
Abstract
We developed a novel and convenient method for rapidly identifying CRISPR/Cas9-based genome-edited biallelic knockout (KO) cells/individuals carrying insertions or deletions of a few nucleotides (indels) by performing PCR on genomic DNA samples under stringent conditions and low MgCl2 concentrations. The biallelic KO samples can be judged as ‘negative’ under these conditions. The sense primer corresponds to the sequence recognised by guide RNA and subsequently cleaved by Cas9 immediately upstream of a target gene’s proto-spacer adjacent motif (PAM), and the reverse primer corresponds to the sequence ~200 bp downstream from the PAM. PCR performed using this primer set under standard MgCl2 concentrations (1.5–2.5 mM) should generate PCR products derived from both mutated and unedited alleles, whereas PCR performed using lower MgCl2 concentrations (0.8–2 mM) should yield products derived from unedited alleles. This enables high-throughput screening of biallelic mutants among cells/embryos having ≥1 indels at a region within 5 bp upstream of the PAM (where more than 94% of indels are known to appear). We performed proof-of-principle analyses of this novel approach using genome-edited Et1, Tyr, Ramp1, Ramp3, and Rosa26 mouse samples carrying various types of indels, and demonstrate that this new technique allows rapid identification of biallelic KO mutants among samples carrying various types of indels and mosaic mutations with 100% accuracy. We name this system detection of biallelic KO mutants harbouring indels using PCR (Bindel-PCR).
Similar content being viewed by others


Introduction
Advances in the development of genome-editing systems, such as zinc-finger nuclease (ZFN), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease (Cas9) (CRISPR/Cas9), provide breakthroughs in life science-related research1,2,3. The CRISPR/Cas9 system is now recognised as one of the most powerful tools for engineering genes of interest; this is because of the simplicity of the system and the high specificity it provides in targeted genome engineering. CRISPR/Cas9-based mutation induction commences with specific cleavage of double-stranded DNA 3–5 bp upstream of the proto-spacer adjacent motif (PAM) in a target locus4,5. The resulting cleavage can be repaired through homology-directed repair (HDR) (which requires homologous recombination with a homology arm), non-homologous end-joining (NHEJ) [which involves no homology or only 1–3 nucleotides (nt) of homology at the junction]6, or microhomology-mediated end-joining (MMEJ) [which involves alignment of microhomologous sequences (1–4 nt) internal to the broken ends before joining]7. Consequently, a DNA fragment can be inserted into the target locus precisely through HDR, and this generates insertion/deletion mutations (‘indels’) through NHEJ and MMEJ8.
Currently, gene-edited mice can be created readily and with high efficiency through one-step generation, which involves microinjection or in vitro electroporation of embryos in the presence of CRISPR/Cas9 reagents9,10. However, together with the development of this process, the demand for selecting biallelic knockout (KO) mutants has been increasing, because phenotypic alteration resulting from the dysfunction of a gene of interest can be readily detected. Therefore, the development of a simple and rapid method for screening biallelic mutants is eagerly awaited.
Genome-edited organisms and samples such as cells/embryos obtained through HDR can be readily identified because their rapid detection is possible through PCR performed using primers corresponding to the specific sequence in the inserted DNA fragment and the use of restriction enzymes that can selectively recognise the resulting PCR products11,12. By comparison, it is more challenging to identify organisms carrying indels generated through the NHEJ- or MMEJ-based repair pathway, because PCR-based amplification of an area showing alteration of a few nucleotides frequently fails to distinguish this sequence from the wild-type (WT) sequence. To date, the T7 endonuclease 1 (T7E1)-based assay and the Surveyor enzyme mismatch cleavage assay have been most frequently used to scan for indels triggered by engineered nucleases13. These methods are based on the identification of heteroduplex DNA formed after melting and hybridizing mutant and WT alleles, and the methods exploit the use of enzymes that can cleave heteroduplex DNA at mismatches formed by single or multiple nucleotides. However, to find the biallelic mutants with indels, these approaches occasionally require an additional in vitro assay such as Cas9RNP cut assay, in which the WT alleles are cleaved by a Cas9/ribonuclear protein complex prepared through in vitro complex formation between guide RNA (gRNA) and Cas9 protein14. Furthermore, the sensitivity of the methods must be increased to detect the presence of samples carrying mosaic mutations. When numerous samples must be precisely genotyped, these processes are expensive, laborious, and frequently time-consuming.
