
Abstract
The enzyme kynurenine aminotransferase (KAT) catalyses the conversion of kynurenine (KYN) to kynurenic acid (KYNA). Although the isozymes KAT1–4 have been identified, KYNA is mainly produced by KAT2 in brain tissues. KNYA is an antagonist of N-methyl-D-aspartate and α-7-nicotinic acetylcholine receptors, and accumulation of KYNA in the brain has been associated with the pathology of schizophrenia. Therefore, KAT2 could be exploited as a therapeutic target for the management of schizophrenia. Although currently available KAT2 inhibitors irreversibly bind to pyridoxal 5′-phosphate (PLP), inhibition via this mechanism may cause adverse side effects because of the presence of other PLP-dependent enzymes. Therefore, we identified novel selective KAT2 inhibitors by screening approximately 13,000 molecules. Among these, glycyrrhizic acid (GL) and its analogues, glycyrrhetinic acid (GA) and carbenoxolone (CBX), were identified as KAT2 inhibitors. These compounds were highly selective for KAT2 and competed with its substrate KYN, but had no effects on the other 3 KAT isozymes. Furthermore, we demonstrated that in complex structures that were predicted in docking calculations, GL, GA and CBX were located on the same surface as the aromatic ring of KYN. These results indicate that GL and its analogues are highly selective and competitive inhibitors of KAT2.
Similar content being viewed by others


Introduction
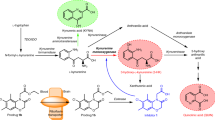
Dietary tryptophan is predominantly metabolised via the kynurenine pathway1 (Fig. 1), which produces neuroactive metabolites such as kynurenic acid (KYNA). KYNA reportedly acts as an antagonist of N-methyl-D-aspartate receptors2,3 and the α-7-nicotinic acetylcholine receptor (α7nAChR)4,5. Furthermore, previous studies in animal models show that elevated KYNA levels in the brain impair cognitive function6, spatial working memory7 and auditory sensory gating8 in rats. Abnormally high KYNA levels have been observed in brain tissues and cerebral spinal fluid (CSF) from patients with schizophrenia9,10,11. Therefore, physiological control of brain KYNA levels is likely critical to the prevention of these conditions1.

Schematic overview of the tryptophan-kynurenine pathway. Kynurenic acid (KYNA) is formed from kynurenine (KYN) by kynurenine aminotransferase (KAT) enzymes. TRP, tryptophan; AA, anthranilic acid; 3-HK, 3-hydroxy kynurenine; 3-HAA, 3-hydroxy anthranilic acid; QUIN, quinolinic acid; TDO, tryptophan 2,3-dioxygenase; IDO1, indoleamine 2,3-dioxygenase 1; IDO2, indoleamine 2,3-dioxygenase 2; KYNU, kynureninase; KMO, kynurenine 3-monooxygenase; NAD+, nicotinamide adenine dinucleotide.
KYNA is produced from kynurenine (KYN) by kynurenine aminotransferase (KAT) (Fig. 1). KAT enzymes require pyridoxal 5′-phosphate (PLP) coenzymes, and four KAT isoforms (KAT1–4) have been identified. In the brain, KYNA is predominantly generated by KAT2, which is expressed in astrocytes12,13,14. Accordingly, KAT2 plays essential roles in regulating KYNA levels in the brain.
Currently, various KAT2 inhibitors, including PF-0485998915, BFF-12216, BFF-81617,18 and S-ESBA19, have been shown to decrease KYNA levels in the brain. In animal models, these KAT2 inhibitors improve cognitive function by releasing dopamine, acetylcholine and glutamate20,21,22,23. PF-04859989 and BFF-122 also act as KAT2 inhibitors by irreversibly binding to the PLP cofactor15,16. However, because over 300 PLP-dependent enzymes and proteins have been identified, irreversible binding to PLP may cause side effects. For example, carbidopa is used for the treatment of Parkinson’s disease, but causes adverse effects due to irreversible binding to PLP24,25.
In this study, we identified novel KAT2 inhibitors with different inhibitory mechanisms using a microplate fluorescence assay for kynurenine aminotransferase as reported previously26 with minor modifications. We then screened for KAT2 inhibitors from approximately 13,000 compounds using a high-throughput screening assay. These experiments identified approximately 20 candidate KAT2 inhibitors (data not shown), including glycyrrhizic acid (GL) and its analogues, glycyrrhetinic acid (GA) and carbenoxolone (CBX) (Fig. 2). The basic structures of these three compounds are similar. GL comprises GA and two glucuronic acid molecules27,28, whereas CBX is a derivative of GA29. GL is also the main component of liquorice, which is a component of the traditional Japanese medicine yokukansan30. Previous reports show that yokukansan alleviates psychological symptoms of schizophrenia31,32 and the behavioural and psychological symptoms of dementia in patients with Alzheimer’s disease and Lewy bodies33 and improves cognitive function in patients with senile dementia34,35. Thus, GL, GA and CBX may be KAT2 inhibitors with efficacy in treatment of mental disorders.

Chemical structures of (a) glycyrrhizic acid (GL), (b) glycyrrhetinic acid (GA) and (c) carbenoxolone (CBX). Chemical structures of the known KAT2 inhibitors (d) PF-04859989, (e) BFF-122, (f) BFF-816 and (g) S-ESBA are also presented.
