
Abstract
The concentration of high-density lipoprotein-cholesterol (HDL-C) in humans is partially determined by genetic factors; however, the role of these factors is incompletely understood. The aim of this study was to examine the prevalence and characteristics of CETP, LIPC, and SCARB1 variants in Korean individuals with extremely high HDL-C levels. We also analysed associations between these variants and cholesterol efflux capacity (CEC), reactive oxygen species (ROS) generation, and vascular cell adhesion molecule-1 (VCAM-1) expression. Of 13,545 participants in the cardiovascular genome cohort, 42 subjects with HDL-C levels >100 mg/dL were analysed. The three target genes were sequenced by targeted next-generation sequencing, the functional effects of detected variants were predicted, and CEC was assessed using a radioisotope and apolipoprotein B-depleted sera. We observed two rare variants of CETP in 13 individuals (rare variant c.A1196G [p.D399G] of CETP was discovered in 12 subjects) and one rare variant of SCARB1 in one individual. Furthermore, all subjects had at least one of four common variants (one CETP and three LIPC variants). Two additional novel CETP variants of unknown frequency were found in two subjects. However, the identified variants did not show significant associations with CEC, ROS generation, or VCAM-1 expression. Our study provides additional insights into the role of genetics in individuals with extremely high HDL-C.
Similar content being viewed by others


Introduction
Plasma concentrations of cholesterol are generally known to be influenced by the combined effects of diverse genetic variants1. Individuals with extremely high lipid levels have a greater chance of being affected by monogenic syndrome in comparison to those with lipid levels closer to the normal values2,3,4. In addition, research into lipid phenotypes has played a key role in highlighting therapeutic targets5.
While the concentration of the high-density lipoprotein-cholesterol (HDL-C) is known to be partly determined by genetic factors, these factors are not entirely clear yet. In this regard, genetic variants associated with extremely high levels of HDL-C are under steady investigation6, and it has been shown that more than 40 genes are associated with HDL-C levels7. Interestingly, a recent large study involving a combination of multiple consortia showed that truncation variants of CETP were associated with high HDL-C levels and a low risk of coronary artery disease8. Although several genetic studies on HDL-C have been performed to date in Japan9,10, comprehensive studies on genetic variants that influence HDL-C in Asians are still limited.
The aim of our study was to investigate the prevalence and characteristics of CETP, LIPC, and SCARB1 variants in Korean individuals with extremely high levels of HDL-C using next-generation sequencing, as rare variants of these genes are known to be associated with the above-mentioned HDL-C phenotype2,11. Additionally, we analysed associations between the identified variants and individuals’ cholesterol efflux capacity (CEC), a functional parameter of HDL that can be affected by metabolic changes in variant carriers.
Results
Clinical characteristics of study subjects
The mean age of 42 study subjects was 54 years, of which 16 (38%) were males. Two subjects (5%) had diabetes mellitus, and three (7%) had coronary artery disease. The mean HDL-C level was 110.1 ± 12.6 mg/dL. In comparison to the characteristics of the total cohort population, the study subjects were younger, had a lower prevalence of cardiovascular risk factors, and had lower triglyceride levels (Table 1).
Analysis of candidate genes
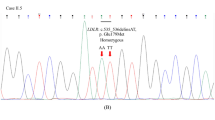
In the 42 subjects, rare and common variants of CETP and common variants of LIPC were frequent. Two rare variants of CETP were present in 13 individuals (31%), whereas one rare variant of SCARB1 was observed in one subject (Table 2). In particular, a rare variant of CETP (c.A1196G [p.D399G]) was discovered in 12 subjects (29%) and the rate was much higher than that in the gnomAD database (Fig. 1A). Conversely, all 42 study subjects had at least one of four common variants of CETP or LIPC. One common variant of CETP (c.G1084A [p.V362I]) and two common variants of LIPC (c.C1068A [p.F356L] and c.A644G [p.N215S]) were highly frequent, and were found in 38, 42, and 42 subjects, respectively (Table 2, Fig. 1A). A summary of the genetic variants found in each individual is presented in Supplementary Table S1, and the locations of the variants in each of the three genes are indicated in Fig. 1B.

