
Abstract
Medical oncologists are challenged to personalize medicine with scientific evidence, drug approvals, and treatment guidelines based on sequencing of clinical samples using next generation sequencer (NGS). Knowledge-based curation systems have the potential to help address this challenge. We report here the results of examining the level of evidence regarding treatment approval and clinical trials between recommendations made by Watson for Genomics (WfG), QIAGEN Clinical Insight Interpret (QCII), and Oncomine knowledge-based reporter (OKR). The tumor samples obtained from the solid cancer patients between May to June 2018 at Kindai University Hospital. The formalin-fixed paraffin-embedded tumor samples (n = 31) were sequenced using Oncomine Comprehensive Assay v3. Variants including copy number alteration and gene fusions identified by the Ion reporter software were used commonly on three curation systems. Curation process of data were provided for 25 solid cancers using three curation systems independently. Concordance and distribution of curated evidence levels of variants were analyzed. As a result of sequencing analysis, nonsynonymous mutation (n = 58), gene fusion (n = 2) or copy number variants (n = 12) were detected in 25 cases, and subsequently subjected to knowledge-based curation systems (WfG, OKR, and QCII). The number of curated information in any systems was 51/72 variants. Concordance of evidence levels was 65.3% between WfG and OKR, 56.9% between WfG and QCII, and 66.7% between OKR and QCII. WfG provided great number of clinical trials for the variants. The annotation of resistance information was also observed. Larger differences were observed in clinical trial matching which could be due to differences in the filtering process among three curation systems. This study demonstrates knowledge-based curation systems (WfG, OKR, and QCII) could be helpful tool for solid cancer treatment decision making. Difference in non-concordant evidence levels was observed between three curation systems, especially in the information of clinical trials. This point will be improved by standardized filtering procedure and enriched database of clinical trials in Japan.
Similar content being viewed by others


Introduction
Medical oncologists are challenged to personalize medicine with rapidly changing scientific evidence, drug approvals, and treatment guidelines. Next generation sequencer (NGS)-based clinical sequencing is approved as in vitro medical diagnostics (IVD) and companion diagnostics for cancer patients.
Clinical sequencing reports are a key component of the clinical sequencing process and in Japan, these are prepared by an expert panel (clinical sequencing team) in each individual institute1. Curation of the annotated gene variants is performed manually by the members of expert panel and is supported by several knowledgebases2,3,4,5. However, the manual curation procedure in the expert panel is time consuming and variation of curation contents often occurs. Efforts to standardize the clinical sequencing report have been undertaken by various Japan cancer related associations and have provided guidance for cancer clinical sequencing6. However, as a result recent technological advances, practical clinical approaches capable of handling robust curated data with automated clinical annotation for the clinician is required. In Japan, the C-CAT (Center for Cancer Genomics and Advanced Therapeutics) database and curation systems has been under construction, and this system operation has started. Gene panel testing that is accompanied by curation results like FoundationOne CDx is uncommon. In addition, the status of drug approval and clinical trials are different among countries.
Manual curation by institutional molecular tumor board is time-consuming, cumbersome and relatively difficult to update in real-time. Information retrieval is an area of expertise in computing and knowledge-based curation systems have the potential to improve and facilitate genetic testing7,8. Furthermore, implementing a global approach will help create and standardized clinical sequencing reporting enabling clinicians to better select therapies for cancer patients and match patients to appropriate clinical trials.
Currently several approaches and are being developed and implemented in genetic testing7,8,9. Herein, we compare three global curation systems; Watson for Genomics (WfG, IBM), Oncomine knowledge-based reporter (OKR, Thermo Fisher Scientific) and QIAGEN Clinical Insight Interpret (QCII, Qiagen) in a cohort of Japanese cancer patients and evaluate them for efficiency in reporting, therapy selection based on levels of evidence, and clinical trial matching.
