an active glucocorticoid, displaying 10-fold more potency than other halogenated cortisone derivatives2. Additionally, the fluorovinblastine analogue exhibits remarkable antitumor activity (IC50 = 300 pM) displaying 30-fold more potency than vinblastine (IC50 = 10 nM)3, and two fluorinated camptothecin analogues showed potent cytotoxicity against the multidrug-resistant KB-VIN cell line, and were more effective than irinotecan. Remarkably, more than 20% of the drugs currently available contain at least one fluorine atom4. For this reason, precursor-directed biosynthesis5,6 and metabolic engineering strategies7,8,9,10,11,12 have been explored in medicinal plants in order to generate novel fluorinated analogues. Precursor-directed biosynthesis procedures with Catharanthus roseus plants and tissue have already demonstrated that is it possible to produce unnatural monoterpene indole alkaloids5,6. The spirocyclic monoterpene oxindole alkaloids from Uncaria, which are biosynthetically related to the monoterpene indole alkaloids of C. roseus, have been investigated due to the therapeutic potential of the oxindole nucleus that is found in numerous natural products13,14. Uncaria (Rubiaceae) is a prolific producer of the bioactive spirocyclic oxindole alkaloids, and in the Brazilian Amazon two economically important Uncaria species are prevalent; Uncaria tomentosa (Willd. ex Schult.) DC. and Uncaria guianensis (Aublet) Gmell. These species are popularly known as “cat’s claw” and have attracted attention for their value in medicine, displaying a wide range of pharmacological activities, such as anticancer15, anti-inflammatory16,17 and immunomodulation effects18. In spite of many pharmacological studies, little work has been reported about the biosynthetic aspects of these compounds; only one 14C-feeding experiment has reported the elucidation of the oxindole alkaloid biosynthesis in Mitragyna parvifolia (Roxb.) Korth (Rubiaceae)19. Thus, here we develop a process for substrate feeding to plantlets of U. guianensis. We use this methodology first to perform a precursor-directed biosynthesis approach by feeding tryptamine analogs (a:5-methyl-tryptamine, b:7-methyl-tryptamine, and c:6-fluoro-tryptamine) to U. guianensis plantlets to obtain unnatural spirocyclic oxindole alkaloids. Two of the most abundant alkaloid analogues were isolated and subjected to 1D and 2D NMR and HRMS. Moreover, in our continuous efforts to unravel the biosynthetic details of the oxindole alkaloid biosynthetic pathway, we also applied this feeding strategy with 13C-precusors (1-13C-D-glucose, 2-13C-tryptophan, 1-13C-glyceraldehyde. and methyl-13C-D-methionine).
Results
Unnatural feeding experiments
Shoots of U. guianensis (2 months old) were transferred to sterile liquid media supplemented with the tryptamine analogs a-c (Fig. 1). A ethanolic extract from fresh shoots of U. guianensis was prepared after 30 days of culture with the tryptamine analogues and assessed by UPLC-DAD-MS. U. guianensis shoots produce natural pentacyclic oxindole alkaloids (POA) 1 and 3, and tetracyclic oxindole alkaloids (TOA) 2 and 4 (Figs 2 and S1). After precursor-directed biosynthesis using a, in addition to the four natural alkaloids (1–4); 1 (m/z 369.1820 [M + H]+) and epimer 3 (m/z 369.1823 [M + H]+), 2 (m/z 385.2140 [M + H]+) and epimer 4 (m/z 385.2142 [M + H]+); just two additional peaks corresponding to either 5-methyl-mitraphylline (1a) or 5-methyl-isomitraphylline (3a) (m/z = 383.1982 [M + H+]) and either 5-methyl-rhynchophylline (2a) or 5-methyl-isorhynchophylline (4a) (m/z = 399.2291 [M + H+]) were observed. Similar results were obtained by incubation with b precursor, corresponding to either 7-methyl-mitraphylline (1b) or 7-methyl-isomitraphylline (3b) (m/z = 383.1985 [M + H+]) and either 7-methyl-rhynchophylline (2b) or 7-methyl-isorhynchophylline (4b) (m/z = 399.2295 [M + H+]). After incubation with c, the extracted-ion chromatograms showed the presence of four natural alkaloids (1–4), as well as four new compounds with masses corresponding to 6-fluoro-mitraphylline (1c) (m/z 387.1734 [M + H]+), 6-fluoro-isomitraphylline (3c) (m/z = 387.1730 [M + H+]), 6-fluoro-rhynchophylline (2c) (m/z 403.2025 [M + H]+) and 6-fluoro-isorhynchophylline (4c) (m/z = 403.2023 [M + H+]) (Figs S2–S6 and Table S1). Having established that U. guianensis could produce unnatural alkaloids, these precursor-directed biosynthesis approaches were performed on a large scale using b-c precursors. After 30 days of culture, ethanolic extracts were prepared and submitted to chromatographic procedures and semi-preparative HPLC. Compounds 3b and 3c were purified and characterized by 1D and 2D NMR (Figs S27–S36 and Table S2) and HRMS (Fig. S37 and Table S3). NMR data from alkaloid 3 showed five important cross signal correlations by HMQC; one related to double bond and four related to aromatic hydrogens (Fig. S20). The unnatural alkaloids 3b and 3c showed four cross signal correlations, showing that one position was substituted by either methyl or fluoride groups (Figs S30 and S35). Additionally, main product ions of natural and unnatural oxindole alkaloids 3, 3b and 3c were confirmed by ESI-QTOF-MS/MS (Fig. 3). All NMR data and ESI-QTOF-MS/MS allowed us to establish the structure of these two novel oxindole alkaloids in comparation with our NMR data and literature data from natural oxindole alkaloids (1 and 320,21,22; 2 and 423), and confirmed the capacity for efficient unnatural oxindole alkaloids production using precursor directed biosynthesis.

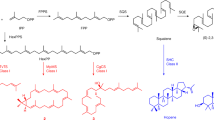
General strategies for oxindole alkaloid analogues design using precursor-directed biosynthesis in U. guianensis plantlets.

Natural and unnatural oxindole alkaloids biosynthesized by U. guianensis plantlets. Square (dashed lines) represent complete characterization of new-to-nature oxindole alkaloids.

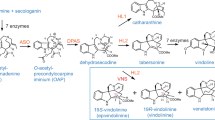
Formation of the main product ions of natural and unnatural oxindole alkaloids 3, 3b and 3c.
Labeled feeding experiments
Given the success of feeding of unnatural substrate analogues, and given that an understanding of the biosynthetic pathway can help to identify intermediates and enzymes involved in oxindole alkaloid biosynthesis, we used this platform to perform labelling studies of the alkaloids of U. guianensis. Segments of U. guianensis were inoculated in liquid medium supplemented with 1-13C-D-glucose (3% m/v), 2-13C-tryptophan (1 mM), 1-13C-DL-glyceraldehyde (1 mM) and methyl-13C-D-methionine (0.3% m/v). Additionally, a control group of plantlets that did not receive 13C-precursors was also prepared. After ten weeks of culture, an ethanolic extract from fresh shoots of U. guianensis, where the oxindole alkaloids 1–4 are the major constituents, was prepared. This ethanolic extract was fractioned by dowex® resin and semi-preparative HPLC procedures to yield both labeled and non-labeled 1, 3 and 4. Incorporation patterns of labeled oxindole alkaloids were determined by quantitative 13C NMR (Figs S38–S45) by comparing the relative intensities of the labeled and non-labeled STL signals for 1, 3 and 4 (Tables S4–S6). DL-glyceraldehyde and D-methionine 13C-precursors were not incorporated into oxindole biosynthesis by U. guianensis shoots. The 13C enrichment patterns of 1, 3 and 4 after 1-13C-D-glucose metabolism indicated that the secologanin scaffold was predominantly formed by the MEP pathway, and oxindole moiety is derived from the shikimate pathway in both the POA (1 and 3) and TOA (2 and 4) framework. The 13C NMR spectrum of 13C-labeled 1 and 3 revealed enhancement of the signals at C-6, C-9, and C-13 derived from tryptamine metabolism; and C-14, C-17, C-18, and C-21, thus indicating incorporation of a label into the corresponding C-1 and C-5 of IPP derived from the MEP pathway (Fig. 4). The 13C NMR spectrum of labeled 4 presented a similar enrichment pattern for the corresponding carbon atoms at C-6, C-9, C-14, C-17, C-18, and C-21 from the TOA. Position C-23 from 1 and 3, and positions C-23 and C-24 from 4 showed high enrichment, corresponding to SAM action. Isotopic labeling from the 2-13C-tryptophan experiment led to enrichment at positions C-2 of 1, 3 and 4, therefore indicating operation of the tryptamine into the oxindole alkaloids.