Here, we present a rapid and convenient assay system, named detection of biallelic KO mutants harbouring indels using PCR (Bindel-PCR), for the identification of CRISPR/Cas9-induced indels by employing only a general PCR apparatus and Thermus aquaticus-derived PCR DNA polymerase [recombinant Taq (rTaq) DNA polymerase or HiDi DNA polymerase]. Bindel-PCR requires 2 rounds of PCR to detect biallelic editing events in both alleles. The first round of PCR was carried out under standard concentrations of Mg2+ (1.5–2.5 mM) for confirming successful amplification of a target region. The second round of PCR was performed on the first PCR products under low MgCl2 concentrations. Biallelic KO samples are considered negative under these conditions. We show proof-of-principle analyses of this novel approach using genome-edited Et1, Tyr, Ramp1, Ramp3, and Rosa26 mouse samples carrying various types of indels.
Results
Survey of the sites frequently showing CRISPR/Cas9-induced indels and primer design used for Bindel-PCR
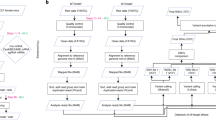
Bindel-PCR principle and flowchart are shown in Fig. 1a,b and described in its corresponding figure legend. To design the primers used for Bindel-PCR for detecting biallelic mutants, we first scanned for the sites that had been frequently genome-edited after recognition by gRNA and subsequent cleavage by Cas9; for this, we used data obtained from our recent study [for mouse endothelin-1 gene (Et1) (Sakurai et al., unpublished data)] and other studies [for rat tyrosinase gene (Tyr), Rosa26 (Gt(ROSA)26S), signal regulatory protein-α gene (Sirpa), and cytochrome P450 family 2 subfamily D gene (Cyp2d)15, and for mouse fibroblast growth factor-10 gene (Fgf10)11]. In Fig. 1c, various types of indels in mouse Et1 are shown as examples. The gRNAs used were designed to correspond to the 20-bp sequence (shown by green colour) preceding the PAM. The nt immediately before the PAM was designated as nt (−1), and the nt immediately after the PAM as nt (+1). For example, the sample with one nt deletion at nt (−4) (shown as Example 1 in Fig. 1c) can be shown as “1 nt deletion at nt (−4)”. The sample with one nt insertion at nt (−3) (shown as Example 2 in Fig. 1c) can be shown as “1 nt insertion at nt (−3)”. The sample with more than one nt at nt (−1) (shown as Example 3 in Fig. 1c) can be shown as “>2 nt deletion at nt (−1)”. The sample containing indels of >2 nt spanning the PAM (shown as Example 4 in Fig. 1c) was defined as PAM (−1), which was classified as >2 nt deletion at nt (−1). In Fig. 1d, data from the present study (for mouse Et1) and previous studies (for rat Tyr, Rosa26, Sirpa, and Cyp2d as well as mouse Fgf10) are provided in brief. The site cleaved by the CRISPR/Cas9 system is widely recognised to occur around 3 bp upstream of the PAM frequently4,5. Corresponding to this notion, the accumulated data indicated that indels occurred frequently at the sites [corresponding to nt (−6) to (−1)] before PAM (Fig. 1d). Notably, 97.6% of indels (100% for mouse Et1; 99% for rat Tyr, Rosa26, Sirpa, and Cyp2d; 94% for mouse Fgf10) were generated at nt (−5) to PAM (−1). Based on these results, a region at nt (−5) to PAM (−1) of a target gene was considered as an important target site for setting Bindel-PCR primers.

Principle of Bindel-PCR for identifying CRISPR/Cas9-induced biallelic mutants. (a) Outline of Bindel-PCR. 1st PCR is carried out under standard concentrations of Mg2+ (1.5–2.5 mM) for confirming successful amplification of a target region. For the 2nd PCR, the sense primer was set corresponding to the sequence that is recognised by gRNA and cleaved by Cas9 immediately upstream of the PAM of the target gene, whereas the reverse primer was set on a downstream sequence distant from the PAM. PCR performed using this primer set and the 1st PCR products as template under standard conditions should generate PCR products (~200 bp) derived from both indel-mutated alleles and unedited alleles. Contrastingly, when the concentration of MgCl2 used for PCR is lowered, annealing between a primer and the mutated target sequence becomes unstable, and under this condition, PCR products derived from unedited alleles, such as WT allele, should be generated preferentially. Mg2+ is recognised as not only a cofactor for DNA polymerase activity, but also a promoter of complex formation between primers and DNA template26. Blue boxes, sequences recognised by forward and reverse primers used for 1st PCR; green box, sequence recognised by crRNA; red box, PAM sequence. (b) Flowchart of detection of biallelic KO mutants by using Bindel-PCR. (c) The evaluation rule for indels appearing in the target sequence is shown using mouse Et1 gRNA-recognizing nt region as an example. Green nt correspond to the sequence recognised by gRNA-Et1. Red nt correspond to the PAM sequence. nt −1 to –23 correspond to the 5′ upstream region of the PAM. nt +1 to +15 correspond to the 3′ downstream region of the PAM. Arrows indicate the site of indels classified. WT, wild-type. (d) Analyses of sites frequently showing CRISPR/Cas9-induced indels. Data shown are from our previous study (Sakurai et al., unpublished data) on mouse Et1 and from other studies [Yoshimi et al. (2016) for rat Tyr, Rosa26, Sirpa, and Cyp2d and Hashimoto and Takemoto (2015) for mouse Fgf10]. *Includes the samples with indels at nt (−1) as well as those with indels including a part of the PAM. Et1, endothelin-1; Tyr, tyrosinase; Rosa26, Gt(ROSA)26S; Sirpa, signal regulatory protein-α; Cyp2d, cytochrome P450, family 2, subfamily d; Fgf10, fibroblast growth factor-10.