Results
Targeted sequencing and annotation
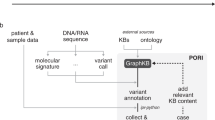
Tumor tissue samples were obtained from the patients with various types of solid cancers were performed using OCAv3, and annotated treatment information by three knowledge systems (Fig. 1). The age of enrolled 31 patients (male 16, female 15) was 10–70’s. Types of cancer are shown in Table 1. The samples were obtained before drug therapy except for one case. Tissue specimen of the exceptional case were obtained at progressive disease to molecularly targeted therapy. The most frequent cancer type was lung cancer. All samples were sequenced successfully. The average depth was ranged from 373 to 2206 in 31 cases. Analyses of mutations, fusions, and copy number alterations were performed using the Ion Reporter software (ver.5.8). Nonsynonymous variants (n = 58), copy number gain (n = 12), or gene fusions (n = 2) were detected in 25/31 (80.6%) cases. The average number of variants in the 25 cases were 2.8 (1 to 7). The frequently called variants was TP53 mutation which was detected in 12 tumor samples.

CONSORT diagram for the study. FFPE specimens obtained from 31 solid tumor patients were subjected to Oncomine Comprehensive Assay v3. Data of variant calling as non-synonymous mutations and copy number variants by Ion reporter were filtered out based on following cut off values; quality score >100, germline allele frequency <1% (ExAC, HGVD), variant allele frequency >5%, and copy number gain threshold >1.5 (log2 ratio). Calling data of fusion transcripts were uploaded unfiltered. Variant data of 25 cases in which at least one variant was detected was analyzed by WfG, OKR, and QCII. Cancer type and gender of the patients, in addition to the filtered data, were uploaded to the curation systems.
Curated contents by three curation systems
The results were compiled by three knowledge-based report systems. The vcf files of 25 case sequencing data with one or more nonsynonymous variants were curated using WfG, OKR, and QCII. The curated contents of the report were classified; Evidence levels of curated contents of pathogenic (or likely pathogenic) variants were classified as (i) approved for the said cancer type (level I), (ii) approved for other cancer type (level II), (iii) clinical trials (level III), (iv) none (Supplementary Table 1). Regarding clinical trials, WfG, OKR, QCII annotated clinical studies in any countries, Asian countries (Japan, Korea, China, Taiwan, and Singapore), and Japan, respectively. Number of evidence level in three curation systems was counted. Curated contents on variant unknown significance (VUS) and gene alteration designated as benign or likely benign in the report was not counted.
WfG annotated therapeutic information in 8/72 (11.1%) as level I, 3/72 (4.2%) as level II, and 31/72 (43.1%) as possible clinical trials (Fig. 2A). OKR annotated therapeutic information in 5/72 (6.9%) as level I, 1/72 (1.4%) as level II, and 21/72 (29.2%) as level III as possible clinical trials (Fig. 2B). QCII annotated therapeutic information in 11/72 (15.3%) as level I and 8/72 (11.1%) as level III (Fig. 2C). EGFR mutation and EML4-ALK fusion gene for lung cancer were common variants described in three systems with the same evidence levels (level I). Different evidence level was described in 46/72 variants among three systems (Fig. 2D). The number of genes described by one system only was 5 for WfG (ATM mutation, CREBBP mutation, TP53 mutation, CCND1 amp, CCNE1 amp), 4 for OKR (SLX4 mutation, FANCl mutation, SETD2 mutation, ERBB3 mutation), and 4 for QCII (BRCA2 mutation, PDGFRA mutation, CDK6 mutation, ESR1 amp). Druggable alterations were annotated in 11 variants by WfG, OKR, and QCII systems. In particular, EGFR ex19 and EML4-ALK fusion transcript were annotated in 4 cases. These gene alterations were recognized as actionable changes for the non-small cell lung cancer and classified as level I in most of the world. Four called variants (4/11, 36.3%) were classified into level I were common in all systems. The remaining 7 variants were classified into different evidence levels between WfG, OKR, and QCII systems. These variants are EGFR and MET copy number gains, and PIK3CA, ERBB2, KRAS and TSC1 non-synonymous mutations. On the other hand, actionable alterations were annotated in 15 variants by any of two systems. Only one variant was classified into class I (1/15, 6.7%) and 25 variants were annotated by only of the systems and were classified into class III. These results suggest that the variants with high evidence levels (level I) were commonly annotated in all curation systems.