Proportion of variant carriers and locations of variants in each gene. (A) Proportion of carriers who had variants in each of the three genes identified in the 42 study subjects. With regard to CETP, 13 subjects carried rare variants, and 38 subjects had common variants. Two variants of unknown frequency in CETP were discovered in two individuals. No rare variant was identified, while common variants of LIPC were found in all subjects. Conversely, with regard to SCARB1, one rare variant was discovered in one subject. (B) Locations of CETP, LIPC, and SCARB1 variants identified in the study subjects.
CETP
Two rare variants of CETP were found in 13 subjects: c.A1196G (p.D399G) in 12 individuals and c.G1195T (p.D399Y) in one individual. One carrier of c.A1196G (p.D399G) was homozygous for this variant, while the others were heterozygous. All rare variants of this gene observed in the present study have been previously reported and were predicted to be damaging. Conversely, one common variant, c.G1084A (p.V362I), was identified in 38 subjects, of which 15 carriers were homozygous for this variant. This common variant was suspected to be benign according to the prediction programs used here. Furthermore, two novel variants of unknown frequency—c.T974C (p.V325A) and c.G537A (p.W179X)—were discovered in two individuals (Table 2).
LIPC
No rare variants of LIPC were identified, and three common variants of LIPC were found among the study subjects: c.C1068A (p.F356L), c.A655G (p.N215S), and c.G283A (p.V95M) in 42, 42, and 16 individuals, respectively. In most cases, the carriers of the first two variants were homozygous, whereas all carriers of the last variant were heterozygous. These three variants have also been previously reported. Of the variants, only c.A655G (p.N215S) was predicted to be disease-causing, whereas the others were suspected to be tolerated (Table 2).
SCARB1
One rare variant of SCARB1 was discovered in one subject: c.G745A (p.D249N). The carrier was heterozygous for this previously reported variant. According to in silico analyses, the effect of this variant was predicted to be uncertain. No common variants of SCARB1 were found in our study subjects (Table 2).
Associations of the variants with CEC, reactive oxygen species (ROS), and vascular cell adhesion molecule-1 (VCAM-1)
When subjects were categorized according to the quartile CEC values, there were no significant differences in the numbers of variants of all target genes or of CETP or LIPC alone. Owing to statistical limitations, the relationship of the SCARB1 variant with CEC could not be analysed. In addition, the numbers of variant carriers were compared between the four groups of subjects as categorized by quartile CEC values. For each of the five variants of CETP, three variants of LIPC, and one variant of SCARB1, there were no differences in the number of carriers between the four quartile groups (Table 3). CEC values were compared between the carriers and non-carriers of three variants, c.A1196G (p.D399G) and c.G1084A (p.V362I) in CETP, and c.G283A (p.V95M) in LIPC. However, the values were similar between the carriers and non-carriers of these three variants (Supplementary Table S4). For other variants, it was not statistically appropriate to compare carriers and non-carriers because the sample sizes were too small.
Likewise, when subjects were categorized according to the quartile ROS generation, there was no significant difference in the numbers of variants of all target genes or of CETP or LIPC alone. For each of variant of three target genes, there were no differences in the number of carriers between the four quartile groups (Supplementary Table S2). ROS generation were similar between the carriers and non-carriers of c.A1196G and c.G1084A in CETP, and c.G283A in LIPC (Supplementary Table S4).
When subjects were categorized according to the quartile VCAM-1 expression, there was no significant difference in the numbers of variants of all target genes or of CETP or LIPC alone. For each of variant of three target genes, there were no differences in the number of carriers between the four quartile groups. However, the number of carriers of c.A1196G in CETP tended to be higher in lower quartile group of VCAM-1 expression (Supplementary Table S3). Although VCAM-1 expression were not significantly different between the carriers and non-carriers of c.A1196G and c.G1084A in CETP, and c.G283A in LIPC, the carriers of c.A1196G showed tendency of lower VCAM-1 expression (Supplementary Table S4).
Discussion
In our study population with extremely high HDL-C levels, rare or common variants of CETP and common variants of LIPC were frequent. All the study subjects were carriers of more than one common variant of CETP or LIPC. Only one individual possessed a SCARB1 variant. All rare or common variants had been previously reported, while two novel CETP variants of unknown frequency were observed in two individuals. In our study population, we did not identify any significant associations between the identified variants and the CEC values of the subjects. These results provide rare and informative data on the genetic spectra of these three genes in East Asian individuals with extremely high levels of HDL-C.