Determination of optimal conditions for Bindel-PCR performed using rTaq DNA polymerase
Based on the above findings that indels occurred more frequently within an area of 5 bp 5′ upstream of the PAM, we decided to use the samples derived from F0 offspring (Sakurai et al., unpublished data) with CRISPR/Cas9 based genome-edited Et1 gene for exploring optimal conditions of Bindel-PCR. We used a PCR primer set (Et1ch-2S/-2A) (Fig. 2a) to amplify a portion of Et1 locus spanning a sequence recognised by gRNA-Et1 (corresponding to the sequence shown by green colour in Fig. 2a), and employed a series of Et1 plasmids carrying various types of indels (Fig. 2b). For example, the plasmids containing inserts with 1 nt deletion at nt (−10), (−5), (−3), and (−1) were named D1-10, D1-5, D1-3, and D1-1, respectively. Similarly, plasmids containing inserts with 1 nt insertion at nt (−10), (−5), (−3), and (−1) were named I1-10, I1-5, I1-3, and I1-1, and those with 3 nt deletion at nt (−10), (−5), and (−1) were named D3-10, D3-5, and D3-1, respectively. Next, using these plasmids carrying various types of Et1 indels, we explored optimal conditions for Bindel-PCR to identify biallelic indels using rTaq DNA polymerase. For this, a sense PCR primer [Bindel-PCR(Et1)1S: 5′-GCGCGTCGTACCGTATGGAC-3′] and a nested sense primer [Bindel-PCR(Et1)2S: 5′-CGCGTCGTACCGTATGGACT-3′] (Fig. 2a) were used. The melting temperature (Tm) of these primers ranged from ~67 °C to 70 °C. We selected the site recognised by the antisense primer [Bindel-PCR(Et1)1A: 5′-GTTCTTTTCCTGCTTGGCAGAAATT-3′: Tm, 70 °C] approximately 200 bp away from the positions recognised by these sense primers (Fig. 2a). In Bindel-PCR, the mutated samples failed to generate amplified products. To avoid the formation of false-positive PCR products, the portion of Et1 spanning the sequence recognised by gRNA-Et1 was first amplified as the 1st PCR (Fig. 2c). The resultant PCR products were next subject to nested PCR to ascertain the optimal conditions that allow successful amplification of a target sequence from the WT but not mutated allele.

Bindel-PCR to identify biallelic KO mutants for Et1. (a) nt sequence in mouse Et1 locus. The location of sequences recognised by gRNA and PCR primers is shown. nt in blue correspond to forward and reverse primers used for 1st PCR. Green nt correspond to the sequence recognised by gRNA-Et1. Red nt correspond to the PAM. Expected sizes of PCR products obtained using different primer sets are shown at the bottom. (b) nt sequences in mouse Et1 locus that are recognised by gRNA-Et1. Various types of indels found in a series of Et1 plasmids are shown. nt in green correspond to the sequence recognised by gRNA-Et1. nt in red correspond to the PAM sequence. Shading indicates modification of Nt. Dashed lines indicate Nt deletion. (c) Gel electrophoretic pattern of PCR products (450 bp) obtained through 1st PCR performed using Et1ch-2S/−2A primer set and a series of Et1 plasmids harbouring various types of indels as template. M, 100-bp-ladder markers. WT, wild-type. The images of these full-length gels are presented in Supplementary Fig. S2c. (d) (e) Gel electrophoretic pattern of products from the 2nd PCR (corresponding to Et1 sequence recognised by gRNA-Et1) obtained after performing PCR on a series of Et1 plasmids [shown in (b) and (c)] under reduced concentrations of MgCl2. Representative results from assays performed using rTaq DNA polymerase are shown in (d). Left panel, 2nd PCR products (155 bp) obtained using Bindel-PCR(Et1)-1S/-1A primer set; right panel, 2nd PCR products (153 bp) obtained using Bindel-PCR(Et1)-2S/-1A primer set. Representative results from assays performed using HiDi DNA polymerase are shown in (e). Left panel, products from the 2nd PCR (155 bp) obtained using Bindel-PCR(Et1)-1S/-1A primer set; right panel, 2nd PCR products (153 bp) obtained using Bindel-PCR(Et1)-2S/-1A primer set. M, 100-bp-ladder markers; WT, wild-type. The images of these full-length gels are presented in Supplementary Fig. S2d,e.