Distribution of evidence level for treatment choice and clinical trials reported by curation systems. (A) The pie chart shows the distribution of levels of the curated therapeutic information curated by WfG. Green; approved for the said cancer type (evidence level 1), orange; approved for other cancer type (evidence level II), blue; information of clinical trials in any countries (level III). (B) The pie chart shows the distribution of levels of the curated therapeutic information curated by OKR. Green; approved for the said cancer type (level 1), orange; approved for other cancer type (evidence level II), blue; information of clinical trials in Asian countries (Japan, Korea, China, Taiwan, Singapore) (level III). (C) The pie chart shows the distribution of levels of the curated therapeutic information curated by QCII. Green; approved for the said cancer type (level 1), orange; approved for other cancer type (level II), blue; information of clinical trials in Japan (level III). (D) Venn diagram shows the commonality of clinically informative genetic variants (across all evidence levels, see Supplementary Table 1) between curation systems.
We compared all annotated therapeutic information between three knowledge systems for 72 variants in 25 cases. The rate of concordance of annotated information of three systems were 65.3% (κ = 0.33, 95% CI 0.32–0.35) for WfG and OKR, 56.9% (κ = 0.21, 95% CI 0.19–0.22) for WfG and QCII, and 66.7% (κ = 0.24, 95% CI 0.23–0.26) for OKR and QCII (Table 2). When focusing on level I and II, the concordance of annotated information of three systems were 87.5% (κ = 0.41, 95% CI 0.37–0.44) for WfG and OKR, 86.1% (κ = 0.46, 95% CI 0.43–0.50) for WfG and QCII, and 93.1% (κ = 0.67, 95% CI 0.62–0.72) for OKR and QCII (Table 3). Increased concordance rate was observed in the annotation of higher levels. In other words, the information of clinical trials was varied between three systems.
Difference in curated contents between mutation, copy number alteration, and gene fusion
To know whether there is difference in curated contents for types of gene alteration, we compared concordance in clinical information levels (informative or non-informative) for mutation or copy number alteration.
Concordance of informative content for non-synonymous variants was 65.5% between WfG and OKR, 58.6% between WfG and QCII, and 69.0% between OKR and QCII. Concordance of informative content for copy number alteration was 58.3% between WfG and OKR, 41.7% between WfG and QCII, and 50.0% between OKR and QCII. Rate of informative information seems to be higher in non-synonymous variants than copy number alteration. However, there is no significant difference in three systems (data not shown).
Curated contents of resistance information
Resistance information was defined in OKR and QCII but not WfG. In 30 out of 31 cases, the details from curated data indicated intrinsic (not acquired) resistance. In one case (S02), tumor specimens were obtained after disease progression on an ALK tyrosine kinase inhibitor. EGFR mutation, KRAS mutation, and EML4-ALK in lung cancer case were commonly annotated resistance information by OKR and QCII. KRAS mutation in endometrial cancer and NF1 mutation in lung cancer case were annotated resistance information only in the OKR system. KIT amp, EGFR amp, MET amp and ALK mutation in lung cancer, and CCNE1 amp in breast cancer were curated as resistant information only in the QCII. The rate of concordance of curated resistance information between OKR and QCII were 90.3% (κ = 0.58, 95% CI 0.54–0.62) (Table 4).
Discussion
In this study, we compared different clinically reporting methods using data obtained in real world clinical practice. The panel was used consisted of 161 genes selected to provide a “comprehensive” overview of data across different solid cancers. Variant data from non-synonymous mutations, copy number gains, and fusion transcripts called by Ion Reporter proceeded through a filtering process in which the final number of called variants per patient passed through and were processed by three state of the art knowledge-based curation systems. From this, actionable information, including clinical trial information was harvested and compared between three curation systems. It is clinically important evidence that clinically relevant variants (level I etc.) was detected uniformly by three curators (so it is safe, it is not missing). On the other hand, the overall concordance of evidence levels of three curation systems are relatively low (56.9%~66.7%), although lack of concordance does not necessarily provide evidence regarding which is ‘correct’ in its recommendation. There is no ‘gold standard’ beyond expert panel. In addition, there was no significant difference for clinical information rate between non-synonymous variants and copy number alteration. We did not calculate it for gene fusions, because number of gene fusion was small. Recently, the highly concordance between in house report and Watson for Oncology for breast cancer patients10. It will be necessary to assess concordance of curation contents between in house expert panel and curation systems in the next study.
