
Abstract
Isolated human islets do not always meet the quality standards required for transplant survival and reliable functional in vitro studies. The formation of pseudoislets, i.e. the reaggregation of a defined number of islet cells after dissociation, improves insulin secretion. We present a simple method of pseudoislet formation from human islet cells and assess the transcriptome and function of isolated human islets and pseudoislets from the same organ donors. Following pseudoislet formation, insulin content/DNA and mRNA/RPS13 resembled that of islets. In pseudoislets, glucose-stimulated insulin secretion (GSIS) was significantly higher (8–13-fold) than in islets (2–4-fold). GSIS of pseudoislets was partly inhibited by the glucagon-like peptide-1 receptor (GLP-1R) antagonist exendin-9. The stimulatory effects of palmitate and forskolin at 12 mM glucose were also significantly higher in pseudoislets than in islets. Further analysis of pseudoislets revealed that regulation of secretion and insulin and glucagon content was maintained over a longer culture period (6–14 d). While adrenaline inhibited GSIS, adrenaline together with palmitate stimulated glucagon secretion 2-fold at low glucose, an effect suppressed by high glucose. Transcriptome analysis revealed that, unlike islets, pseudoislets were deprived of exocrine and endothelial cells. In conclusion, pseudoislet formation restores functional integrity of human islet cells and allows long-term in vitro testing.
Similar content being viewed by others
Introduction
Up to now, isolated human islets have been used for transplantation with only limited success1. Besides transplanted islets being subjected to immunological attacks, other undesired adverse effects also impact islet function2,3. Firstly, due to prolonged hypoxia, necrotic cell death occurs in the centre region of large isolated islets4,5. Secondly, following transplantation, a defective revascularization may hinder hormone dissemination6. Thirdly, the age and metabolic memory of human islets could have an impact on islet function after transplantation7. Jean-Claude Henquin’s careful evaluation of glucose-responsiveness of isolated human islets using a perifusion setting revealed no dependence of glucose-stimulated insulin secretion (GSIS) on donor age and islet size8. While an elevated body mass index (BMI) of the donor correlated with higher insulin content and secretion, the major adverse effect was the contamination of the islet preparation with exocrine tissue. Not only an incomplete purification of islets from exocrine tissue but also the dense packing of islets into a capsule for transplantation may affect islet function9. The use of reaggregated pseudoislets for transplantation was recently shown to result in improved glucose homeostasis10. Upon transplantation, revascularization remains defective in islets but is initiated in reaggregated pseudoislets11. Dissociated isolated islet cells are less glucose responsive than islet cells in clusters12. However, human islet cells exhibit improved glucose-responsiveness when attached to a matrix that enables them to spread13. The role of matrix and spreading for endocrine cell survival and function of human islet transplants has already been summarized elsewhere14,15. Another approach to improve glucose-responsiveness is the formation of pseudoislets11. In pseudoislets, hypoxia-damaged cells are eliminated and the superfusion with the stimulatory solution is ameliorated. Indeed, the limited oxygenation of the core of large islets favours hypoxia-induced apoptotic cell death and dysfunction5,16.
The fact that small islets display a better GSIS than large islets has been documented using isolated islets from rat and mouse17,18,19,20. Human islets vary also in their size and cell composition21,22. A higher percentage of glucagon-producing cells within the islet improves the glucose responsiveness23,24. By contrast, somatostatin exerts inhibitory effects on insulin and glucagon secretion25,26. A uniform distribution of α-, β- and δ-cells therefore reduces islet heterogeneity and variations for in vitro testing of islet function.
This study was initiated to evaluate the improvement of GSIS upon the formation of pseudoislets. This entailed comparing the glucose-responsiveness of the isolated human islets prior to dissociation with that of pseudoislets originating from the same organ donors. In addition, the transcriptomes of islets and pseudoislets were compared to determine whether differential expression underlies functional improvement. The results reveal a specific restitution of GSIS, together with a higher expression of β-cell markers, glucose transporter type 2 (GLUT-2), paired box gene 4 (PAX4) and islet-amyloid-polypeptide (IAPP) and a simultaneous reduction of acinar, ductal and endocrine cell markers. The pronounced inhibition of GSIS by adrenaline in the absence of endothelial cells suggests that adrenaline exerts a direct effect on β-cells and that insulin is released by regulated exocytosis. Furthermore, we ascertained that glucagon secretion is highest at low glucose in the presence of both palmitate and adrenaline, a stimulation inhibited by high glucose.