First, the 1st PCR was performed using plasmids (5 ng/tube) containing various types of indels at Et1 locus (PCR protocol: 95 °C for 1 min, 35 cycles of 95 °C for 15 s, 68 °C for 10 s, and 72 °C for 40 s) in a PCR buffer [10 mM Tris (pH 8.3), 50 mM KCl, 200 μM dNTPs, and 1.5 mM MgCl2] containing rTaq DNA polymerase (0.2 U/25 μL) and Et1ch-2S/-2A primers. The expected 450-bp amplification band was obtained from all the plasmid samples used (Fig. 2c).
Next, as the 2nd PCR, nested PCR was performed using Bindel-PCR (Et1)-1S/-1A or Bindel-PCR (Et1)-2S/-1A primer set (Fig. 2a). We first tested the annealing temperature, various dNTP concentrations, 2-step PCR (shuttle PCR), and 3-step touchdown PCR with annealing at 72 °C (decreasing 1 °C per cycle to 61 °C), which are considered as a simple and rapid means to optimise PCR21, for the potential establishment of Bindel-PCR, but failed to obtain a reproducible condition for Bindel PCR (data not shown). Consequently, we found that Mg2+ concentration and the location of primers as one of the critical factors for Bindel-PCR. Thus, the 1st PCR products (0.5–1 ng) (Fig. 2c) were subject to PCR under these conditions: 95 °C for 1 min, 40–45 cycles of 95 °C for 15 s, 68 °C for 10 s, and 72 °C for 40 s; the annealing temperature of 68 °C was employed according to the Tm (~70 °C) of the primers used (Fig. 2d). PCR was repeated thrice for each condition to confirm reproducibility. The amplification efficiency of a target sequence tended to decrease with a decrease in Mg2+ concentration, but the results clearly indicated that the inability to detect the sequence carrying indels depended on the Mg2+ concentration (Fig. 2d). For example, no amplification product was obtained from either the WT or mutated allele when PCR was performed, using Bindel-PCR(Et1)-1S/-1A primer set (Fig. 2d), in the presence of 0.75 mM MgCl2; by contrast, PCR in the presence of 1.5 mM MgCl2 yielded amplification products from these samples and from samples (D3-5 and D3-1) carrying a 3-bp deletion (Fig. 2d, left panel), and PCR [using Bindel-PCR(Et1)-1S/-1A primer set] in the presence of 0.85 mM MgCl2 clearly yielded products derived only from the samples WT, I1-10, and I1-1 (Fig. 2d, left panel). Sample I1-1 was positively identified here probably because it contained 1 nt added to the sequence type of nt (−1) (Fig. 2a,b). Therefore, Bindel-PCR(Et1)-2S/-1A primer set (Fig. 2a) was adopted. The PCR-amplification mode used was the same as that used for amplifying various samples (Fig. 2b). The results showed that D1-10 reacted weakly under this PCR condition; as expected, the sample I1-1 was negative, but sample I1-10 was still positive. These results indicate that it is possible to identify biallelic mutants by Bindel-PCR among the samples with indels having 1 ≥ nt deletion and insertion at nt (−5) to PAM (−1) of a target gene. However, when these results were combined with the data shown in Fig. 1d, the frequency of detection of samples carrying indels, as exemplified by the samples I1-10 and D1-10, was found to be very low. Based on these results, we determined the optimal PCR conditions that enable detection of almost all the samples carrying indels at nt (−5) to PAM (−1) of a target gene (coverage of indels ranged 94–100%; see Fig. 1d).