Results
Inhibitory activity of GL and its analogues
To evaluate the inhibitory activities of GL, GA and CBX against KAT2, we measured half-maximal inhibitory concentrations (IC50) and estimated inhibition constants (Ki). GL (IC50, 4.51 ± 0.20 μM; Ki, 10.42 ± 1.62 μM), GA (IC50, 6.96 ± 0.37 μM; Ki, 6.92 ± 0.60 μM) and CBX (IC50, 3.90 ± 0.37 μM; Ki, 4.11 ± 0.37 μM) showed high inhibitory activity against human KAT2 (Figs 3 and 4). In investigations of the selectivity of GL, GA and CBX for KAT isoforms, all three compounds showed inhibitory activity against mouse KAT2, but none inhibited the other 3 KAT isozymes (Table 1), indicating selectivity for KAT2.

Dose dependent inhibitory activities of GL and its analogues on human KAT2. Half inhibitory concentrations (IC50) of (a) GL, (b) GA and (c) CBX were analysed using nonlinear curve fitting. All experiments were performed in triplicate and data are presented as means ± standard errors of the mean (SEM).

Lineweaver–Burk plots of the inhibitory kinetics of PF-04859989, GL and its analogues towards KAT2. Inhibition kinetics of (a) PF-04859989, (b) GL, (c) GA and (d) CBX were evaluated using Lineweaver–Burk analyses. All experiments were performed in triplicate and data are presented as means ± SEM. Analyses were performed in the presence of varying concentrations of KYN and KAT2 inhibitor. Inhibitor concentrations: (a) PF-04859989, 300 nM; (b) GL, 4.5 μM; (c) GA, 6.0 μM; (d) CBX, 3.5 μM; Ki values were calculated using the global fit formula (GraphPad Prism v6.07).
Inhibitory mechanisms of the compounds against human KAT2
To investigate inhibitory mechanisms of GL, GA and CBX, we measured KAT2 activity in the presence of varying KYN concentrations and inhibitory compounds at close to their IC50 (GL, 4.5 μM; GA, 6.0 μM; CBX, 3.5 μM). Additionally, we then compared inhibitory properties of these compounds with those of the KAT2 inhibitor PF-04859989, which binds PLP irreversibly. PF-04859989 was used at its IC50 value of 300 nM, as determined in our assays. Because PF-04859989 binds to PLP, maximum reaction rates (Vmax) varied but the Michaelis–Menten constant (Km) did not change (Fig. 4a). In contrast, Km values for GL, GA and CBX changed with substrate concentrations, whereas respective Vmax values did not vary under these conditions (Fig. 4b–d). These results showed that GL, GA and CBX do not bind PLP, but act as competitive inhibitors of KYN.
Computational docking of GL and its analogues to KAT2
Three-dimensional structures of KAT2-ligand complexes were obtained using the docking calculations shown in Fig. 5. For comparison, we also present a crystal structure from the protein data bank (PDB; PDB ID, 2R2N) in Fig. 5a. All ligands were located in the ligand-binding pocket of two KAT2 molecules. To elucidate the interactions between KAT2 and these ligands, we magnified the structures of ligand-binding pockets (Fig. 6), and identified residues that are involved in hydrogen-bond formation and stacking, as shown in Fig. 7. In these figures, the subunits A and B are shown in green and cyan, respectively. In the crystal structure of 2R2N, KYN forms hydrogen bonds with Asn202(B) and Arg399(B) (Figs 6a and 7a), and the binding structure is stabilised by a π-π interactions between the aromatic ring of KYN and the Tyr74(A) side chain. Although GL formed a hydrogen bond with Ser77(A), no hydrogen bonds were observed between GL and Asn202(B) or Arg399(B) (Figs 6b and 7b). Yet CH-π stacking interactions with Tyr74(A) may stabilise the binding structure. The hydrophobic moiety of GL was located on hydrophobic surfaces comprising Leu40(A), Tyr74(A), Pro76(A) and Gln73(A) and the carbohydrate chain of GL was located outside of the ligand-binding pocket. Similarly, the hydrophobic moiety of GA was located on the hydrophobic surface of Leu40(A), Tyr74(A), Pro76(A) and Gln73(A) (Fig. 6c), and the binding structure of GA favours stabilisation by CH-π stacking interactions with Tyr74(A) (Fig. 7c). Although GA formed no hydrogen bonds with Asn202(B), Arg399(B) or Ser77(A), hydrogen bonds with Gly39(A) and Tyr142(B) were observed. The docking pose of CBX, which had a similar IC50 value to that of GL, was also similar to that of GL (Figs 6d and 7d). Moreover, the residues required for stabilisation of ligand binding by hydrophobic effects and stacking interactions were conserved among GL and its analogues (Table 2). In particular, Tyr74 is considered one of the most important residues for ligand binding.

The complex structures of KAT2 with GL and its analogues. (a) Crystal structure of KAT2-KYN (PDB ID, 2R2N) and (b) predicted structures of KAT2-GL, (c) KAT2-GA and (d) KAT2-CBX. In the homodimer of KAT2, the two subunits A and B are shown in green and cyan, respectively. KYN is presented in grey, GL is shown in pink, GA is shown in purple, CBX is shown in orange, PMP and PLP are shown in yellow.

Docking models of GL and its analogues with the ligand-binding pocket of KAT2. Ligand binding pockets in (a) the crystal structure of 2R2N; (b) predicted structures for GL, (c) GA and (d) CBX are illustrated. Colour coordinates are the same as in Fig. 5. The residues that are involved in interactions with ligands are shown in stick models. Atom colour; H (white), O (red) and N (blue). Dotted lines represent hydrogen bonds.

Key KAT2 residues for binding of GL and its analogues. Residues that are involved in hydrogen-bonding formations and stacking interactions in (a) the crystal structure of 2R2N, (b) predicted structures for GL, (c) GA and (d) CBX are illustrated. The colour coordinates are as in Fig. 5. Residues are shown with stick models with the atoms H (white), O (red) and N (blue). Black dotted lines represent hydrogen bonds. Red dotted lines show possible stacking interactions.