Results
Functional comparison of human islets and pseudoislets
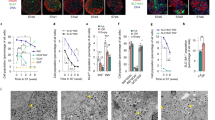
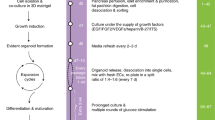
The ultimate functional test for β-cells is GSIS. Using a standard test of GSIS – static incubation for 1 h and increasing glucose concentrations – a high variation of both basal secretion and glucose responsiveness was observed in isolated human islets from non-diabetic organ donors (Suppl. Fig. 1A,B and Table 1). The mean glucose responsiveness did not become significant between 2.8 and 12 or 20 mM glucose (Suppl. Fig. 1C). In only 50% of the preparations did glucose augment insulin secretion more than 2-fold. Hypoxia prior to and during isolation and a prolonged shipping time is known to be detrimental to the islet core and function4,5. We therefore sought a method which improves islet cell function, ameliorates GSIS and allows long term testing. To this end, the human islets were dissociated into single cells and reaggregated to pseudoislets using the hanging drop method (Fig. 1A). To estimate the cell number, the DNA contents of islets and pseudoislets were measured (Fig. 1B). While the DNA content of islets was highly variable (5–20 ng), the DNA content of the pseudoislets was less variable (5–6 ng), reflecting a uniform pseudoislet size (Fig. 1B). The mean insulin content of islets was also very variable between the donors (Suppl. Fig. 1D). The insulin content per cell (i.e. DNA) varied by 20–40% between islets and pseudoislets of the same donor, indicating that pseudoislet formation did not affect insulin content of β-cells (Suppl. Fig. 1E,F). Pseudoislet formation initiated from 1000, 2000 or 4000 cells resulted in higher glucose responsiveness (8–13-fold stimulation) than non-dispersed islets cultured overnight (2-fold stimulation) from the same donor (Fig. 1C,D). This improvement in β-cell function is the result of lower basal secretion as well as of higher glucose-stimulation and was observed in every pseudoislet preparation (Fig. 1C–N). When non-dispersed isolated islets were cultured for the same length of time as the pseudoislets, the difference in GSIS remained significant (Suppl. Fig. 1G).

Reaggregation of human islet cells to pseudoislets specifically improves GSIS. (A) Experimental design of pseudoislet formation. Isolated human islets were cultured overnight. Non-dispersed islets were partly used for insulin secretion. The others (1000–2000 islets) were digested into single cells using trypsin. After digestion, a defined number of single cells were reaggregated in hanging drops of 20 µl over 3 days. After 2 days or longer culture in non-adhesive multi-well plates, the pseudoislets were used in the experiments. (B) DNA content of single islets and pseudoislets (formed from 2000 cells and cultured for 2 days) from the same donors (n = 2) were measured as described under Materials and Methods. (C,D) Comparison of GSIS of islets and pseudoislets reaggregated from 1000, 2000 and 4000 cells as indicated expressed as (C) fold-stimulation (12 mM glucose over 2.8 mM glucose). (D) % of insulin content (white bars, 2.8 mM glucose and grey bars, 12 mM glucose). Results are expressed as means + s.e.m. of n = 5 independent preparations. (E–N) GSIS of individual preparations of islets (black signs) and pseudoislets (red signs) expressed as (E–I) fold-stimulation and (J–N) % of insulin content. *p < 0.05, ***p < 0.0002 denotes significant GSIS; #p < 0.05 denotes significant difference between secretion at 12 mM glucose of islets and the respective pseudoislet. §p < 0.05 denotes significance to secretion of islets at 2.8 mM glucose. €p < 0.05 significance between secretion at 12 mM glucose of 1000-cells and 4000-cells pseudoislets.
In addition to glucose, palmitate-stimulated insulin release was more efficient in pseudoislets than in islets at 12 mM glucose (€p < 0.05; Fig. 2A). At 2.8 mM glucose, palmitate augmented secretion in both islets and pseudoislets (Fig. 2A). Forskolin evoked a 2-fold enhancement of GSIS in islets and pseudoislets. Again, secretion was significantly higher in pseudoislets than in islets at 12 mM glucose (€p < 0.05, Fig. 2A). This effect was not mimicked by exendin-4 (Fig. 2B,C). However, the GLP-1R antagonist exendin-9 reduced GSIS by 50% in pseudoislets, suggesting that, under glucose stimulation, GLP-1R was already activated (Fig. 2C). These results indicate that pseudoislet formation improves GSIS which is partly antagonised by exendin-9, a GLP-1R inhibitor.