Labelling patterns obtained by biosynthetic precursor 1-13C-D-glucose and 2-13C-tryptophan into POA (1 and 3) and TOA (2 and 4).
Discussion
Our studies showed that precursor-directed biosynthesis strategies using analogue precursors produce successful unnatural oxindole alkaloids in U. guianensis shoots. Four unnatural fluoro-oxindole alkaloids were detected using 6-fluoro-tryptamine derivative (c); two TOA and two POA, while after incubation with methyl-precursors (a-b) only two unnatural methylated-oxindole alkaloids were detected; one TOA and one POA scaffold. These data suggest that the fluorinated c precursor is more acceptable to the biosynthetic enzymes in U. guianensis compared to a-c precursors as previously reported in C. roseus7. It is important to point out that U. guianensis produces structurally dissimilar alkaloids compared to those found in C. roseus5. These results showed a precursor-directed biosynthesis strategy could provide the opportunity to rationally design both oxindole and vinca analogues, perhaps to improve their pharmacological properties. The biosynthetic pathways of POA and TOA from U. guianensis are in agreement with the previously described monoterpene indole alkaloid biosynthesis in C. roseus. After formation of the indole and terpenoid precursors (Fig. S46), secologanin and tryptamine precursors undergo a Mannich (Pictet-Spengler) reaction to form the tetrahydro-β-carboline strictosidine intermediate24. The intermediate dehydrogeissochizine6 is the key precursor for synthesis of both POA and TOA scaffolds. For POA, the ajmalicine (heteroyohimine-type alkaloid) likely undergoes an oxidation reaction which could be stereospecific, and yields intermediate I and II as described in Strychnos, Aspidosperma and Iboga alkaloids25. Our proposal involves a pinacol-pinacolone rearrangement, which is common in limonoids biosynthesis, as occurs in limonoids from Khaya senegalensis26. Intermediate II, which undergoes a rapid pinacol-pinacolone rearrangement, loses H2O from intermediate I, and is the key precursor of spirocyclic oxindole scaffold in order to produce either mitraphylline or isomitraphylline (POA). For TOA, the same mechanism observed for POA was proposed, mediated by dehydrogeissochizine which led to intermediate III, it then undergoes an oxidation step, yielding either rhynchophylline or isorhynchophylline from intermediate V.
Our results, together with previously reported data in C. roseus, seem to suggest that terpene indole and oxindole alkaloids are biosynthesized by similar pathways. Our studies showed that precursor-directed biosynthesis strategies using 5-methyl-, 7-methyl- and 6-fluoro-tryptamine precursors produce successful unnatural oxindole alkaloids in U. guianensis shoots. These results showed that a precursor-directed biosynthesis strategy could provide the opportunity to rationally design a broad variety of new molecules. Our work applies biosynthetic knowledge to advance the art of making new molecules which will inevitably trigger new potential drug candidates. We have also established the biosynthetic pathway of 1–4 which are derived from the MEP and shikimate pathways. This experiments also allowed us to establish the biosynthetic pathway for spirocyclic oxindole alkaloids with 13C-precursors for the first time in the literature.