Discussion
In this study, we examined the inhibitory effects of GL, GA and CBX on KAT2 using recombinant proteins. Our data show that GL, GA and CBX are strong KAT2 inhibitors and compete with the substrate KYN.
Previous studies have indicated that increased KYNA concentrations in the brain impair cognitive function6, spatial working memory7 and auditory sensory gating8 in rats. Moreover, abnormal increases in KYNA have been observed in brain tissues and CSF from patients with schizophrenia9,10,11. Because KYNA production in the brain is predominantly catalysed by KAT212,13,14, KAT2 inhibitors effectively control KYNA excesses in the brain, leading to improved cognitive function and spatial memory in rats23,36. Moreover, KAT2-knockout mice displayed improvements in cognitive function, as observed with KAT2 inhibitors37.
Several KAT2 inhibitors are currently available. Among these, PF-04859989 and BFF-122 irreversibly bind PLP, which is a cofactor of all KAT isoforms15,16. Irreversible binding to PLP may lead to undesirable inhibition of over 300 PLP-dependent enzymes and proteins. S-ESBA and NS-1502 are KAT2 inhibitors that show selective and reversible inhibition of KAT238,39. But, reported IC50 values for these compounds with human KAT2 are very high (S-ESBA, approximately 1000 μM; NS-1502, 315 μM)38,39. In studies with the recently developed KAT2 inhibitor BFF-81617,18, oral administration prevented increases in KYNA in the brain after systemic kynurenine injections and attenuated the resulting glutamate release in the rat prefrontal cortex18. The mechanisms of action (competitive or non-competitive) and selectivity of BFF816 for KAT isoforms remains unclear.
In this study, we identified novel KAT2 inhibitors with strong inhibitory effects. The IC50 values of these compounds (GL, GA and CBX) were low μM range on human KAT2 (Table 1). Although these IC50 values were determined at the basic conditions (pH9.5) to obtain the optimal fluorescence26, at the physiological conditions (pH 7.4), we confirmed that the IC50 values of GL, GA and CBX on human KAT2 were almost the same as those of pH9.5 (7.93 ± 0.17 μM, 9.87 ± 0.32 μM, and 3.34 ± 0.25 μM, respectively). We also showed that these compounds are highly selective for KAT2 among KAT isozymes and compete with its substrate KYN. Furthermore, we predicted complex structures using docking calculations and located GL, GA and CBX on the same surface as the aromatic ring of KYN. These docking structures suggest that the high inhibitory activities of GL and its analogues reflect larger hydrophobic interfaces with KAT2 than with KAT2-KYN. In agreement, the hydrophobic effects of Leu40 and Tyr74 are common among KAT2 inhibitors that are described in previous studies15,16,40,41. Furthermore, Gly39 forms a hydrogen bond with GA and with BFF-12216 and the hydrogen bond between the inhibitor and Tyr142 of the KAT2-GA complex was also formed in the complex of KAT2 and NS-15024. In contrast, the hydrogen bond with Ser77 is unique to docking structures of GL and CBX. Yet the carbohydrate chain of GL, which is located outside of the ligand-binding pocket, is not likely to affect the affinity for KAT2. Because these inhibitors merely alter PLP binding, the risks of side effects may be minimised in comparison with those of PF-04859989 and BFF-122, which inhibit KAT2 by forming covalent adducts with PLP. In addition, compared with S-ESBA and NS-1502, which exhibit reversible inhibitory effects, GL, GA and CBX have potent inhibitory activities. Therefore, we successfully identified three new KAT2 inhibitors that are superior to other KAT2 inhibitors.
The present experiments show that the inhibitory effects of GL, GA and CBX on KAT2 are less potent in mice than in humans (Table 1). These differences may arise from differences in conserved residues among species. Accordingly, in the sequence alignments of mouse, rat and human KAT2 reported by Pellicciari et al., sequence variants in human and rat KAT2 were associated with relative inhibitory activities of S-ESBA38. Our computational docking study shows that the hydrophobic effects of Leu40 in human KAT2 are important for its interactions with GL and its analogues (Table 2). Among residues that participate in binding of human KAT2 to GL and its analogues, Leu40 is substituted to Ser in mouse KAT2. We speculate that this difference undermines the inhibitory effects of GL and its analogues on mouse KAT2.
We also demonstrated that among the four KAT isoenzymes (KAT1–4), GL, GA and CBX selectively inhibit KAT2 (Table 1). Because KYNA is predominantly generated by KAT2 in brain tissues12,13,14, specific inhibitors of KAT2 are eagerly awaited. Rossi, F. et al. solved the crystal structures of human KAT1 and KAT2, and showed important structural differences that could be exploited in the rational design of highly selective KAT2 inhibitors42,43,44,45. In particular, human KAT1 and KAT2 share 15% overall sequence identity, and structural analyses of their active sites revealed striking differences in substrate binding pockets42,45 (for review see43,44). We also demonstrated that hydrophobic interactions between Leu40(A) and Tyr74(A) with GL and its analogues are similar to those of the KAT2 inhibitors reported in previous studies. But we suggest that Pro76(A) and Gln73(A) are additional unique residues that interact with GL and its analogues (Table 2). These residues may be involved in the selective inhibitory effects of GL and its analogues for human KAT2. Although the mechanisms through which GL and its analogues specifically inhibit KAT2 remain unknown, these structural differences between KAT isozymes and the unique residues that interact with GL and its analogues are likely mediators of specificity. Hence, the present observations can be used to inform the rational design of specific and competitive KAT2 inhibitors.