FFAR1 and GLP-1R mediated augmentation of GSIS. Isolated islets and pseudoislets (reaggregated from 2000 cells) were prepared, cultured for 2 days and incubated as described under Materials and Methods. Insulin secretion (A,B) of non-dispersed isolated islets (overnight culture) and (A,C) of the pseudoislets was stimulated as indicated. Results are expressed as means + s.e.m. of n = 3–5 independent preparations. §p < 0.05 denotes significance to 2.8 mM glucose; #p < 0.05 denotes significance to 12 mM glucose and &p < 0.05 denotes significance between the two groups as indicated. €p < 0.05 denotes significant difference between pseudoislets and islets at the same condition. White bars represent secretion at 2.8 mM glucose, grey bars at 12 mM glucose. Abbreviations used: Glc (glucose), Pal (palmitate), Fors (forskolin), Ex-4 (exendin-4), Ex-9 (exendin-9).
Receptor effects after long term culture of pseudoislets: regulation of insulin and glucagon secretion by adrenaline
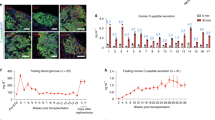
A stable functional integrity of pseudoislets over a period of weeks is a prerequisite for subchronic tests, such as toxicological testing. Insulin and glucagon content did not change during 8 days of culture (19.6 ± 2.9 ng and 19.8 ± 3.6 ng of insulin n = 5, 1.8 ± 0.3 ng and 1.5 ± 0.2 ng of glucagon n = 3 at 6 days and 14 days of culture, respectively). Glucose- and palmitate-dependent stimulation of insulin secretion was maintained in pseudoislets, even after an additional 8-days culture period under standard conditions (Fig. 3A). Secretion occurred in a regulated manner and receptors were functionally expressed in pseudoislets. This was endorsed by the pronounced inhibition of insulin secretion by adrenaline. Both, glucose- and palmitate-stimulated insulin secretion were significantly inhibited by adrenaline. Interestingly, adrenaline augmented glucagon secretion at low glucose, an effect which was more pronounced in the presence of palmitate (Fig. 3B). High glucose prevented the stimulatory effect of adrenaline on glucagon secretion. These results indicate that adrenaline is a potent regulator not only of insulin but also of glucagon secretion in human endocrine cells.

Long term functional integrity of pseudoislets: Opposing effects of glucose and adrenaline on insulin and glucagon secretion. Pseudoislets were cultured and incubated with test substances as described under Materials and Methods. (A) Insulin and (B) glucagon were measured in the same samples and are expressed as means + s.e.m. of n = 5 independent preparations. §p < 0.05 denotes significance to 2.8 mM glucose, #p < 0.05 significance to 12 mM glucose and &p < 0.05 significance to 2.8 mM glucose of the respective group containing the same test substances. 2.8 mM glucose (white bars), 12 mM glucose (grey bars). Abbreviations used: Glc (glucose), Adr (adrenaline), Ex-4 (exendin-4), Pal (palmitate).
Differential expression of β-cell specific genes in pseudoislets
Transcriptome analyses of islets and pseudoislets were performed to determine the functional improvement observed in pseudoislets. This revealed a higher abundance of 198 mRNAs and a lower abundance of 367 mRNAs in pseudoislets than in islets from the same donors (2-fold change, Padj < 0.05; Suppl. Table 1). The comparatively high levels of insulin mRNA in islets (HI) and pseudoislets (PI) were confirmed by reverse transcriptase polymerase chain reaction (RT-PCR; Fig. 4A,E). The fact that islets and pseudoislets have similar amounts of insulin mRNA facilitates a comparison between the two with regard to the expression of other β-cell-specific genes. The mRNA levels of other β-cell-specific genes, in particular IAPP and PAX4, were significantly higher in pseudoislets than in islets, whereas mRNA levels of MAF BZIP transcription factor A (MAFA) and neuronal differentiation 1 (NEUROD1) remained unchanged (Fig. 4A). The augmented expression of GLUT-2 (SLC2A2), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 (PFKFB2) and fatty acid receptor 1 (FFAR1) in pseudoislets could explain the increased responsiveness to glucose and palmitate, respectively (Fig. 4B–D). Other mRNA levels of proteins involved in GSIS such as glucokinase (GCK), ATP binding cassette subfamily C member 8 (SUR1, ABCC8), potassium voltage gated channel subfamily J member 11 (KCNJ11) and calcium voltage-gated channel subunit alpha1A (CACNA1A) as well as GLP1R did not differ in the two preparations. In addition, relative mRNA levels of inhibitory receptors, such as the α2-adrenergic receptor (ADRA2A) and somatostatin receptors (SSTR1 and SSTR3), were more abundant in pseudoislets than in islets (Fig. 4C). Other than higher mRNA levels of some of the enzymes involved in glucose metabolism, 2,6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) and insulin processing enzyme proprotein convertase subtilisin/Kexin type 1 (PCSK1), mRNA levels of the other endocrine cell markers remained unchanged (Fig. 4 and Table 2).