Methods
Chemicals
The labeled precursors (1-13C-D-glucose, 1-13C-DL-glyceraldehyde, and methyl-13C-D-methionine) and analogue precursors (a-5-methyl-tryptamine, b-7-methyl-tryptamine and c-6-fluoro-tryptamine) were purchased from Sigma-Aldrich®. The 2-13C-tryptophan was obtained from Cambridge Isotope Laboratories, Inc.
General procedure from in vitro shoot cultures of U. guianensis
The plant material was identified by Dr. Piero Giuseppe Delprete, of Herbier de Guyane, Institut de Recherche pour le Développement. A voucher specimen from U. guianensis was deposited in the Herbarium of Medicinal Plants at UNAERP (HPMU 2411), Ribeirão Preto, SP, Brazil. The collection of U. guianensis specimens investigated in this study was previously authorized by the Brazilian Council for the Administration and Management of Genetic Patrimony (CGEN) of the Brazilian Ministry of the Environment (MMA) via the National Council for Scientific and Technological Development (CGEN/MMA Process number: A2578BA). Disinfected explants (by 1% (w/v) cercobin and captan solutions) were added to MS medium27 with 3% (w/v) glucose, 0.2% phytogel®, 1 mM of benzylaminopurine (BAP), pH adjusted to 6.0 and maintained at 25 ± 2 °C (55–60% relative humidity with a 16-h photoperiod of 40 μmol m−2 s−1 intensity, provided by 85 W cool-white GE fluorescent lamps) and micropropagated every two months.
Precusor-directed biosynthesis experiments
Typtamine analogues (a–c) were dissolved in 500 µL of DMSO (1 mM5), syringe filtered through a 0.2 µm filter to sterilize, and added to MS medium with 3% (w/v) glucose, 1 mM of BAP, pH adjusted to 6.0. In vitro plantlets (n = 50 shoots; 2 months old) were then transferred individually to the MS medium described above and maintained at 25 ± 2 °C for 30 days. After incubation, the aerial parts from fresh U. guianensis shoots were extracted with ethanol in order to obtain the crude extracts.
HPLC-DAD and UPLC-DAD-QTOF analyses
The crude extracts were dissolved in ACN-H2O (8:2) and assessed by HPLC-DAD and UPLC-DAD-QTOF using modified chromatographic conditions from literature28. For UPLC-DAD-QTOF, samples were ionized by ESI with a MicrOTOF Bruker Daltonics mass spectrometer (Milford MA, USA). Mass spectra were obtained in positive ionization mode and the following parameters were applied: capillary energy of 3.5 kV, nitrogen was used as nebulization, drying gas (5.5 bar and 10 L/h), and temperature drying time of 220 °C. UPLC conditions were carried out using the isocratic system: 0–18′ (60:40; A:B), 18–32′ (50:50; A:B), and 32–40′ (100% of B %), 0.3 mL.min−1 flow rate and detection at 245 nm (A-triethylammonium acetate buffer 35 mM, 0.2% v/v, pH 6.9; B-ACN); and an Agilent Zorbax Eclipse XDB-C18 column: 3.5 µ, 4.6 × 150 mm. For HPLC-DAD, the samples were analyzed with the same chromatographic conditions described above, except for the flow rate, which was 0.8 mL.min−1.