Determination of optimal conditions for HiDi DNA polymerase-based Bindel-PCR
T. aquaticus-derived HiDi DNA polymerase is known to discriminate between mismatched primers16. To investigate whether this enzyme can be used for selecting biallelic KO samples harbouring indels, we examined the optimal conditions for HiDi DNA polymerase-based Bindel-PCR (Fig. 2e). The 1st PCR was the same as that shown in Fig. 2c, and the PCR products generated by the 1st PCR were used as templates (0.5–1 ng/tube) for the nested PCR. The nested PCR was performed in PCR solutions that were composed of 50 mM Tris (pH 9.2), 16 mM (NH4)2SO4, MgCl2 (ranging from 2.5 to 1.5 mM), 0.1% Tween-20, and 200 μM dNTPs and contained the primer set Bindel-PCR(Et1)-1S/-1A (or Bindel-PCR(Et1)-2S/-1A) and HiDi DNA polymerase (0.2 U/25 μL) (PCR protocol: 95 °C for 1 min, 40–45 cycles of 95 °C for 15 s, 68 °C for 10 s, and 72 °C for 40 s). PCR was performed thrice for each condition to confirm reproducibility. When HiDi DNA polymerase was used, the PCR-amplification efficiency of a target sequence tended to decrease with a decrease in Mg2+ concentration, indicating again that the inability to detect sequences carrying indels was dependent on the Mg2+ concentration. In the presence of 2 mM MgCl2, the reaction amplified none of the samples except WT and I1-1 (Fig. 2e, left panel). The sample I1-1 showed positive reaction probably because of the same reason it was positive when rTaq DNA polymerase was employed (preceding subsection; Fig. 2d, left panel). Notably, with Bindel-PCR(Et1)-2S/-1A primer set, the samples except for WT, D1-10, and I1-10 were PCR negative, when MgCl2 concentrations ranged between 2.25 and 2 mM (Fig. 2e, right panel). This might be solely the result of the ability of HiDi DNA polymerase16 to discriminate between mismatched primers or a result of the combined use of the polymerase and Bindel-PCR(Et1)-2S primer. PCR performed in the presence of 1.5 mM MgCl2 produced no positive products, not even from the WT sample (Fig. 2e, right panel). Notably, under the stringent conditions (where Mg2+ concentration was lowered), HiDi DNA polymerase tended to show superior performance relative to rTaq DNA polymerase in terms of the inability to amplify samples harbouring indels. Thus, we successfully determined the optimal conditions that lead to the inability to amplify almost all samples carrying indels at the PAM (−10) site and its downstream portion.
Evaluating sensitivity of Bindel-PCR for identifying biallelic mutants among mixed samples containing various indels through comparison with T7 endonuclease 1 (T7E1) assay and polyacrylamide gel electrophoresis (PAGE)
We performed Bindel-PCR using samples mixed with various types of plasmids (shown in Fig. 2b). In the upper panel of Fig. 3a, the content of plasmid in each mixed sample is shown. For example, sample numbered 1 and 6 contain WT. Sample numbered 2 is mixed and contains D1 series (D1-10, −5, −3 and −1), I1 series (I1-10, −5, −3 and −1) and D3 series (D3-10, −5 and −1). Simultaneously, T7E1 assay and PAGE were performed using the same samples used for Bindel-PCR. Identification of indels in the mouse Et1 locus was based on the method shown in Fig. 2d,e. Bindel-PCR in the presence of 1.5 mM MgCl2 yielded clear PCR products in the samples numbered 5 and 7 (all of which lack D1-10 and L1-10), but failed in the presence of 0.85 mM MgCl2 (lower low of Fig. 3a), which appears to be consistent with the results shown in Fig. 2d,e. These results indicate that Bindel-PCR can confer identification of biallelic mutants among the mixed samples with various indels, at least at nt (−5) to PAM (−1) of a target gene. The results obtained after T7E1 assay are shown in Fig. 3b. The mixed samples numbered 1 and 6 were wild-type controls. As expected, they exhibited resistance against T7E1 cleavage. Notably, the cleavage pattern in the mixed samples numbered 5 and 7, which had been identified as biallelic mutation after Bindel-PCR-based analysis, was identical to those numbered 2, 3, 4, 7, and the WT sample. Consequently, this suggests that T7E1-based identification of biallelic mutation requires an additional in vitro assay (e.g., Cas9RNP cut assay). The results obtained after PAGE are shown in Fig. 3c. General PAGE/TBE system based analysis of the mixed samples revealed that there were no additional bands throughout the samples used. The mixed samples used in this study contain indels with 1 or 3 nt deletion or those with 1 nt insertion, these are also considered as “mosaic samples”. In this context, Bindel-PCR is useful to identify samples with biallelic mutations among the samples comprised by complicated mosaic mutations.