Transcriptome analyses revealed higher mRNA abundance of proteins expressed in β-cells in human pseudoislets than in isolated human islets from the same donors. (A–D) Islet (HI) and pseudoislet reaggregated from 2000 cells (PI) mRNA levels were measured by RNAseq as described under Materials and Methods. The data of 4 different donors are presented as log2 reads. (E) Relative mRNA levels of INS, GCG, SST, PDX1 and FFAR1 in islets and pseudoislets were measured by RT-PCR. Asterisks denote significant differences between HI and PI, (A–D) padj < 0.05, R package; (E) t-test.
Since insulin (INS), glucagon (GCG) and somatostatin (SST) mRNAs are highly abundant, the mRNA levels were also estimated by RT-PCR (Fig. 4E). Interestingly, GCG mRNA levels were significantly higher in pseudoislets than in islets. The mRNA levels of proteins related to apoptosis and inflammation as well as the components of extracellular matrix (ECM) and endothelial cell markers were less abundant in pseudoislets than in islets (Table 2). As anticipated, pseudoislets were depleted of mRNAs of exocrine and ductal cell markers, indicative of the efficient purification of pseudoislets from contaminating exocrine tissue (Suppl. Table 2).
In conclusion, the pseudoislets prepared from adult human islets represent a purified and functional cell system suitable for long term in vitro studies.
Discussion
Functional integrity of isolated human islets from organ donors is a major concern not only for clinical but also for scientific purposes. One problem of organ donors is the advanced age and organ fibrosis which have an impact on organ function. This study presents evidence that the preparation of pseudoislets allows selection of highly functional endocrine cells from organ donor islets. Most significantly, unlike for isolated islets, the regulation of insulin secretion was amended. In parallel, relative mRNA levels of β-cell markers were significantly higher in pseudoislets than in the originating islets. It is well known that hypoxia induce islet damage, particularly in the core of larger islets4,5. These damaged cells are removed upon dissociation and reaggregation of intact islet cells. Cell death may be one reason, why basal secretion is higher and GSIS lower in islets than in pseudoislets. Following isolation, human islets can often only be used after a prolonged shipping period, causing further damage to the islet preparation. Here, we present a simple method for the preparation of pseudoislets with functional integrity, even from human islets without proper GSIS.
Our results further suggest that pseudoislets reaggregated from 1000-cells respond better to glucose than pseudoislets from 4000-cells (Fig. 1D). It has already been observed that smaller islets have a functional superiority over larger ones18. Interestingly, the size of neighbouring islets within the human pancreas is highly variable and small islets prevail. However, the overall β-cell mass is generally determined by large islets20,27. Of note, the isolation procedure is selective for bigger islets, while small islets are often digested and therefore lost. We therefore speculate that in vivo within the pancreatic tissue the small islets are relevant for islet function, whereas large islets predominantly account for β-cell mass. New data suggest that early stages in the development of diabetes are accompanied by dysfunctional hormone secretion leading to hyperglycemic episodes, albeit not by a decline of β-cell mass11. This observation attaches only minor importance to the reduction of β-cell mass in the development of type 2 diabetes mellitus (T2DM).
The preparation of pseudoislets not only enables us to generate spheroids identical in size and functional response and, therefore, useful for in vitro studies, but also to control the composition of pseudoislets, i.e. the ratio of β-:α-:δ-cells28 and the addition of other cell types such as endothelial cells, smooth muscle cells, precursor cells and components of the extracellular matrix. A standardized functional test, i.e. GSIS, is both simple and sensitive on account of its dependence on intact metabolism. In pseudoislets, GSIS and insulin content remain stable over 8-days cultures. This renders them useful for long-term toxicological studies, e.g. for the identification of drug-induced dysfunctions and structural alterations of native pancreatic endocrine cells. The pseudoislet preparation may also be suitable to examine the capacity of human β-cell to synthesize sufficient insulin upon repeated stimulation. The use of human pseudoislets has therefore an advantage over the use of isolated native human islets which is hampered by a highly variable cell composition and necrotic islet core, and also over cell lines which have an unphysiological high proliferation rate.