In the present study, 31 and 100% of the subjects possessed rare or common variants, respectively, of the three genes. These rates are higher than the values for rare or common variants (11 and 19% respectively) reported in a previous study performed in Canada7. However, while the present study included only individuals with HDL-C values > 100 mg/dL, the aforementioned study included those with HDL-C values of 54–70 mg/dL. Thus, phenotypic differences between the study populations may be related to the discrepancy in variant frequencies. Furthermore, another previous study using targeted next-generation sequencing reported that 5.2% of participants with extremely high levels of HDL-C (mean HDL-C: 96 mg/dL) possessed variants that were either causing or probably causing12. In contrast, the majority of the subjects in the present study possessed causing variants. The underlying reason for this difference remains to be clarified. In the Canadian study, large-effect variants were most frequent in LIPC in individuals with extremely high levels of HDL-C. However, most rare variants in the current analysis were variants of CETP.
It has been previously reported that homozygous loss-of-function mutations in CETP are associated with 80–100% higher levels of HDL-C13,14. Moreover, common loss-of-function variants of this gene are related to 20–30% higher values of HDL-C14. To date, a few rare CETP gene defects have been identified, such as the D442G variant in exon 1515 and G181X in exon 109. According to a study using in vitro point mutagenesis, mutations of the hydrophobic amino acids at positions 454–475 are associated with a reduction in cholesteryl ester transfer activity10. Notably, two of the variants found in the present study were located at position 399. One of the most studied CETP polymorphisms is Taq1B, which is known to affect HDL-C levels in homozygous carriers16. We also found a common variant at position 362 of CETP that was present in most study subjects, though it was predicted to be benign. In addition, while we identified two novel variants of CETP in our subjects, further study is needed to determine their functional and clinical relevance.
With regard to LIPC, the S267F and 480C > T variants have been reported to be associated with high HDL-C levels17,18. In addition, several common variants were reported to be correlated with high HDL-C levels19,20. In our study, only common variants were found in LIPC, and of these, only N215S was predicted to be causing. Accordingly, rare variants of LIPC seem to contribute very little to the phenotype of extremely high HDL-C in our population.
Recently, it has been shown that the P376L variant of SCARB1 is associated with elevated HDL-C levels and risk of coronary heart disease21. This study analysed individuals with extremely high levels of HDL-C, with a mean of 107 mg/dL. Of 328 subjects, five subjects (1.5%) carried homozygous or heterozygous variants, and when three cohorts were combined, the prevalence was 1.8%. In this regard, we could assume that variants of SCARB1 are very rare in the general population. In a prior study, c.889C > T (P297S), a causing variant, was discovered in one of 162 Caucasians with HDL-C levels above the 95th percentile22. In another study, two out of 120 Caucasians (1.7%) with HDL-C levels above the 90th percentile were found to have S112F (C588T) or T175A (A776T), which are both rare causing variants23. Conversely, in a large data of whole-genome sequenced Icelanders, three rare variants (combined allelic frequency of 0.2%), one low-frequency variant, and three common variants of SCARB1 were found to be associated with elevated HDL-C24. Furthermore, an American study that analysed subjects with HDL-C levels in the highest and lowest deciles found three common and novel SCARB1 variants associated with HDL-C levels25. In our study, we identified only the D249N variant of SCARB1 in one of the 42 subjects (2.4%). These results suggest that SCARB1 variants may be diversely distributed in various populations.
Although studies on genetic variants associated with CEC have been very limited26, variants of genes affecting HDL metabolism have been suggested as candidates27. In a previous study, eight individuals with variants of CETP or LIPC and HDL-C values ≥ 105 mg/dL exhibited elevated CEC values. This finding was reported to be the result of increased HDL2 and the enhanced intrinsic capacity of HDL328. In a recent study, one LIPC and two CETP variants revealed associations with ATP transporter-dependent CEC. However, the significance disappeared after adjusting for HDL-C and triglyceride levels29. Conversely, CEC values were similar between carriers and non-carriers of the P376L variant of SCARB121. Nevertheless, since the carriers of the P297S variant had low CEC values in spite of high HDL-C levels22, the relationship between SCARB1 variants and CEC values appear to be inconsistent. In the current study, CEC was not associated with the numbers of variants of the target genes or the presence of any specific variant. Furthermore, while we compared individuals according to genotype, the phenotypes of our subjects—particularly their HDL-C values—were very similar, which could have influenced our results.