Analysis of sensitivity of Bindel-PCR for identifying biallelic mutants among mixed samples containing various indels. (a) Bindel-PCR of mixed Et1 indel samples. Top row: the list of mixed samples containing plasmids carrying various indels shown in Fig. 2b. Both D1-10 and I1-10 (all of which is shown by red colour) are the samples that are proven not to detect the indels after analysis using Bindel-PCR (see Fig. 2d,e). WT, wild-type genomic DNA used as a positive control; M, 100-bp-ladder markers. Upper panel: 1st PCR (450 bp) of Bindel-PCR using Et1ch-2S/-2A primer set. Middle panel: 2nd PCR (153 bp) of 1st Bindel PCR products (upper panel) under 1.5 mM MgCl2 concentration and using Bindel-PCR(Et1)-2S/-1A primer set. Lower panel: 2nd PCR (153 bp) of 1st Bindel PCR products (upper panel) under 0.85 mM MgCl2 concentration and using Bindel-PCR(Et1)-2S/-1A primer set. (b) The results of T7E1 assay of 1st Bindel-PCR products [see upper panel of (a)]. (c) The results of 5% PAGE of 2nd Bindel-PCR products [see middle panel of (a)]. The images of these full-length gels are presented in Supplementary Figs S3a–c.
Sensitivity of Bindel-PCR for identifying biallelic KO mice among genetically mosaic animals
CRISPR/Cas9 is a powerful system for inducing genome editing in various organisms, but this technique frequently creates individuals carrying mosaic mutations, with various types of indels and WT sequences being included17. We considered it critical to test whether Bindel-PCR can detect WT alleles present in small numbers in a genome-edited animal carrying larger numbers of mutated alleles; a failure in this would lower the usability of Bindel-PCR. Here, we sought to measure the sensitivity for detecting the presence of WT alleles in mice carrying mosaic mutations. For this purpose, we used a set (Fig. 2b) that included various types of indels for Et1, and we prepared several samples in which certain types of indels were mixed at appropriate DNA:DNA ratios. For example, we prepared Indel Mix 1 (D1-5:I1-3 = 1:1), Indel Mix 2 (D1-3:D3-5 = 1:1), and Indel Mix 3 (D3-3:I1-3 = 1:1), and then added WT DNA to Indel Mix 3 at ratios of 1:1, 1:3, 1:7, 1:15, 1:31, 1:61, and 1:127; these were termed, respectively, WT:Indel Mix = 1:1, 1:3, 1:7, 1:15, 1:31, 1:61, and 1:127 (Fig. 4a). These ratios were tested based on the assumption of early mouse embryos being edited by CRISPR/Cas9-related components at these stages: zygote, 2-cell, 4-cell, 8-cell, morula (comprising 16 cells), early blastocyst (32 cells), and blastocyst (64–128 cells) (Fig. 4b). Each WT:Indel Mix template (~5 ng/tube) was subject to PCR by using the previously determined optimal conditions. For example, when rTaq DNA polymerase was used, PCR was performed using Bindel-PCR(Et1)-2S/-1A primer set in the presence of 0.85 mM Mg2+ (Fig. 2d, right panel), whereas when HiDi DNA polymerase was used, PCR was performed using Bindel-PCR(Et1)-2S/-1A primer set in the presence of 2 mM Mg2+ (Fig. 2e, right panel). Both DNA polymerases yielded positive bands for samples containing the WT allele, but they failed to amplify the samples with Indel Mix 1-3, as expected (Fig. 2d,e and 3); more specifically, both DNA polymerases yielded positive bands for the samples corresponding to WT:Indel Mix = 1:1 to 1:127 (Fig. 4a). Generally, when zygotes are transfected with genome-editing components through microinjection or electroporation, genome editing at target loci is largely completed in the zygote stage or at least by the 2- or 4-cell stage17,18. Our results obtained using Bindel-PCR indicate that the presence of the WT allele at a target locus can discerned when mosaic mutations occur by the 2-to-4-cell stage; thus, Bindel-PCR enables identification of biallelic KO mutants among animals harbouring mosaic mutations.

Analysis of sensitivity for detecting the presence of WT alleles in samples containing various ratios of mutant-type templates. (a) PCR of the 1st PCR templates (shown in Fig. 2c) mixed at appropriate ratios. For example, Indel Mix 3 was prepared at the ratio of D3-5:I1-1 = 1:1. For preparing WT:Indel M3, WT DNA was added to Indel Mix 3 (D3-5:I1-1 = 1:1) at ratios of 1:1, 1:3, 1:7, 1:15, 1:31, 1:63, and 1:127. Upper panel: Results of 2nd PCR performed using rTaq DNA polymerase and Bindel-PCR-2S/1A primer set in the presence of 0.85 mM MgCl2. Lower panel: Results of 2nd PCR performed using HiDi DNA polymerase and Bindel-PCR(Et1)-2S/-1A primer set in the presence of 2 mM MgCl2. Each gel lane was loaded with 10 μL of PCR products. M, 100-bp-ladder markers. (b) WT:Indel samples prepared based on assuming that early mouse embryos are edited by CRISPR/Cas9-related components at these stages: zygote (FE, fertilised egg), 2-cell (2C), 4-cell (4C), 8-cell (8C), morula (M; comprising 16 cells), early blastocyst (B; 32 cells), and blastocyst (64–128 cells). The images of these full-length gels are presented in Supplementary Fig. S4a.