In contrast to published observations, the activation of GLP1R by exendin-4 did not enhance GSIS in islets and pseudoislets29. Interestingly, in pseudoislets improved GSIS was inhibited by 50% by exendin-9, an antagonist of GLP-1R. This observation suggests that GLP-1R signalling is already activated in pseudoislets. The importance of GLP-1R for GSIS has already been demonstrated with mouse islets30. Using in situ perfusion of mouse pancreas, proper GSIS was shown to depend on the paracrine effects of glucagon which are exerted not only through glucagon receptor but also through GLP-1R31. Here, our data suggest that a similar mechanism may apply to human islet cell interactions, and that glucagon released from α-cells activated β-cell GLP-1R and contributes to the improvement of GSIS in pseudoislets compared to islets. In humans, the main site of action of GLP-1 that improves glucose homeostasis is not completely understood32. The concentration of GLP-1 that augments GSIS in vitro is much higher than the concentration in blood. In contrast to exendin-4, forskolin augmented GSIS 2-fold in both preparations. The direct stimulation of adenylyl cyclase by forskolin leads firstly to an excessive augmentation of cellular cAMP levels and, secondly, bypasses receptor regulation of adenylyl cyclase33. Not only is the enzyme activity stimulated by a variety of different receptors, it is also potently inhibited through receptors present in β-cells, such as adrenoceptors and somatostatin receptors34.
The direct inhibitory effect of adrenaline on insulin secretion has been well studied in rodents35,36. In humans, an inhibitory action through α2-adrenoceptor (ADRA2) activation has been documented, although sympathetic nerve endings have been found only in association with endothelial cells37,38. Of note, the receptor is activated both by sympathetic release of noradrenaline and by humoral action of adrenaline. Our RNAseq data suggest that ADRA2A is the main receptor subtype expressed in human islet cells. ADRA2C and ADRA2B mRNA levels were much lower than ADRA2A (baseMeans were 1030 (DESeq2 normalized counts) for ADRA2A, 2.5 for ADRA2B, 45 for ADRA2C). Single islet cell analysis suggests that ADRA2A is expressed mainly in β-cells39.
Adrenaline not only suppressed GSIS but also increased glucagon secretion at low, 2.8 mM glucose, an effect accentuated by palmitate. The stimulatory effect of adrenaline on α-cells has already been described using mouse islets and purified rat α-cells40,41,42. The levels of fatty acids and adrenaline in blood are at their highest during fasting and stress, conditions dominated by glucagon-induced mobilisation of glucose43. Beta-1 adrenergic receptor (ADRB1) mRNA was found exclusively in α-cells, while ADRB2 in α- as well as in a number of other endocrine islet cells39. FFAR1 is expressed in β- and α-cells. Our results suggest that adrenaline is an important regulator of glucagon secretion and that its stimulatory effect is inhibited by high glucose. The effect of glucose on glucagon release seems to be insulin independent since insulin secretion is suppressed in the presence of adrenaline (Fig. 3A,B, compare the last two columns).
Islet transplantation is mainly required for type 1 diabetes mellitus (T1DM) therapy1. The major concern with regard to the transplanted islets is the autoimmune destruction of β-cells. A certain degree of protection of transplanted islets is effected by encapsulation of the transplant9. Such an approach, however, leads to a tight packing of islets which, in turn, may not be conducive to islet function and survival. Revascularization, an approach with which transplant survival can be improved44, was recently observed to be suppressed in intact islets due to vessel debris but improved in reaggregated small pseudoislets6. In this sense, small reaggregates survive thanks to a better oxygenation and perfusion but also due to a better revascularization prognosis.
In conclusion, using pseudoislets reaggregated from isolated human islet cells, we show a GLP1R-dependent improvement of GSIS and identify adrenaline not only as a potent inhibitor of GSIS but also as an activator of glucagon secretion. Pseudoislets are suitable for functional studies of native human endocrine cells. They are a promising tool for toxicological studies to investigate drug-induced functional and structural adverse effects and may also play an important role in future transplantations.
Materials and Methods
Human islet cell isolation and pseudoislet preparation
The human islets used in this study were procured through the European Consortium for Islet Transplantation (ECIT Centers in Geneva, Lille and Milano, JDRF award 31-2008-416 for basic research program) or purchased from Tebu-Bio (Offenbach, Germany). The donor characteristics are provided in Table 1. Donors gave informed consent for the use of their islets preparations in scientific research. Ethical approval for the use and for the procedures and protocols involved in handling of human islets isolated from organ donors was obtained from the Ethics Commission of the Medical Faculty of the Eberhard-Karls-University and the University Hospital of Tübingen (533/2010BO2 and 098/2017BO1). All experiments and methods involving human islets and islet cell aggregates were performed in accordance with the above mentioned approvals, guidelines and regulations.