In our study, the c.A1196G (p.D399G; rs2303790) variant of CETP showed a tendency, although not significant, of lower VCAM-1 expression. In previous studies, this variant showed association with eye pathologies including retinal disease30,31. The relationship between this variant and cardiovascular disease has been inconsistent30,31. Interestingly, it has been reported that HDL from CETP deficient individuals could have differential impact on VCAM-1 inhibition32. However, further studies are needed to clarify our findings regarding the effect of this CETP variant on VCAM-1.
While our study provides important genetic information of extremely elevated levels of HDL-C in Koreans, our study has some potential limitations. First, we did not collect data on family history or acquire samples from the family members of our subjects. An analysis of variants using co-segregation might have provided additional insight into their functionality. Second, the prediction of the causality of variants using publicly available tools for the analyses can be imperfect, and this may be another limitation. Third, we attempted to identify relationships between the genetic variants and CEC. However, as previously mentioned, the phenotypes of our subjects were quite homogeneous, and thus it was likely more challenging to identify the differential effects of the variants. Finally, we could not include a control group with normal HDL-C levels in our study. Having such a control group might have provided clearer insights about the effect of variants identified in the study.
In conclusion, rare or common variants of CETP and common variants of LIPC were frequently found in the study population with extremely high levels of HDL-C, while SCARB1 variants were very uncommon. The presence of the identified variants was not associated with CEC, ROS generation, and VCAM-1 expression in the subjects of the present study. Our results provide comprehensive data regarding the spectrum of genetic variants of three target genes in East Asians with this HDL-C phenotype.
Methods
Study population
The Institutional Review Board of Severance Hospital approved the methods of the present study and all subjects and/or their legal guardians provided written informed consent. All research was performed in accordance with relevant guidelines/regulations. Between November 2000 and March 2011, 13,545 subjects were enrolled in the Cardiovascular Genome Center Cohort, Yonsei University College of Medicine, Seoul, Korea. Individuals ≥18 years of age were recruited into this cohort when they visited Severance Hospital for cardiovascular diseases, the control of risk factors, or health check-ups. Participants were interviewed regarding their medical history, after which they underwent a physical examination and laboratory evaluation. Of these individuals, 42 subjects whose HDL-C levels were >100 mg/dL were analysed. These subjects were not undergoing lipid-lowering therapy prior to enrolment in the current study. Pregnant women, individuals affected by cancer or thyroid, liver, or kidney disease, or patients undergoing pharmacotherapy that could affect lipid profiles (such as lipid-modifying agents, corticosteroids, or oral oestrogen) were excluded.
Assessment of laboratory values, CEC, ROS, and VCAM-1
The levels of total cholesterol, triglyceride, HDL-C, and LDL-C were measured in all study participants. The participants fasted and avoided alcohol for at least 12 h prior to blood sampling. Samples were analysed within 4 h by a laboratory certified by the Korean Society of Laboratory Medicine. We analysed the potential relationships between the variants we sequenced here and CEC, a functional parameter of HDL. Assays for CEC, ROS, and VCAM-1 are described in the Supplementary Information.
Targeted sequencing and analyses of variants
Three target genes were sequenced: CETP (MIM 118470), LIPC (MIM 151670), and SCARB1 (MIM 601040). Genomic DNA was extracted from blood using the Qiagen Dneasy kit (Qiagen, Valencia, CA, USA). For mutation analyses, a panel for targeted DNA capture and sequencing was developed by Celemics, Inc. (Seoul, Korea). Targeted sequencing and variant analyses were performed as follows. DNA fragments that contained all coding exons and exon-intron junctions were enriched by solution-based hybridization capture, followed by sequencing using the Illumina HiSeq. 2000 platform (Illumina, Inc., San Diego, CA, USA). The quality of next-generation sequencing data, including coverage information, is presented in Supplementary Fig. S1.
An analysis of sequencing data was conducted using an in-house analysis pipeline as previously described3. Briefly, sequencing reads from the HiSeq. 2000 raw data were sorted by index and barcode sequences. Sorted FASTQ files were aligned to the hg19 reference genome using the Burrows-Wheeler Aligner (BWA; ver. 0.7.12) BWA-MEM algorithm. Output files in SAM format were converted into BAM files and sorted using SAMtools (ver. 1.1). Duplicate removal was performed with Picard tools (ver. 1.128) MarkDuplicates. Realignment around known indel sites and base quality score recalibration (BQSR) were conducted using GATK (v3.3.0) to create the final BAM files. Variants were called using the GATK v3.3.0 Unified Genotyper algorithm for loci with a sequencing depth greater than or equal to 50×. An analysis of splice site regions, including sufficient intronic bases, was performed using Human Splicing Finder.