WfG reported the evidence levels of the greater numbers of the variants7,11. In the curation process to inform the clinical trials, WfG and QCII used the global database. WfG was reported clinical trials without limiting the regions. QCII filtered the data to be limited to the Asian region. OKR used Asian database. WfG presented many US clinical trials (Fig. 1). The discrepancy between three systems is likely to be due to difference in the filtering procedure of regional clinical trials. In fact, when excluding the data of clinical trials (level III), they showed higher concordance rate (86.1%~93.1% for levels I and II) in three systems. It is necessary to discuss with physicians in Asian countries including Japan whether it is necessary to inform clinical trials in US and European regions. The template of OKR can be created in several languages including Japanese, Chinese, and Korean. This approach is convenience for physicians in Asian. It can be also considered to create the reports in the Asian languages in other systems.
Although the sample of this study is small, it is still adequate since purpose of this study was not to analyze statistically significant of the differences between the panels, but rather it aimed to compare the implementation of different clinically reporting methods using data obtained in real world clinical practice. In addition, actionable information, including clinical trial information, was compared between three curation systems. Actionable information for one or several variants was provided for each case and only limited information was available for use for comparative analysis. Thus for the purpose of comparing the output results of annotated alterations with regards to therapeutic indication, based on evidence level, and clinical trial matching, we considered the sample size to be adequate. Information management challenges in cancer care are occurring in a practice environment where there is little time available for tracking and accessing relevant information at the point of care12,13. For example, a study that surveyed 1,117 oncologists reported that on average 4.6 h per week were spent keeping current in the field; while 53 h per week were spent on patient care and administrative tasks14. The situation is same in Japan. Importantly, it took ten minutes or shorter to complete the curation from uploading the vcf files to getting the report using any curation system.
Methods
Clinical specimens
A total of 31 solid tumor patients enrolled to this study between May to June 2018 at Kindai University Hospital. All patients provided written informed consent to participation in the study, including the collection of tumor tissue for analysis. This study was conducted in compliance with the Helsinki Declaration and the Ethical Guidelines for Medical and Health Research Involving Human Subjects by the Japanese government and has been approved by the ethics committee of Kindai University Faculty of Medicine (approved no 29–203).
Tissue processing
The collected formalin-fixed, paraffin-embedded tumor specimens underwent histological review, and only those containing sufficient tumor cells (at least 10%) as revealed by hematoxylin-eosin staining were subjected to nucleic acid extraction. DNA and RNA were isolated from the tissue with the use of an AllPrep DNA/RNA Mini Kit (Qiagen, Valencia, CA). The quality and quantity of the nucleic acid were verified with the use of a NanoDrop 2000 device, PicoGreen dsDNA Reagent, and RiboGreen RNA Reagent (all from Thermo Scientific, Wilmington, DE).
Next-generation sequencing
Tumor DNA and RNA were subjected to analysis with Oncomine Comprehensive assay v3 (OCAv3) for detection of mutations, copy number gain, and gene fusions. For DNA library preparation, tumor DNA (20 ng) was subjected to multiplex polymerase chain reaction (PCR) amplification with the use of an Ion AmpliSeq Library Kit 2.0 and DNA OCAv3 (all from Thermo Fisher Scientific). The PCR products were ligated to Ion Xpress Barcode Adapters (Thermo Fisher Scientific) and purified with the use of Agencourt AMPure XP beads (Beckman Coulter, Brea, CA). The purified libraries were pooled and then sequenced with an Ion Torrent Proton instrument, Ion PI Hi-Q Chef Kit, and Ion PI v3 Chip (all from Thermo Fisher Scientific). DNA sequencing data was accessed through the Torrent Suite ver.5.8 program (Thermo Fisher Scientific). Reads were aligned with the hg19 human reference genome, and potential mutations and copy number alteration were called with the use of Ion Reporter™ Software ver.5.8. Raw variant calls were filtered with quality score of <100, and were manually checked using the integrative genomics viewer (IGV; Broad Institute, Cambridge, MA). Germline mutations were excluded with the use of the Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB)15 and Exome Aggregation Consortium database (http://exac.broadinstitute.org/). For RNA library preparation, tumor RNA (20 ng) was subjected to reverse transcription with the use of a SuperScript IV VILO Master Mix (Thermo Fisher Scientific) followed by library generation with the use of an Ion AmpliSeq Library Kit 2.0 and RNA OCAv3 (all from Thermo Fisher Scientific). The PCR products were ligated to Ion Xpress Barcode Adapters (Thermo Fisher Scientific) and purified with the use of Agencourt AMPure XP beads (Beckman Coulter, Brea, CA). The purified libraries were pooled and then sequenced with an Ion Torrent Proton instrument, Ion PI Hi-Q Chef Kit, and Ion PI v3 Chip (all from Thermo Fisher Scientific). RNA sequencing data were accessed through the Torrent Suite ver.5.8 program (Thermo Fisher Scientific). Reads were aligned with the hg19 human reference genome, and potential fusions were analyzed with the use of Ion Reporter™ Software ver.5.8.