To reduce exocrine contamination, the islets were cleaned immediately upon arrival by hand picking under a stereo dissecting microscope in Hank’s balanced salt solution supplemented by 0.25% bovine serum albumin (BSA). After an overnight culture in CMRL1066 medium containing (in mM): 5 glucose, 10 Hepes, 2 L-glutamine supplemented with 10% (v/v) fetal calf serum (FCS, Serva, Heidelberg, Germany) and antibiotics (100 U/ml), the islets were dissociated into single cells using trypsin (42.5 U/mL) in phosphate buffered saline containing 2% (w/v) ethylenediaminetetraacetic acid (EDTA-PBS) and gentle shaking. Following dissociation, cell viability (>95%) was assessed by trypan blue staining. Drops (20 µl of medium) containing 1000, 2000 or 4000 cells were placed onto the top of a petri dish and cultured as hanging drops for 3 days (Fig. 1). Medium was added to the bottom of the petri dishes to avoid drying out. The cells reaggregated within the hanging drop to form one pseudoislet. Each pseudoislet was then placed into a non-adhesive well of a 24-well-plate containing 0.5 ml medium and cultured for another 2 days. Pseudoislets purchased from InSphero (InSphero AG, Schlieren, Switzerland) were cultured for 6 days and 14 days before functional testing (Fig. 3).
Measurement of insulin and glucagon secretion
Isolated islets and pseudoislets were tested in parallel for insulin secretion. The islets or pseudoislets were carefully washed and preincubated for 1 h in modified Krebs-Ringer buffer (KRB) containing (in mM): 135 NaCl, 4.8 KCl, 2.6 CaCl2, 1.17 KH2PO4, 1.18 MgSO4, 5 NaHCO3, 10 HEPES, 2.8 glucose and 0.5% BSA.
Thereafter, isolated islets (5 islets/0.5 ml) and pseudoislets (1 pseudoislet/0.1 ml) were incubated for 1 h in fresh KRB containing test substances at appropriate concentrations. Secreted insulin or glucagon in the supernatant and insulin or glucagon content after extraction in acid ethanol (1.5% (v/v) HCl in 75% (v/v) ethanol) was measured using a radioimmunoassay (Millipore, USA) or a sensitive enzyme-linked immunosorbent assay (ELISA; Mercodia, Sweden).
DNA measurements
DNA of samples containing 10 islets or 10 pseudoislets was extracted using QIAamp DNA-Mico Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions. DNA concentration was measured by fluorescence using the Qubit® dsDNA HS Assay Kit and Qubit® 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, U.S.).
Transcriptome profiling
Aliquots of cultured human islets and their respective pseudoislets were used for RNA extraction using NucleoSpin® RNA XS (Macherey-Nagel, Düren, Germany). RNA integrity (RIN ≥ 9) was measured with Bioanalyzer 2100 (Agilent Technologies). Sequencing was performed as described previously at the Center for Molecular and Cellular Bioengineering (Andreas Dahl, CMCB, Technical University Dresden, Germany)45. In short, mRNA was isolated from 120 ng RNA by poly-dT enrichment using the NEBNext Poly(A) mRNA Magnetic Isolation Module. After fragmentation, the samples were subjected to the workflow for strand-specific RNASeq library preparation (Ultra Directional RNA Library Prep II, NEB) and 75 bp single read sequencing was performed on Illumina NextSeq500. After sequencing, FastQC was used to perform quality control. Differential expression between islets and pseudoislets was tested with DESeq R package (v.1.10.1). The complete analysis is available in Supplementary File 2 and under GEO code GSE133903.
RT-PCR
RNA (100 ng) was transcribed using a recombinant reverse transcriptase (Transcriptor first strand cDNA synthesis kit #4897030001, Roche Diagnostics, Rotkreuz, Switzerland). PCR was performed with the LightCycler480 Probes Master system (Roche Diagnostics GmbH, Mannheim, Germany). Specific primers used were: hINS2 up: 5′-AGGCTTCTTCTACACACCCAAG-3′, down: 5′-CACAATGCCACGCTTCTG-3′, probe #27; for hGCG up: 5′-TCTGTTCTACAGCACACTACCAGA-3′, down: 5′-AGCTTGCCTTGTACCAGCATT-3′, probe #5; for hSST up: 5′-ACCCCAGACTCCGTCAGTTT-3′, down: 5′- ACAGCAGCTCTGCCAAGAAG-3′, probe #38; for hPDX1 up: 5′-AAGCTCACGCGTGGAAAG-3′, down: 5′-GCCGTGAGATGTACTTGTTGAA-3′, probe #78; for hFFAR1 up: 5′-GTGTCACCTGGGTCTGGTCT-3′, down: 5′-CCAGGGAGGTGTTGCTGT-3′, probe #20. The housekeeping gene used was hRPS13 up: 5′-GGTTGAAGTTGACATCTGACGA-3′, down: 5′-CTTGTGCAACACCATGTGAA-3′, probe #68. Gene expression was quantified by the 2−(ΔCt) method relative to the housekeeping gene.