Isolation of unnatural oxindole alkaloids and NMR analysis
Crude extracts from aerial parts (0.8 g) were solubilized in ethanol-water solution (4:6), added with 500 mg of strong anionic resin (Dowex Marathon, Sigma-Aldrich) and submitted to magnetic agitation for 30 minutes. After this, a fraction enriched in oxindole alkaloid was obtained by a Dowex resin wash using ethanol-ammonium hydroxide (99.9:0.1) followed by magnetic agitation for 30 minutes and rota-evaporated at 37 °C. The fractions (8–10 mg) were then submitted to semi-preparative HPLC-DAD using the solvent system: 0–18′ (65:35; A:B), 18–32′ (50:50; A:B), and 32–40′ (100% of B %), 3 mL.min−1 flow rate, and detection at 245 nm (A-triethylammonium acetate buffer 35 mM, 0.2% v/v, pH 6.9; B-ACN); and an Agilent Zorbax Eclipse XDB-C18 column, 5 µ, 250 × 9.4 mm, to yield 7-methyl-isomitraphylline (3b, 0.7 mg) and 6-fluoro-isomitraphylline (3c, 0.8 mg). Isolated unnatural oxindole alkaloids were analyzed by NMR 1D and 2D (using DMSO-d6 as the solvent) with a 600 MHz Bruker Avance III HD equipped with a cryogenically cooled 5 mm dual probe optimized for 13C and 1H.
ESI-QTOF-MS/MS analysis
The precursor ions of 3, 3b and 3c were analyzed with a Triple TOF 5600 + DualSpray Ion Source AB SCiex (Massachusetts, U.S.A.) mass spectrometer. The ESI interface conditions were as follows: ion spray floating voltage = 4.0 kV, declustering potential = 70 V, gases 1 and 2 = 15 psi, and curtain gas = 25 psi. The sample was introduced into the mass spectrometer with the aid of a syringe pump coupled to the instrument operating at a flow rate of 5.0 μL/min. The tandem mass spectrometry experiments (MS/MS) with collision-induced dissociation were carried out by using N2 as collision gas on the selected precursor ion [M + H]+, at collision energy values ranging from 10 to 100 V. The mass analyzer was calibrated with a tuning solution that was supplied in the Chemical Standards Kit shipped with the system, to give a resolution of approximately 40,000. The mass data were processed with Analyst TF software.
Feeding studies using 13C-precusors
The feeding experiments with 1-13C-D-glucose (3% m/v)29, 1-13C-DL-glyceraldehyde (1 mM), methyl-13C-D-methionine (0.3% m/v)30, and 2-13C-tryptophan (1 mM) were performed in liquid culture medium. U. guianensis plantlets (n = 30 shoots; 2 months old) were inoculated in liquid Murashige & Skoog medium with 1 mM of BAP supplemented with13-C-precursors (in four separate experiments), transferred into glass tubes (8.5 cm × 2.5 cm) containing 2.5 mL of liquid culture medium and incubated for 8 weeks. After this period, the aerial parts from U. guianensis shoots were extracted with ethanol for 12 h to obtain the crude extract.
Isolation of 13C-labeled and non-labeled oxindole alkaloids (1–4) and NMR analysis
Chromatographic procedures for the isolation of 13C-labeled and non-labeled oxindole alkaloids were carried out using the procedure described above by anionic Dowex resin and semi-preparative HPLC to yield enriched mitraphylline (1, 2.0 mg derived from 1-13C-D-glucose, 2.0 mg derived from 2-13C-tryptophan, 2.0 mg derived from 1-13C-DL-glyceraldehyde, and 2.0 mg derived from methyl-13C-D-methionine); isomitraphylline (3, 3.0 mg derived from 1-13C-D-glucose, 3.0 mg derived from 2-13C-tryptophan, 3.0 mg derived from 1-13C-DL-glyceraldehyde, and 3.0 mg derived from methyl-13C-D-methionine); and isorhynchophylline (4, 2.0 mg derived from 1-13C-D-glucose). 13C NMR spectra of 13C-labeled, non-labeled, and unnatural oxindole alkaloids isolated were recorded with a 600 MHz Bruker Avance III HD using DMSO-d6. The relative 13C enrichments were obtained by comparing the relative intensity of both the labeled and non-labeled signals of oxindole alkaloids (1, 3 and 4).