The functional annotation of genetic variants was performed by ANNOVAR (ver. 2014-11-12). The predictions of the functional effects of single-nucleotide variants were acquired using SIFT, PolyPhen-2, and MutationTaster, and were matched against the Korean population exome data (n = 476) and a public database of variants (dsSNP 138, Exome Variant Server, and 1000 Genome Project SNP [April 2012 release] from both Asian and all-population databases). We then prioritized variants according to the following criteria: (1) variants that were reported to be disease-causing in the Human Gene Mutation Database, (2) disruptive variants (nonsense, splice-site [two nucleotides on either side of the intron/exon boundary], and frameshift) that were novel or rare, and (3) novel or rare missense variants that were predicted to be deleterious by any of the three prediction programs. Variants that met these criteria were validated by bidirectional Sanger sequencing of PCR amplicons. Databases used for confirming the identity and frequency of the variants included 1000 Genome Project, Exome Sequencing Project 6500, and gnomAD browser (http://gnomad.broadinstitute.org/). Variants with a minor allele frequency (MAF) of <1% were classified as rare, whereas those with a MAF of ≥5% according to public databases were classified as common.
Statistical analyses
Continuous variables are presented as the mean ± standard deviation or median (interquartile range) and were compared using an independent t-test or Kruskal-Wallis test depending on the distribution of the data. Categorical variables were compared using a chi-square test or Fisher’s exact test. To analyse associations between genetic variants and the CEC of the test subjects, individuals were divided into four groups according to the quartile values of CEC. All statistical analyses were performed using R software version 3.5.0 (R Foundation for Statistical Computing, Vienna, Austria). For all analyses, differences with p < 0.05 were considered to be significant.
References
Teslovich, T. M. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 466, 707–13 (2010).
Hegele, R. A. Plasma lipoproteins: genetic influences and clinical implications. Nat. Rev. Genet. 10, 109–121 (2009).
Lee, C. J. et al. Rare and common variants of APOB and PCSK9 in Korean patients with extremely low low-density lipoprotein-cholesterol levels. PloS One. 12, e0186446 (2017).
Lee, C. J. et al. Variants of lipolysis-related genes in Korean patients with very high triglycerides. Yonsei. Med. J. 59, 148–153 (2018).
Khera, A. V. & Rader, D. J. Discovery and validation of new molecular targets in treating dyslipidemia: the role of human genetics. Trends Cardiovasc. Med. 19, 195–201 (2009).
Khetarpal, S. A. et al. Multiplexed targeted resequencing identifies coding and regulatory variation underlying phenotypic extremes of high-density lipoprotein cholesterol in humans. Circ. Genom. Precis. Med. 11, e002070 (2018).
Oldoni, F., Sinke, R. J. & Kuivenhoven, J. A. Mendelian disorders of high-density lipoprotein metabolism. Circ. Res. 114, 124–142 (2014).
Nomura, A. et al. Protein-truncating variants at the cholesteryl ester transfer protein gene and risk for coronary heart disease. Circ. Res. 121, 81–88 (2017).
Weissglas-Volkov, D. & Pajukanta, P. Genetic causes of high and low serum HDL-cholesterol. J. Lipid Res. 51, 2032–2057 (2010).
Dron, J. S. et al. Polygenic determinants in extremes of high-density lipoprotein cholesterol. J. Lipid Res. 58, 2162–2170 (2017).
Sadananda, S. N. et al. Targeted next-generation sequencing to diagnose disorders of HDL cholesterol. J. Lipid Res. 5, 1993–2001 (2015).
Rosenson, R. S. et al. HDL and atherosclerotic cardiovascular disease: genetic insights into complex biology. Nat. Rev. Cardiol. 15, 9–19 (2018).
Takahashi, K. et al. A missense mutation in the cholesteryl ester transfer protein gene with possible dominant effects on plasma high density lipoproteins. J. Clin. Invest. 92, 2060–2064 (1993).
Arai, T. et al. A novel nonsense mutation (G181X) in the human cholesteryl ester transfer protein gene in Japanese hyperalphalipoproteinemic subjects. J. Lipid Res. 37, 2145–54 (1996).