Identification of biallelic KO mutants among F0 offspring after genome editing targeting the loci Et1, Tyr, and Ramp1 (receptor activity modifying protein-1) and among 12.5 dpc foetal samples after genome editing targeting the loci Ramp3 and Rosa26
As a proof-of-principle experiment to test the feasibility of Bindel-PCR, we performed 1st PCR by using genomic DNA samples (10) obtained from the genome-edited F0 offspring that had already been demonstrated to be biallelic KO for Et1 locus (Fig. 5a; Sakurai et al., unpublished data). After agarose-gel electrophoresis-based confirmation that the 1st PCR products were successfully produced (Fig. 5b), the 2nd PCR was performed using the 1st PCR products as templates (Fig. 5c): With both rTaq and HiDi DNA polymerases, PCR products were generated from all the samples tested in the 1st PCR, but almost all the samples (except for the WT samples) were judged as negative when the 2nd PCR was conducted. These results indicate that Bindel-PCR is useful for rapid and convenient identification of biallelic KO mutants among F0 offspring carrying various types of indels and mosaic mutations with 100% accuracy.

Identification of biallelic KO mutants among F0 offspring obtained after CRISPR/Cas9-mediated introduction of indels at Et1 locus. (a) Sequencing results of genomic DNA samples (10) obtained from the genome-edited F0 offspring that had already been demonstrated to be biallelic KO for Et1 locus (Sakurai et al., unpublished data). Green nt correspond to the sequence recognised by gRNA-Et1. Red nt correspond to the PAM sequence. Dashed lines indicate nt deletion. Frequency = no. clones detected/total no. clones sequenced. (b) Result of 1st PCR of genomic DNA samples 1–10, performed using Et1ch-2S/-2A primer set. WT, wild-type genomic DNA used as a positive control; M, 100-bp-ladder markers. (c) Upper panel: Results of 2nd PCR performed using rTaq DNA polymerase and Bindel-PCR(Et1)-2S/-1A primer set in the presence of 0.85 mM MgCl2; lower panel: results of 2nd PCR performed using HiDi DNA polymerase and Bindel-PCR(Et1)-2S/-1A primer set in the presence of 2 mM MgCl2. WT, wild-type genomic DNA used as a positive control; M, 100-bp-ladder markers. The images of these full-length gels are presented in Supplementary Fig. S5b,c.
We also performed Taq DNA polymerase-based Bindel-PCR by using F0 mice in which Tyr and Ramp1 loci were targeted (Fig. 6a,b). For Tyr, we used 3 F0 mice (Nos 1, 2, and 3), which were obtained by electroporating C57BL/6J (B6) fertilised eggs in a solution containing Tyr-targeting gRNA and Cas9 protein, and one F1 offspring (No. 4) derived from mating between a B6 female and No. 1 F0 male (Fig. 6a and Supplementary Table S1). The coat colour of the 3 F0 mice was white, which indicated that they were biallelic KO for Tyr locus, whereas No. 4 F1 offspring was black, which indicated that it was heterozygous KO for Tyr locus. In the 1st PCR, we used a PCR primer set (Tyrch-1S/-1A in Fig. 6a and Table S3) to amplify a portion of Tyr locus spanning a sequence recognised by gRNA-Tyr (Fig. 6a). The 1st PCR thermal-cycling protocol was 95 °C for 1 min, 35 cycles of 95 °C for 15 s, 68 °C for 10 s, and 72 °C for 40 s, and the reaction was performed in a PCR buffer [10 mM Tris (pH 8.3), 50 mM KCl, 200 μM dNTPs, and 1.5 mM MgCl2] containing rTaq DNA polymerase (0.5 U/25 μL). Next, 2nd PCR was performed using Bindel-PCR(Tyr)-1S/-1A primer set (Tm, ~69 °C; Fig. 6a and Table S3), and the reaction was in a solution containing the 1st PCR products (0.5–1 ng) as the template; the PCR protocol was 95 °C for 1 min, 40–45 cycles of 95 °C for 15 s, 68 °C for 10 s, and 72 °C for 40 s. All tested samples (except No. 2 F0 sample) generated a 443-bp PCR product (Fig. 6a, upper right panel). Subsequent sequencing analysis revealed that No. 2 F0 sample contained an ~200-nt deletion around the target site of Tyr locus (Supplementary Fig. S2a). When the 2nd PCR was performed, almost all samples (except No. 4 F0 and WT samples) were identified as negative (Fig. 6a, lower right panel). These results suggested that the 3 F0 mice (Nos 1–3) were biallelic KO mice, and sequencing of the samples confirmed this notion (Supplementary Fig. S2a).