Watson for genomics
The following information was uploaded to Watson for Genomics (WfG): cancer type, a list of variants as a variant calling file, copy number alterations as linear copy-number values file, and fusion status as gene name (Supplementary Table 2). After the above information was uploaded, the WfG returns detailed annotation of 1) mutation profile (benign, likely benign, likely pathogenic, pathogenic, and variant of unknown significance, VUS) and 2) drug (evidence level, approval status in U.S. Food and Drug Administration, clinical trials, potential therapeutic options, and literature). Benign and likely benign variants were removed from the report (Supplementary Table 3). A report was generated by WfG showing the variants alongside potential targeted drugs. An example of the interface is shown in supplemental data. Software version was V38.159 (01-JUN-2018) and V39.135 (20-JUL-2018) in this study.
Oncomine knowledgebase reporter
The vcf files generated by Ion Reporter™ Software with the information of cancer type was uploaded to Oncomine Knowledgebase Reporter (OKR) (Supplementary Table 2). After the above information was uploaded, the OKR returns detailed annotation of 1) mutation profile (level of evidence based on a Joint Consensus Recommendation in ASCO, AMP, and CAP)16 and 2) drug (approval status in U.S. Food and Drug Administration, clinical trials, potential therapeutic options, and literature) (Supplementary Table 3). The location of clinical trial was set at Japan, Korea, and China. A report was generated by OKR showing the variants alongside potential targeted drugs. An example of the interface is shown in supplemental data. Software version was V3.2 (2018.03 (006)) and V3.3 (2018.06(004)) in this study.
QIAGEN clinical insight interpret
The vcf files generated by Ion Reporter™ Software with the information of cancer type was uploaded to QIAGEN Clinical Insight Interpret (QCII) (Supplementary Table 2). After the above information was uploaded, the QCII returns detailed annotation of 1) mutation profile (computed classification according to previous recommendation)16 and 2) drug (phase in U.S. Food and Drug Administration, clinical trials, potential therapeutic options, and literature) (Supplementary Table 3). The location of clinical trial was set at Japan. A report was generated by QCII showing the variants alongside potential targeted drugs. An example of the interface is shown in supplemental data. Software was used from 19th June 2018 to 31th July 2018 in this study.
Statistical analysis
The kappa statistic and associated 95% confidence intervals were used to measure agreement among curation methods.
Conclusion
Watson for Genomics (WfG), QIAGEN Clinical Insight Interpret (QCII), and Oncomine knowledge-based reporter (OKR) could work in the NGS based clinical sequencing with expert panel in routine use for patients with solid cancers. Region-specific automatic curation algorithms are necessary in a global curation system.
References
Takeda, M. et al. Clinical application of amplicon-based next-generation sequencing to therapeutic decision making in lung cancer. Ann Oncol 26, 2477–2482, https://doi.org/10.1093/annonc/mdv475 mdv475 [pii] (2015).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862–868, https://doi.org/10.1093/nar/gkv1222 gkv1222 [pii] (2016).
Chakravarty, D. et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017, https://doi.org/10.1200/PO.17.00011 (2017).
Rivera-Munoz, E. A. et al. ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum Mutat 39, 1614–1622, https://doi.org/10.1002/humu.23645 (2018).