KAT2 inhibitors with therapeutic potential for psychiatric disorders must pass through the blood brain barrier (BBB). CBX failed to pass through the BBB in previous reports46, precluding further consideration. In contrast, GL, which is the main constituent of liquorice and comprises a molecule of GA and two glucuronic acid molecules27,28, is metabolised to GA by microbiota in the intestine following oral administration, and GA is then absorbed into the bloodstream47,48 and through the BBB30. Hence, further studies are warranted to clarify the KAT2 inhibitory effects of GL in animal models of psychiatric diseases.
Liquorice is an ingredient of yokukansan, which is a traditional Japanese Kampo medicine. Yokukansan is effective as a treatment for patients with symptoms of schizophrenia31,32 and in patients with behavioural or psychological symptoms of dementia due to Alzheimer disease and Lewy bodies33,34,35. Yet, yokukansan has multiple ingredients and it is not clear which of these are responsible for the therapeutic effects. Yokukansan reportedly inhibits glutamate release in zinc-deficient rats49 and exerts neuroprotective effects by accommodating dysfuntional glutamate transporters in astrocytes50. The inhibitory effects of yokukansan on KAT2 have not, however, been clarified. Further studies are required to evaluate the involvement of KAT2 inhibition in the neuroprotective and therapeutic effects of yokukansan in patients with psychiatric disorders.
As a pharmacological agent, GL has anti-inflammatory51,52 and antiviral activities53 and inhibits hepatic fibrosis54 and tumour growth55. Following improvements in solubility, Glycyrrhizin salts, such as diammonium glycyrrhizinate and dipotassium glycyrrhizinate, have anti-inflammatory effects that are similar to those of GL56,57. Furthermore, drugs containing GL, such as stronger neo-minophagen C and glycyron tablets (Minophagen Pharmaceutical, Tokyo, Japan), are commercially available and have been used to treat chronic liver disease58,59. Thus, the use of GL as a KAT2 inhibitor for the treatment of schizophrenia represents a drug repositioning process through which new therapeutic uses for existing drugs are discovered.
In conclusion, we demonstrate that GL, GA and CBX are inhibitors of KAT2. These inhibitors were selective and potent, and operated through mechanisms of substrate competition. Thus, our findings suggest that drugs containing GL are excellent candidates for drug repositioning in the treatment of schizophrenia.
Methods
Chemicals
GL, GL dipotassium salt, GA and dimethyl sulfoxide (DMSO) were purchased from FUJIFILM Wako Pure Chemical, Ltd. (Osaka, Japan). CBX was purchased from Abcam (UK). All compounds were dissolved in DMSO.
Enzyme production
All methods were carried out in accordance with relevant guidelines and regulations. The protocol for all animal experiments was approved by the Institutional Animal Care and Use Committee of Fujita Health University. Human KAT1, KAT2 and KAT4 cDNAs were synthesised from human blood peripheral leukocytes total RNA (TaKaRa, Japan) using a ReverTra Ace Kit (Toyobo, Osaka, Japan). Mouse KAT2 cDNA was synthesised from total RNA that was extracted from whole brains of mice using a ReverTra Ace Kit. All cDNAs were amplified using polymerase chain reactions with specific primers.
Amplified cDNAs were cloned into the pFastBac HTC vector (Invitrogen, Carlsbad, CA, USA), which was transformed into Escherichia coli DH5α cells. The pFastBac HTC vector containing the target gene was transformed into E. coli DH10Bac cells and a baculovirus transfer vector was transfected into insect Sf9 cells. Recombinant enzymes were expressed by infection of Sf9 cells with a high-titre baculovirus.
Sf9 cells were pelleted by centrifugation and were then dissolved in 50 mM phosphate buffer (pH 8.0) containing 300 mM NaCl and 10 mM imidazole. After sonication, cell lysates were centrifuged at 10,000 × g for 20 min at 4 °C, and recombinant enzymes in supernatants were added to pre-equilibrated Ni-NTA resin (Qiagen). Enzyme/resin complexes were transferred to columns, were washed with buffer containing 300 mM NaCl and 20 mM imidazole in 50 mM phosphate buffer (pH 8.0), and recombinant enzymes were eluted with buffer containing 300 mM NaCl, 250 mM imidazole and 50 mM phosphate (pH 8.0). Enzyme fractions were pooled based on purity, as determined using sodium dodecyl sulfate polyacrylamide gel electrophoresis, and were then desalted using PD-10 columns (GE Healthcare, UK). Recombinant human KAT3 was purchased from OriGene Technologies, Inc. (USA).
High-throughput screening assays for inhibitors of human KAT2
High-throughput screening assays for inhibitors of human KAT2 were conducted using a microplate fluorescence assay for kynurenine aminotransferase26 with minor modifications. In these assays, KAT2 enzyme activities were measured in black 384-well untreated plates. The human KAT2 reaction mixture (20 μL) contained 10 ng/μL recombinant human KAT2, 1 mM L-KYN, 1 mM α-ketoglutaric acid, 500 μM PLP, 0.005% Tween 20 and 150 mM AMP buffer (pH 9.5), and was added to 384-well plates containing compounds using a Multidrop dispenser (Thermo Fisher Scientific, USA). The compound library comprised about 13,000 compounds from the Drug Discovery Initiative at the University of Tokyo. The compound library includes about 9,600 diverse compounds for pilot screening and about 3,400 known bioactive compounds. All compounds were dissolved and diluted in DMSO to a final concentration of 10 μM. Reaction mixtures were incubated for 2 h at room temperature, and 20 μL aliquots of 300 mM zinc acetate (pH 5.5) were then added directly using a Multidrop dispenser. Fluorescence intensities of KYNA were measured using an ARVO X Multi Label Reader (PerkinElmer, USA) at an excitation wavelength of 340 nm and an emission wavelength of 460 nm. Assay quality was validated by calculating signal–background and Z’ factor. These assays identified approximately 20 candidate KAT2 inhibitors with potent inhibitory activity from about 13,000 compounds. Candidate compounds were validated in quadruplicate KAT2 enzyme activity assays.