Yamashita, S., Hirano, K., Sakai, N. & Matsuzawa, Y. Molecular biology and pathophysiological aspects of plasma cholesteryl ester transfer protein. Biochim. Biophys. Acta. 1529, 257–75 (2000).
Boekholdt, S. M. & Thompson, J. F. Natural genetic variation as a tool in understanding the role of CETP in lipid levels and disease. J. Lipid Res. 44, 1080–1093 (2003).
McCaskie, P. A. et al. The C-480T hepatic lipase polymorphism is associated with HDL-C but not with risk of coronary heart disease. Clin. Genet. 70, 114–121 (2006).
Johannsen, T. H. et al. Hepatic lipase, genetically elevated high-density lipoprotein, and risk of ischemic cardiovascular disease. J. Clin. Endocrinol. Metab. 94, 1264–1273 (2009).
Isaacs, A., Sayed-Tabatabaei, F. A., Njajou, O. T., Witteman, J. C. & van Duijn, C. M. The -514 C->T hepatic lipase promoter region polymorphism and plasma lipids: a meta-analysis. J. Clin. Endocrinol. Metab. 89, 3858–3863 (2004).
Hodoglugil, U., Williamson, D. W. & Mahley, R. W. Polymorphisms in the hepatic lipase gene affect plasma HDL-cholesterol levels in a Turkish population. J. Lipid Res. 51, 422–30 (2010).
Zanoni, P. et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 351, 1166–1171 (2016).
Vergeer, M. et al. Genetic variant of the scavenger receptor BI in humans. N. Engl. J. Med. 364, 136–145 (2011).
Brunham, L. R. et al. Novel mutations in scavenger receptor BI associated with high HDL cholesterol in humans. Clin. Genet. 79, 575–581 (2011).
Helgadottir, A. et al. Rare SCARB1 mutations associate with high-density lipoprotein cholesterol but not with coronary artery disease. Eur. Heart J. 39, 2172–2178 (2018).
Niemsiri, V. et al. Impact of genetic variants in human scavenger receptor class B type I (SCARB1) on plasma lipid traits. Circ. Cardiovasc Genet. 7, 838–847 (2014).
Cheon, E. J. et al. Novel association between CDKAL1 and cholesterol efflux capacity: replication after GWAS-based discovery. Atherosclerosis. 273, 21–27 (2018).
Villard, E. F. et al. Genetic determination of plasma cholesterol efflux capacity is gender-specific and independent of HDL-cholesterol levels. Arterioscler. Thromb. Vasc. Biol. 33, 822–828 (2013).
El Khoury, P. et al. Improved plasma cholesterol efflux capacity from human macrophages in patients with hyperalphalipoproteinemia. Atherosclerosis. 234, 193–199 (2014).
Low-Kam, C. et al. Variants at the APOE/C1/C2/C4 locus modulate cholesterol efflux capacity independently of high-density lipoprotein cholesterol. J. Am. Heart Assoc. 7, e009545 (2018).
Cheng, C. Y. et al. New loci and coding variants confer risk for age-related macular degeneration in East Asians. Nat. Commun. 6, 6063 (2015).
Milwood, I. Y. et al. Association of CETP gene variants with risk for vascular and nonvascular dieseases among Chinese adults. JAMA. Cardiol. 3, 34–43 (2018).
Gomaraschi, M. et al. eNOS activation by HDL is impaired in genetic CETP deficiency. PloS. One. 9, e95925 (2014).
Acknowledgements
Funding: This research was financially supported by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (2015M3A9B6029138). The funder had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, or approval of the manuscript; and decision to submit the manuscript for publication
Author information
Authors and Affiliations
Contributions
S.-H.L. proposed the study design and provided funding; M.S.P. and J.H.L. analysed and interpreted the genetic data; M.K. performed the assessment of CEC; S.-J.A. performed the ROS experiments; J.L. performed VCAM-1 experiments; C.J.L. and S.-H.L. analysed and interpreted the clinical data; S.P., S.M.K., Y.J. and S.-H.L. provided clinical data; C.J.L., J.H.L. and S.-H.L. wrote the manuscript. All authors revised the manuscript and approved the final article.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, C.J., Park, M.S., Kim, M. et al. CETP, LIPC, and SCARB1 variants in individuals with extremely high high-density lipoprotein-cholesterol levels. Sci Rep 9, 10915 (2019). https://doi.org/10.1038/s41598-019-47456-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47456-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