References
Pye, C. R., Bertin, M. J., Lokey, R. S., Gerwick, W. H. & Linington, R. G. Retrospective analysis of natural products provides insights for future discovery trends. PNAS 114(22), 5601–5606 (2017).
Wang, J. et al. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev. 114(4), 2432–2506 (2014).
Sears, J. E. & Boger, D. L. Total synthesis of vinblastine, related natural products, and key analogues and development of inspired methodology suitable for the systematic study of their structure–function properties. Acc. Chem. Res. 48(3), 653–662 (2015).
Yang, C.-J. et al. Design, semisynthesis and potent cytotoxic activity of novel 10-fluorocamptothecin derivatives. Bioorg. Med. Chem. Lett. 27(20), 4694–4697 (2017).
McCoy, E. & O’Connor, S. E. Directed Biosynthesis of Alkaloid Analogs in the Medicinal Plant Catharanthus roseus. J. Am. Chem. Soc. 128(44), 14276–14277 (2006).
Bernhardt, P., McCoy, E. & O’Connor, S. E. Rapid identification of enzyme variants for reengineered alkaloid biosynthesis in periwinkle. Chem. Biol. 14(8), 888–897 (2007).
Runguphan, W. & O’Connor, S. E. Metabolic reprogramming of periwinkle plant culture. Nat. Chem. Biol. 5(3), 151–153 (2009).
Runguphan, W., Maresh, J. & O’Connor, S. E. Silencing of tryptamine biosynthesis for production of nonnatural alkaloids in plant culture. PNAS 106(33), 13673–13678.
Lee, H.-Y., Yerkes, N. & O’Connor, S. E. Aza-tryptamine substrates in monoterpene indole alkaloid biosynthesis. Chem. Biol. 16(12), 1225–1229 (2009).
Glenn, W. S., Nims, E. & O’Connor, S. E. Reengineering a tryptophan halogenase to preferentially chlorinate a direct alkaloid precursor. J. Am. Chem. Soc. 133(48), 19346–19349 (2011).
Runguphan, W. & O’Connor, S. E. Diversification of monoterpene indole alkaloid analogs through cross-coupling. Org. Lett. 15(11), 2850–2853 (2013).
Brown, S. & O’Connor, S. E. Halogenase engineering for the generation of new natural product analogues. ChemBioChem. 16(15), 2129–2135 (2015).
Yu, B., Yu, D.-Q. & Liu, H.-M. Spirooxindoles: Promising scaffolds for anticancer agentes. Eur. J. Med. Chem. 97, 673–698 (2015).
Kaur, M., Singh, M., Chadha, N. & Silakari, O. Oxindole: A chemical prism carrying plethora of therapeutic benefits. Eur. J. Med. Chem. 123, 858–894 (2016).
Heitzman, M. E., Neto, C. C., Winiarz, E., Vaisberg, A. J. & Hammond, G. B. Ethnobotany, phytochemistry and pharmacology of Uncaria (Rubiaceae). Phytochemistry 66(1), 5–29 (2005).
Rojas-Duran, R. et al. Anti-inflammatory activity of mitraphylline isolated from Uncaria tomentosa bark. J. Ethnopharmacol. 143(3), 801–804 (2012).
Urdanibia, I., Michelangeli, F., Ruiz, M.-C., Milano, B. & Taylor, P. Anti-inflammatory and antitumoural effects of Uncaria guianensis bark. J. Ethnopharmacol. 150(3), 1154–1162 (2013).
Montserrat-de la Paz, S. et al. Pharmacological effects of mitraphylline from Uncaria tomentosa in primary human monocytes: skew toward M2 macrophages. J. Ethnopharmacol. 170, 128–135 (2015).
Shellard, E. J. & Houghton, P. J. The Mitragyna species of Asia. XXVI. Further ‘ in vivo’ studies using C-alkaloids, in the alkaloidal pattern in young plants of Mitragyna parvifolia (Roxb.) Korth grown from seed obtained from Sri Lanka (Ceylon). Planta Med. 25(1), 80–87 (1974).