Enzyme inhibition and kinetics assays
Inhibitory activities of the identified compounds against KAT1 and KAT2 were measured using the enzyme activity assays described above. KAT1 reaction mixtures (20 μL) contained 10 ng/μL recombinant human KAT1, 1 mM L-KYN, 1 mM sodium pyruvate, 100 μM PLP, 0.005% Tween 20 and inhibitors at various concentrations in 150 mM 2-amino-2-methyl-1-propanol (AMP) buffer (pH 7.4 or 9.5). Human and mouse KAT2 reaction mixtures (20 μL) contained 10 ng/μL recombinant human or mouse KAT2, 1 mM L-KYN, 1 mM α-ketoglutaric acid, 100 μM PLP, 0.005% Tween 20 and inhibitors at various concentrations in 150 mM AMP buffer (pH 9.5). Following the addition of 20 μL aliquots of 300 mM zinc acetate, fluorescence intensities of KYNA were measured using an ARVO X Multi Label Reader (PerkinElmer, USA).
KAT3 and KAT4 enzyme activities were measured using high performance liquid chromatography (HPLC) analyses of reaction products. Briefly, KAT3 reaction mixtures (50 μL) contained 10 ng/μL recombinant human KAT3, 1 mM L-KYN, 1 mM sodium pyruvate, 100 μM PLP, 0.005% Tween 20 and inhibitors at various concentrations in 150 mM AMP buffer (pH 9.5). KAT4 reaction mixtures (50 μL) contained recombinant human KAT4, 1 mM L-KYN, 1 mM α-ketoglutaric acid, 100 μM PLP, 0.005% Tween 20 and inhibitors at various concentrations in 100 mM Tris buffer (pH 7.5). Reaction mixtures were incubated for 1 h at 37 °C and reactions were then stopped by adding 3% perchloric acid at a ratio of 1:1. KYNA contents of reaction mixtures were then determined using HPLC (Shimadzu, Kyoto, Japan) following isocratic elution from a reverse-phase column (TSKgel ODS-100V, 3 μm, 4.6 mm [ID] × 150 mm [L]; Tosoh, Tokyo, Japan) using a mobile phase containing 10 mM sodium acetate with 2% acetonitrile (pH adjusted to 4.5) at flow rate of 0.8 mL/min. KYNA was detected using a fluorescence detector (RF-20Axs) at an excitation wavelength of 344 nm and an emission wavelength of 380 nm.
To determine steady state kinetic parameters for the substrate L-KYN, 50 μL reaction mixtures containing 10 ng/μL recombinant human KAT2, 1 mM α-KG, 100 μM PLP, 0.005% Tween 20, L-KYN at concentrations of 10, 5, 2.5, 1.25, 0.62, 0.31 and 0.15 mM and GL, GA or CBX or PF-04859989 at 4.5, 6.0 and 3.5 μM and 300 nM, respectively. Following incubation for 30 min at 37 °C, reactions were stopped by adding 3% perchloric acid at a 1:1 ratio. KYNA concentrations were measured using the same HPLC method as described above. Double reciprocal plots were used to determine types of inhibition. Competitive inhibition constants (Ki) for GL, GA and CBX were calculated using the global fit formula (GraphPad Prism v6.07)60,61 as follows:
where v = initial velocity, Vmax = maximum velocity, [S] = substrate concentration, Km = Michaelis–Menten constant and [I] = inhibitor concentration. The Ki value of PF-04859989 was calculated using the global fit formula as follows:
where Vmaxinh = maximum enzyme velocity for the concentration of inhibitor. IC50 values were determined by nonlinear curve fitting using GraphPad Prism v6.07. All statistical analyses were performed using GraphPad Prism v6.07.
Docking calculations
Computational docking trials were performed using GOLD 5.6 software under default settings62. The three-dimensional (3D) structure of KAT2, which forms a homo-dimer63,64, was retrieved from the Protein Data Bank (PDB; PDB ID, 2R2N). Because this crystal structure includes the substrate KYN and the coenzyme analogue pyridoxamine 5′-phosphate (PMP), KYN was removed, and PLP was substituted for PMP before calculations. For this substitution, the PLP structure was optimised using Gaussian16 (Gaussian Inc., Wallingford CT, 2016) with B3LYP/6-31 + G(d,p). Missing hydrogen atoms in the PDB structure were computationally added using Hermes (https://www.ccdc.cam.ac.uk/). The centre of KYN in 2R2N was defined as the centre of the ligand-binding site, and the binding site radius was set at 10.0 Å. Ligand structures were optimised at the same calculation level as for PLP.
References
Fujigaki, H., Yamamoto, Y. & Saito, K. L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology 112, 264–274, https://doi.org/10.1016/j.neuropharm.2016.01.011 (2017).
Danysz, W., Fadda, E., Wroblewski, J. T. & Costa, E. Kynurenate and 2-amino-5-phosphonovalerate interact with multiple binding sites of the N-methyl-D-aspartate-sensitive glutamate receptor domain. Neuroscience letters 96, 340–344 (1989).
Stone, T. W. & Darlington, L. G. The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders. British journal of pharmacology 169, 1211–1227, https://doi.org/10.1111/bph.12230 (2013).