Statistics
Statistical analysis of insulin and glucagon secretion was performed using GraphPad Prism (GraphPad Software Inc, La Jolla, CA). One way ANOVA and Tukey post testing was used when more than 2 conditions were compared. For glucose stimulation (GSIS) Student’s t-test for unpaired two samples was used and is marked by *. Differences were considered statistically significant at p ≤ 0.05. RNAseq data were analysed using DESeq R package (v.1.10.1).
References
Shapiro, A. M., Pokrywczynska, M. & Ricordi, C. Clinical pancreatic islet transplantation. Nat. Rev. Endocrinol 13, 268–277 (2017).
Andres, A. et al. Macrophage depletion prolongs discordant but not concordant islet xenograft survival. Transplantation 79, 543–549 (2005).
Gill, R. G. Antigen presentation pathways for immunity to islet transplants. Relevance to immunoisolation. Ann N Y Acad Sci 875, 255–260 (1999).
Giuliani, M. et al. Central necrosis in isolated hypoxic human pancreatic islets: evidence for postisolation ischemia. Cell transplantation 14, 67–76 (2005).
Komatsu, H. et al. Oxygen environment and islet size are the primary limiting factors of isolated pancreatic islet survival. PLoS One 12, e0183780 (2017).
Zhao, M. et al. Modification of human islet preparation: an effective approach to improve graft outcome after islet transplantation? Hormone and metabolic research 47, 72–77 (2015).
Niclauss, N. et al. Influence of donor age on islet isolation and transplantation outcome. Transplantation 91, 360–366 (2011).
Henquin, J. C. Influence of organ donor attributes and preparation characteristics on the dynamics of insulin secretion in isolated human islets. Physiological reports 6(5), e13646 (2018).
Hwa, A. J. & Weir, G. C. Transplantation of Macroencapsulated Insulin-Producing Cells. Current diabetes reports 18, 50 (2018).
Zuellig, R. A. et al. Improved physiological properties of gravity-enforced reassembled rat and human pancreatic pseudo-islets. Journal of tissue engineering and regenerative medicine 11, 109–120 (2017).
Yu, Y. et al. Bioengineered human pseudoislets form efficiently from donated tissue, compare favourably with native islets in vitro and restore normoglycaemia in mice. Diabetologia 61, 2016–2029 (2018).
Wojtusciszyn, A., Armanet, M., Morel, P., Berney, T. & Bosco, D. Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia 51, 1843–1852 (2008).
Ris, F. et al. Impact of integrin-matrix matching and inhibition of apoptosis on the survival of purified human beta-cells in vitro. Diabetologia 45, 841–850 (2002).
Stendahl, J. C., Kaufman, D. B. & Stupp, S. I. Extracellular matrix in pancreatic islets: relevance to scaffold design and transplantation. Cell transplantation 18, 1–12 (2009).
Llacua, L. A., Faas, M. M. & de Vos, P. Extracellular matrix molecules and their potential contribution to the function of transplanted pancreatic islets. Diabetologia 61, 1261–1272 (2018).
Komatsu, H., Kandeel, F. & Mullen, Y. Impact of Oxygen on Pancreatic Islet Survival. Pancreas 47, 533–543 (2018).
Guruswamy Damodaran, R., Poussard, A., Cote, B., Andersen, P. L. & Vermette, P. Insulin secretion kinetics from single islets reveals distinct subpopulations. Biotechnology progress 34, 1059–1068 (2018).
Kitahara, A. & Adelman, R. C. Altered regulation of insulin secretion in isolated islets of different sizes in aging rats. Biochemical and biophysical research communications 87, 1207–1213 (1979).
Lehmann, R. et al. Superiority of small islets in human islet transplantation. Diabetes 56, 594–603 (2007).
MacGregor, R. R. et al. Small rat islets are superior to large islets in in vitro function and in transplantation outcomes. American journal of physiology. Endocrinology and metabolism 290, E771–779 (2006).
Kim, A. et al. Islet architecture: A comparative study. Islets 1, 129–136 (2009).
Brissova, M. et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society 53, 1087–1097 (2005).
Cabrera, O. et al. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci USA 103, 2334–2339 (2006).
Trimble, E. R., Halban, P. A., Wollheim, C. B. & Renold, A. E. Functional differences between rat islets of ventral and dorsal pancreatic origin. J Clin Invest 69, 405–413 (1982).
Braun, M. The somatostatin receptor in human pancreatic beta-cells. Vitamins and hormones 95, 165–193 (2014).
Barbieux, C. et al. Asymmetrical distribution of delta and PP cells in human pancreatic islets. J Endocrinol 229, 123–132 (2016).
Williams, S. J., Schwasinger-Schmidt, T., Zamierowski, D. & Stehno-Bittel, L. Diffusion into human islets is limited to molecules below 10 kDa. Tissue &. cell 44, 332–341 (2012).