Toure, H., Babadjamian, A., Balansard, G., Faure, R. & Houghtons, P. J. Complete H and C NMR chemical shift assignments for some pentacyclic oxindole alkaloids. Spectrosc. Lett. 25(2), 293–300 (1992).
Seki, H., Takayama, H., Aimi, N., Sakai, S. & Ponglux, D. A nuclear magnetic resonance study on the eleven stereoisomers of heteroyohimbine-type oxindole alkaloid. Chem. Pharm. Bull. 41(12), 2077–2086 (1993).
Carbonezi, C. A. et al. Determinação por RMN das configurações relativas e conformações de alcalóides oxindólicos isolados de Uncaria guianensis. Quim. Nova 27(6), 878–881 (2004).
Wenkert, E. et al. Carbon-13 nuclear magnetic resonance spectroscopy of naturally occurring substances. XIX. Aspidosperma alkaloids. J. Am. Chem. Soc 95(15), 4990–4995 (1973).
Miettinen, K. et al. The seco-iridoid pathway from Catharanthus roseus. Nat. Commun. 5(3606), 1–11 (2014).
Scott, A. I. & Qureshi, A. A. Biogenesis of Strychnos, Aspidosperma, and Iboga alkaloids. Structure and reactions of preakuammicine. J. Am. Chem. Soc. 91(21), 5874–5876 (1969).
Olmo, L. R. V. et al. Limonoids from leaves of Khaya senegalensis. Phytochemistry 44(6), 1157–1161 (1997).
Murashige, T. & Skoog, F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Plant 15, 473–497 (1962).
Bertol, G., Franco, L. & De Oliveira, B. H. HPLC analysis of oxindole alkaloids in Uncaria Tomentosa: sample preparation and analysis optimization by factorial design. Phytochem. Anal 23, 143–151 (2012).
Lopes, A. A. et al. A biosynthetic pathway of sesquiterpene lactones in Smallanthus sonchifolius and their localization in leaf tissues by MALDI imaging. Chem. Commun. 49, 9989–9991 (2013).
Kutrzeba, L. et al. Biosynthesis of salvinorin A proceeds via the deoxyxylulose phosphate pathway. Phytochemistry 68(14), 1872–1881 (2007).
Acknowledgements
The authors are grateful to the São Paulo Research Foundation (FAPESP, grant #2013/07349-9) for their financial support and to the Biotechnology Unit (UNAERP). A.A.L., B.C. and B.M. Also thanks to FAPESP for the award of scholarships (Grant #2013/22609-7; #2015/18978-2 and #2014/26422-1). The authors would like to thank Prof. Dr. Norberto Peporine Lopes for UPLC-DAD-QTOF analyses and also thank Dr. Sarah E. O’Connor (Department of Natural Product Biosynthesis,Max Planck Institute of Chemical Ecology, Jena, Germany) for many productive discussions and valuable suggestions for this manuscript.
Author information
Authors and Affiliations
Contributions
A.A.L. designed research; A.A.L., B.C., B.M. and E.J.C. performed research; A.A.L., S.C.F., M.F.G.F.S. and A.M.S.P. analyzed data; A.A.L. wrote the manuscript and all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lopes, A.A., Chioca, B., Musquiari, B. et al. Unnatural spirocyclic oxindole alkaloids biosynthesis in Uncaria guianensis. Sci Rep 9, 11349 (2019). https://doi.org/10.1038/s41598-019-47706-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47706-3
This article is cited by
-
Lewis base-catalyzed synthesis of highly functionalized spirooxindole-pyranopyrazoles and their in vitro anticancer studies
Medicinal Chemistry Research (2023)
-
Alkaloid diversification in the genus Palicourea (Rubiaceae: Palicoureeae) viewed from a (retro-)biogenetic perspective
Phytochemistry Reviews (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