Hilmas, C. et al. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. The Journal of neuroscience: the official journal of the Society for Neuroscience 21, 7463-7473 (2001).
Stone, T. W. Kynurenic acid blocks nicotinic synaptic transmission to hippocampal interneurons in young rats. The European journal of neuroscience 25, 2656–2665, https://doi.org/10.1111/j.1460-9568.2007.05540.x (2007).
Alexander, K. S., Wu, H. Q., Schwarcz, R. & Bruno, J. P. Acute elevations of brain kynurenic acid impair cognitive flexibility: normalization by the alpha7 positive modulator galantamine. Psychopharmacology 220, 627–637, https://doi.org/10.1007/s00213-011-2539-2 (2012).
Chess, A. C., Simoni, M. K., Alling, T. E. & Bucci, D. J. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophrenia bulletin 33, 797–804, https://doi.org/10.1093/schbul/sbl033 (2007).
Shepard, P. D., Joy, B., Clerkin, L. & Schwarcz, R. Micromolar brain levels of kynurenic acid are associated with a disruption of auditory sensory gating in the rat. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology 28, 1454–1462, https://doi.org/10.1038/sj.npp.1300188 (2003).
Linderholm, K. R. et al. Increased levels of kynurenine and kynurenic acid in the CSF of patients with schizophrenia. Schizophrenia bulletin 38, 426–432, https://doi.org/10.1093/schbul/sbq086 (2012).
Schwarcz, R. et al. Increased cortical kynurenate content in schizophrenia. Biological psychiatry 50, 521–530 (2001).
Erhardt, S. et al. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neuroscience letters 313, 96–98 (2001).
Guillemin, G. J. et al. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. Journal of neurochemistry 78, 842–853 (2001).
Heyes, M. P., Chen, C. Y., Major, E. O. & Saito, K. Different kynurenine pathway enzymes limit quinolinic acid formation by various human cell types. The Biochemical journal 326(Pt 2), 351–356 (1997).
Guidetti, P., Hoffman, G. E., Melendez-Ferro, M., Albuquerque, E. X. & Schwarcz, R. Astrocytic localization of kynurenine aminotransferase II in the rat brain visualized by immunocytochemistry. Glia 55, 78–92, https://doi.org/10.1002/glia.20432 (2007).
Dounay, A. B. et al. Discovery of Brain-Penetrant, Irreversible Kynurenine Aminotransferase II Inhibitors for Schizophrenia. ACS medicinal chemistry letters 3, 187–192, https://doi.org/10.1021/ml200204m (2012).
Rossi, F. et al. Crystal structure-based selective targeting of the pyridoxal 5′-phosphate dependent enzyme kynurenine aminotransferase II for cognitive enhancement. Journal of medicinal chemistry 53, 5684–5689, https://doi.org/10.1021/jm100464k (2010).
Wu, H. Q. et al. Targeting kynurenine aminotransferase II in psychiatric diseases: promising effects of an orally active enzyme inhibitor. Schizophrenia bulletin 40(Suppl 2), S152–158, https://doi.org/10.1093/schbul/sbt157 (2014).
Bortz, D. M., Wu, H. Q., Schwarcz, R. & Bruno, J. P. Oral administration of a specific kynurenic acid synthesis (KAT II) inhibitor attenuates evoked glutamate release in rat prefrontal cortex. Neuropharmacology 121, 69–78, https://doi.org/10.1016/j.neuropharm.2017.04.023 (2017).
Pellicciari, R. et al. Modulators of the kynurenine pathway of tryptophan metabolism: synthesis and preliminary biological evaluation of (S)-4-(ethylsulfonyl)benzoylalanine, a potent and selective kynurenine aminotransferase II (KAT II) inhibitor. Chem Med Chem 1, 528–531, https://doi.org/10.1002/cmdc.200500095 (2006).
Amori, L. et al. Specific inhibition of kynurenate synthesis enhances extracellular dopamine levels in the rodent striatum. Neuroscience 159, 196–203, https://doi.org/10.1016/j.neuroscience.2008.11.055 (2009).
Zmarowski, A. et al. Astrocyte-derived kynurenic acid modulates basal and evoked cortical acetylcholine release. The European journal of neuroscience 29, 529–538, https://doi.org/10.1111/j.1460-9568.2008.06594.x (2009).
Pocivavsek, A. et al. Fluctuations in endogenous kynurenic acid control hippocampal glutamate and memory. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology 36, 2357–2367, https://doi.org/10.1038/npp.2011.127 (2011).
Kozak, R. et al. Reduction of brain kynurenic acid improves cognitive function. The Journal of neuroscience: the official journal of the Society for Neuroscience 34, 10592–10602, https://doi.org/10.1523/JNEUROSCI.1107-14.2014 (2014).
Hinz, M., Stein, A. & Cole, T. Parkinson’s disease: carbidopa, nausea, and dyskinesia. Clinical pharmacology: advances and applications 6, 189–194, https://doi.org/10.2147/CPAA.S72234 (2014).
Hinz, M., Stein, A. & Cole, T. The Parkinson’s disease death rate: carbidopa and vitamin B6. Clinical pharmacology: advances and applications 6, 161–169, https://doi.org/10.2147/CPAA.S70707 (2014).
Wong, J., Ray, W. J. & Kornilova, A. Y. Development of a microplate fluorescence assay for kynurenine aminotransferase. Analytical biochemistry 409, 183–188, https://doi.org/10.1016/j.ab.2010.10.037 (2011).