Furuyama, K. et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 567, 43–48 (2019).
Kolic, J., Spigelman, A. F., Smith, A. M., Manning Fox, J. E. & MacDonald, P. E. Insulin secretion induced by glucose-dependent insulinotropic polypeptide requires phosphatidylinositol 3-kinase gamma in rodent and human beta-cells. J Biol Chem 289, 32109–32120 (2014).
Serre, V. et al. Exendin-(9-39) is an inverse agonist of the murine glucagon-like peptide-1 receptor: implications for basal intracellular cyclic adenosine 3′,5′-monophosphate levels and beta-cell glucose competence. Endocrinology 139, 4448–4454 (1998).
Svendsen, B. et al. Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell reports 25, 1127–1134 (2018).
Kolic, J. & MacDonald, P. E. cAMP-independent effects of GLP-1 on beta cells. J Clin Invest 125, 4327–4330 (2015).
Ullrich, S. & Wollheim, C. B. Islet cyclic AMP levels are not lowered during alpha 2- adrenergic inhibition of insulin release. J. Biol. Chem 259, 4111–4115 (1984).
Tengholm, A. & Gylfe, E. cAMP signalling in insulin and glucagon secretion. Diabetes, obesity & metabolism 19(Suppl 1), 42–53 (2017).
Peterhoff, M. et al. Inhibition of insulin secretion via distinct signaling pathways in alpha2-adrenoceptor knockout mice. Eur. J. Endocrinol 149, 343–350 (2003).
Ahren, B. Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia 43, 393–410 (2000).
Rodriguez-Diaz, R. et al. Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab 14, 45–54 (2011).
Rosengren, A. H. et al. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science 327, 217–220 (2010).
Segerstolpe, A. et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab 24, 593–607 (2016).
De Marinis, Y. Z. et al. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab 11, 543–553 (2010).
Pipeleers, D. G. et al. Interplay of nutrients and hormones in the regulation of insulin release. Endocrinology 117, 824–833 (1985).
Hamilton, A. et al. Adrenaline Stimulates Glucagon Secretion by Tpc2-Dependent Ca(2+) Mobilization From Acidic Stores in Pancreatic alpha-Cells. Diabetes 67, 1128–1139 (2018).
Kraus-Friedmann, N. Hormonal regulation of hepatic gluconeogenesis. Physiol Rev 64, 170–259 (1984).
Lehmann, V., Andersen, P. L., Guruswamy Damodaran, R. & Vermette, P. Method for Isolation of Pancreatic Blood Vessels, their Culture and Co-culture with Islets of Langerhans. Biotechnology progress 35, e2745 (2019).
Anastasiou, V. et al. Aldehyde dehydrogenase activity is necessary for beta cell development and functionality in mice. Diabetologia 59, 139–150 (2016).
Acknowledgements
We acknowledge the skilled technical assistance of Sieglinde Haug and Elisabeth Metzinger (University Hospital of Tübingen, Tübingen, Germany) and Sandra Daß (Boehringer Ingelheim, Biberach, Germany). We thank Marie Gauder and Stefan Czemmel (QBic, University of Tübingen) for the analysis of RNAseq data. This work was supported by a grant (01GI0925) from the German Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD e.V.). ELG received a fellowship from the DZD e.V.
Author information
Authors and Affiliations
Contributions
S.U., H.-U.H. and M.B. designed and supervised the study. E.L.G., F.G. an M.B.O. designed and performed the experiments. S.U., M.B. and U.D. designed and supervised the long term culture experiments, which were performed at the Boehringer Ingelheim laboratories (Fig. 3). E.L.G., S.U. and M.B. analysed data and prepared figures. S.U. and E.L.G. wrote the manuscript. All authors interpreted and discussed the results and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lorza-Gil, E., Gerst, F., Oquendo, M.B. et al. Glucose, adrenaline and palmitate antagonistically regulate insulin and glucagon secretion in human pseudoislets. Sci Rep 9, 10261 (2019). https://doi.org/10.1038/s41598-019-46545-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46545-6
This article is cited by
-
Sphingolipid subtypes differentially control proinsulin processing and systemic glucose homeostasis
Nature Cell Biology (2023)
-
Effects of adrenergic-stimulated lipolysis and cytokine production on in vitro mouse adipose tissue–islet interactions
Scientific Reports (2022)
-
Islet sympathetic innervation and islet neuropathology in patients with type 1 diabetes
Scientific Reports (2021)
-
FFA2-, but not FFA3-agonists inhibit GSIS of human pseudoislets: a comparative study with mouse islets and rat INS-1E cells
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