Tate, J. G. et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947, https://doi.org/10.1093/nar/gky1015 5146192 [pii] (2019).
Sunami, K. et al. Clinical practice guidance for next-generation sequencing in cancer diagnosis and treatment (Edition 1.0). Cancer Sci 109, 2980–2985, https://doi.org/10.1111/cas.13730 (2018).
Patel, N. M. et al. Enhancing Next-Generation Sequencing-Guided Cancer Care Through Cognitive Computing. Oncologist 23, 179–185, https://doi.org/10.1634/theoncologist.2017-0170 theoncologist.2017-0170 [pii] (2018).
Garofoli, A. et al. PipeIT: Singularity Container for Molecular Diagnostic Somatic Variant Calling on Ion Torrent NGS Platform. J Mol Diagn, S1525-1578(18)30338-6 [pii] https://doi.org/10.1016/j.jmoldx.2019.05.001 (2019).
Takeuchi, S. & Okuda, S. Knowledge base toward understanding actionable alterations and realizing precision oncology. Int J Clin Oncol 24, 123–130, https://doi.org/10.1007/s10147-018-1378-0 [pii] (2019).
Somashekhar, S. P. et al. Watson for Oncology and breast cancer treatment recommendations: agreement with an expert multidisciplinary tumor board. Ann Oncol 29, 418–423, https://doi.org/10.1093/annonc/mdx781 4781689 [pii] (2018).
Itahashi, K. et al. Evaluating Clinical Genome Sequence Analysis by Watson for Genomics. Front Med (Lausanne) 5, 305, https://doi.org/10.3389/fmed.2018.00305 (2018).
Grunfeld, E. et al. Cancer care workers in Ontario: prevalence of burnout, job stress and job satisfaction. CMAJ 163, 166–169 (2000).
Woolhandler, S. & Himmelstein, D. U. Administrative work consumes one-sixth of U.S. physicians’ working hours and lowers their career satisfaction. Int J Health Serv 44, 635–642, https://doi.org/10.2190/HS.44.4.a (2014).
Shanafelt, T. D. et al. Burnout and career satisfaction among US oncologists. J Clin Oncol 32, 678–686, https://doi.org/10.1200/JCO.2013.51.8480 JCO.2013.51.8480 [pii] (2014).
Narahara, M. et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS One 9, e100924, https://doi.org/10.1371/journal.pone.0100924 PONE-D-14-03246 [pii] (2014).
Li, M. M. et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 19, 4–23, S1525-1578(16)30223-9 [pii] https://doi.org/10.1016/j.jmoldx.2016.10.002 (2017).
Acknowledgements
The authors wish to thank A. Yukumoto, M. Kitano, and A. Kurumatani for their support regarding the targeted sequencing. This study was supported by the contract research fund from SBI Life Insurance Co., Ltd.
Author information
Authors and Affiliations
Contributions
K. Sakai and K. Nishio designed and wrote the main manuscript. K. Sakai and M. Takeda assembled the figures, tables, and supporting information. M. Takeda, S. Shimizu, T. Takahama, T. Yoshida, S. Watanabe, T. Iwasa, K. Yonesaka, S. Suzuki, H. Hayashi, H. Kawakami, Y. Nonagase, K. Tanaka, J. Tsurutani, K. Saigoh, A. Ito, T. Mitsudomi, and K. Nakagawa contributed to sample collection, the interpretation of the results, and manuscript review. All authors approved and read the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
This study was supported by the contract research fund from SBI Life Insurance Co., Ltd. All authors have declared no other conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sakai, K., Takeda, M., Shimizu, S. et al. A comparative study of curated contents by knowledge-based curation system in cancer clinical sequencing. Sci Rep 9, 11340 (2019). https://doi.org/10.1038/s41598-019-47673-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47673-9
This article is cited by
-
Introducing AI to the molecular tumor board: one direction toward the establishment of precision medicine using large-scale cancer clinical and biological information
Experimental Hematology & Oncology (2022)
-
JCGA: the Japanese version of the Cancer Genome Atlas and its contribution to the interpretation of gene alterations detected in clinical cancer genome sequencing
Human Genome Variation (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