Sharifzadeh, M. et al. A time course analysis of systemic administration of aqueous licorice extract on spatial memory retention in rats. Planta medica 74, 485–490, https://doi.org/10.1055/s-2008-1074494 (2008).
Li, X. L. & Zhou, A. G. Evaluation of the immunity activity of glycyrrhizin in AR mice. Molecules 17, 716–727, https://doi.org/10.3390/molecules17010716 (2012).
Davidson, J. S. & Baumgarten, I. M. Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap-junctional intercellular communication. Structure-activity relationships. The Journal of pharmacology and experimental therapeutics 246, 1104–1107 (1988).
Tabuchi, M., Imamura, S., Kawakami, Z., Ikarashi, Y. & Kase, Y. The blood-brain barrier permeability of 18beta-glycyrrhetinic acid, a major metabolite of glycyrrhizin in Glycyrrhiza root, a constituent of the traditional Japanese medicine yokukansan. Cellular and molecular neurobiology 32, 1139–1146, https://doi.org/10.1007/s10571-012-9839-x (2012).
Miyaoka, T. et al. Yokukansan (TJ-54) for treatment of very-late-onset schizophrenia-like psychosis: an open-label study. Phytomedicine: international journal of phytotherapy and phytopharmacology 20, 654–658, https://doi.org/10.1016/j.phymed.2013.01.007 (2013).
Miyaoka, T. et al. Yi-gan san as adjunctive therapy for treatment-resistant schizophrenia: an open-label study. Clinical neuropharmacology 32, 6–9, https://doi.org/10.1097/WNF.0b013e31817e08c3 (2009).
Monji, A. et al. Effect of yokukansan on the behavioral and psychological symptoms of dementia in elderly patients with Alzheimer’s disease. Progress in neuro-psychopharmacology & biological psychiatry 33, 308–311, https://doi.org/10.1016/j.pnpbp.2008.12.008 (2009).
Mizukami, K. et al. A randomized cross-over study of a traditional Japanese medicine (kampo), yokukansan, in the treatment of the behavioural and psychological symptoms of dementia. The international journal of neuropsychopharmacology 12, 191–199, https://doi.org/10.1017/S146114570800970X (2009).
Iwasaki, K. et al. A randomized, observer-blind, controlled trial of the traditional Chinese medicine Yi-Gan San for improvement of behavioral and psychological symptoms and activities of daily living in dementia patients. The Journal of clinical psychiatry 66, 248–252 (2005).
Pocivavsek, A., Elmer, G. I. & Schwarcz, R. Inhibition of kynurenine aminotransferase II attenuates hippocampus-dependent memory deficit in adult rats treated prenatally with kynurenine. Hippocampus 29, 73–77, https://doi.org/10.1002/hipo.23040 (2019).
Potter, M. C. et al. Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology 35, 1734–1742, https://doi.org/10.1038/npp.2010.39 (2010).
Pellicciari, R. et al. Sequence variants in kynurenine aminotransferase II (KAT II) orthologs determine different potencies of the inhibitor S-ESBA. Chem Med Chem 3, 1199–1202, https://doi.org/10.1002/cmdc.200800109 (2008).
Nematollahi, A., Sun, G., Jayawickrama, G. S., Hanrahan, J. R. & Church, W. B. Study of the Activity and Possible Mechanism of Action of a Reversible Inhibitor of Recombinant Human KAT-2: A Promising Lead in Neurodegenerative and Cognitive Disorders. Molecules 21, https://doi.org/10.3390/molecules21070856 (2016).
Dounay, A. B. et al. PF-04859989 as a template for structure-based drug design: identification of new pyrazole series of irreversible KAT II inhibitors with improved lipophilic efficiency. Bioorganic & medicinal chemistry letters 23, 1961–1966, https://doi.org/10.1016/j.bmcl.2013.02.039 (2013).
Nematollahi, A., Sun, G., Jayawickrama, G. S., Hanrahan, J. R. & Church, W. B. Crystal structure and mechanistic analysis of a novel human kynurenine aminotransferase-2 reversible inhibitor. Medicinal chemistry research: an international journal for rapid communications on design and mechanisms of action of biologically active agents 26, 2514–2519 (2017).
Rossi, F., Garavaglia, S., Montalbano, V., Walsh, M. A. & Rizzi, M. Crystal structure of human kynurenine aminotransferase II, a drug target for the treatment of schizophrenia. The Journal of biological chemistry 283, 3559–3566, https://doi.org/10.1074/jbc.M707925200 (2008).
Rossi, F., Miggiano, R., Ferraris, D. M. & Rizzi, M. The Synthesis of Kynurenic Acid in Mammals: An Updated Kynurenine Aminotransferase Structural KATalogue. Frontiers in molecular biosciences 6, 7, https://doi.org/10.3389/fmolb.2019.00007 (2019).
Rossi, F., Schwarcz, R. & Rizzi, M. Curiosity to kill the KAT (kynurenine aminotransferase): structural insights into brain kynurenic acid synthesis. Current opinion in structural biology 18, 748–755, https://doi.org/10.1016/j.sbi.2008.09.009 (2008).
Rossi, F., Han, Q., Li, J. & Rizzi, M. Crystal structure of human kynurenine aminotransferase I. The Journal of biological chemistry 279, 50214–50220, https://doi.org/10.1074/jbc.M409291200 (2004).
Leshchenko, Y., Likhodii, S., Yue, W., Burnham, W. M. & Perez Velazquez, J. L. Carbenoxolone does not cross the blood brain barrier: an HPLC study. BMC neuroscience 7, 3, https://doi.org/10.1186/1471-2202-7-3 (2006).
Takeda, S. et al. Bioavailability study of glycyrrhetic acid after oral administration of glycyrrhizin in rats; relevance to the intestinal bacterial hydrolysis. The Journal of pharmacy and pharmacology 48, 902–905 (1996).
Hattori, M. et al. Metabolism of glycyrrhizin by human intestinal flora. II. Isolation and characterization of human intestinal bacteria capable of metabolizing glycyrrhizin and related compounds. Chemical & pharmaceutical bulletin 33, 210–217 (1985).
Takeda, A., Itoh, H., Tamano, H., Yuzurihara, M. & Oku, N. Suppressive effect of Yokukansan on excessive release of glutamate and aspartate in the hippocampus of zinc-deficient rats. Nutritional neuroscience 11, 41–46, https://doi.org/10.1179/147683008X301414 (2008).
Kawakami, Z. et al. Neuroprotective effects of yokukansan, a traditional Japanese medicine, on glutamate-mediated excitotoxicity in cultured cells. Neuroscience 159, 1397–1407, https://doi.org/10.1016/j.neuroscience.2009.02.004 (2009).
Honda, H. et al. Glycyrrhizin and isoliquiritigenin suppress the LPS sensor toll-like receptor 4/MD-2 complex signaling in a different manner. Journal of leukocyte biology 91, 967–976, https://doi.org/10.1189/jlb.0112038 (2012).
Schrofelbauer, B. et al. Glycyrrhizin, the main active compound in liquorice, attenuates pro-inflammatory responses by interfering with membrane-dependent receptor signalling. The Biochemical journal 421, 473–482, https://doi.org/10.1042/BJ20082416 (2009).
Cinatl, J. et al. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 361, 2045–2046 (2003).
Tu, C. T. et al. Glycyrrhizin regulates CD4+ T cell response during liver fibrogenesis via JNK, ERK and PI3K/AKT pathway. International immunopharmacology 14, 410–421, https://doi.org/10.1016/j.intimp.2012.08.013 (2012).
Shiota, G. et al. Inhibition of hepatocellular carcinoma by glycyrrhizin in diethylnitrosamine-treated mice. Carcinogenesis 20, 59–63 (1999).
Zhu, X. et al. Diammonium glycyrrhizinate upregulates PGC-1alpha and protects against Abeta1-42-induced neurotoxicity. PloS one 7, e35823, https://doi.org/10.1371/journal.pone.0035823 (2012).
Vitali, R. et al. Dipotassium Glycyrrhizate Inhibits HMGB1-Dependent Inflammation and Ameliorates Colitis in Mice. PloS one 8, e66527, https://doi.org/10.1371/journal.pone.0066527 (2013).
Zhang, L. & Wang, B. Randomized clinical trial with two doses (100 and 40 ml) of Stronger Neo-Minophagen C in Chinese patients with chronic hepatitis B. Hepatology research: the official journal of the Japan Society of Hepatology 24, 220 (2002).
Suzuki, T., Tsukahara, M., Akasaka, Y. & Inoue, H. A highly sensitive LC-MS/MS method for simultaneous determination of glycyrrhizin and its active metabolite glycyrrhetinic acid: Application to a human pharmacokinetic study after oral administration. Biomedical chromatography: BMC 31, https://doi.org/10.1002/bmc.4032 (2017).
Cianchetta, G. et al. Mechanism of Inhibition of Novel Tryptophan Hydroxylase Inhibitors Revealed by Co-crystal Structures and Kinetic Analysis. Current chemical genomics 4, 19–26, https://doi.org/10.2174/1875397301004010019 (2010).
Mathieu, Y. et al. Characterization of a Phanerochaete chrysosporium glutathione transferase reveals a novel structural and functional class with ligandin properties. The Journal of biological chemistry 287, 39001–39011, https://doi.org/10.1074/jbc.M112.402776 (2012).
Jones, G., Willett, P., Glen, R. C., Leach, A. R. & Taylor, R. Development and validation of a genetic algorithm for flexible docking. Journal of molecular biology 267, 727–748, https://doi.org/10.1006/jmbi.1996.0897 (1997).
Han, Q., Robinson, H. & Li, J. Crystal structure of human kynurenine aminotransferase II. The Journal of biological chemistry 283, 3567–3573, https://doi.org/10.1074/jbc.M708358200 (2008).
Han, Q., Cai, T., Tagle, D. A. & Li, J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cellular and molecular life sciences: CMLS 67, 353–368, https://doi.org/10.1007/s00018-009-0166-4 (2010).
Acknowledgements
This research was partially supported by the Platform Project for Supporting Drug Discovery and Life Science Research from AMED under Grant Number JP17am0101086. The work of the authors was partially supported by JSPS KAKENHI Grant Numbers 15H03086 (K.S.), 16K10195 (A.M.), 17K01969 (H.F.), 17H04252 (T.N.), 17H07222 (Y.Y.), 17K08257 (A.O.) and 18K15377 (K.K.). We would also like to thank Dr. R. Imamura for her excellent technical support.
Author information
Authors and Affiliations
Contributions
Y. Yoshida, H. Fujigaki, K. Kato, K. Yamazaki and S. Fujigaki performed the experiments. Y. Yoshida, H. Fujigaki, K. Kato and A. Oda wrote and T. Nabeshima revised the manuscript. Y. Yoshida., H. Fujigaki, K. Kunisawa, Y. Yamamoto, A. Mouri, A. Oda, T. Nabeshima and K. Saito designed and directed the project.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoshida, Y., Fujigaki, H., Kato, K. et al. Selective and competitive inhibition of kynurenine aminotransferase 2 by glycyrrhizic acid and its analogues. Sci Rep 9, 10243 (2019). https://doi.org/10.1038/s41598-019-46666-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46666-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