Identification of biallelic KO mutants among F0 offspring carrying CRISPR/Cas9-induced indels at Tyr (a) and Ramp1 (b) loci. (a) Location of Tyr sequences recognised by gRNAs and the PCR primers used. Blue, nt sites used as forward and reverse primers for the 1st PCR; green, nt corresponding to gRNA target sequence; red, PAM sequence. Expected sizes of PCR products obtained using different primer sets are shown at the bottom. In the upper right panel, the results of the 1st PCR of genomic DNA samples of three F0 mice (lanes 1–3; Supplementary Table S1) and one F1 offspring (lane 4) are shown when PCR was performed using rTaq DNA polymerase and Tyrch-1S/-1A primer set in the presence of 1.5 mM MgCl2. In the lower right panel, the results of the 2nd PCR using 1st PCR products are shown when PCR was performed using rTaq DNA polymerase and Bindel-PCR(Tyr)-1S/-1A primer set in the presence of 0.85 mM MgCl2. (b) Location of Ramp1 sequences recognised by gRNAs and the PCR primers used. Sites indicated by blue, green, and red correspond to the sites recognised by primers, gRNA, and PAM, respectively. In the upper right panel, the results of the 1st PCR of genomic DNA samples of eight F0 mice (lanes 1–8; Supplementary Table S1) are shown when PCR was performed using rTaq DNA polymerase and Ramp1ch-1S/-1A primer set in the presence of 1.5 mM MgCl2. In the lower right panel, the results of the 2nd PCR of 1st PCR products are shown when PCR was performed using rTaq DNA polymerase and Bindel-PCR(Ramp1)-1S/-1A primer set in the presence of 0.85 mM MgCl2. WT, wild-type genomic DNA used as a positive control; M, 100-bp-ladder markers. Images of these full-length gels are presented in Supplementary Fig. S6a,b.
For Ramp1, we used 8 F0 mice (Nos 1–8) (Supplementary Table S1), which had been obtained by electroporating maternal Cas9-containing sCAT fertilised eggs19 in a solution containing gRNA-Ramp1 alone (Fig. 6b). The 1st PCR conditions were the same as that used for checking mutations in Et1 and Tyr loci. All tested samples were identified as positive (Fig. 6b, upper right panel). For 2nd PCR, the 1st PCR products (0.5–1 ng) were subject to nested PCR using Bindel-PCR(Ramp1)-1S/-1A primer set (Tm, 74 °C–76 °C; Fig. 6b, and Table S3) under these conditions: 95 °C for 1 min, 40–45 cycles of 95 °C for 15 s, 72 °C for 40 s, and 72 °C for 40 s. Sample Nos 4, 5, and 8 (but not WT sample) were identified as negative (Fig. 6b, lower right panel), which suggested that mice Nos. 4, 5, and 8 were biallelic KO mice; subsequent sequencing of the samples confirmed this assessment (Supplementary Fig. S2b).
Furthermore, we performed Taq DNA polymerase-based Bindel-PCR by using 12.5 dpc foetal samples, in which Ramp3 and Rosa26 loci had been targeted (Supplementary Fig. S1a,b). For detection of indels in Ramp3, we used 8 foetuses (nos 1–8), which were derived from C57BL/6J (B6) fertilised eggs electroporated in a solution containing Ramp3-targeting gRNA and Cas9 protein (Table S2 and Supplementary Fig. S1a, left drawing). The 1st PCR conditions were the same as that used for checking mutations in Et1 locus. All tested samples were identified as positive (Supplementary Fig. S1a, upper right panel). For the 2nd PCR, the 1st PCR products (0.5–1 ng) were subject to nested PCR by using Bindel-PCR(Ramp3)-1S/-1A primer set (Tm, 51 °C–52 °C; Supplementary Fig. S1a, left drawing and Table S3) under the following conditions: 95 °C for 1 min, 40–45 cycles of 95 °C for 15 s, 57 °C for 10 s, 72 °C for 40 s, and 72 °C for 40 s, and the reaction was performed in a PCR buffer [10 mM Tris (pH 8.3), 50 mM KCl, 200 μM dNTPs, and 0.8–0.9 mM MgCl2] containing rTaq DNA polymerase (0.5 U/25 μL). Sample numbered 5 was identified as negative (Supplementary Fig. S1a, lower right panel), suggesting that it had biallelic mutations. Subsequent sequencing of the samples confirmed this hypothesis (Supplementary Fig. S2c). Finally, for detection of indels in Rosa26, we used 9 foetuses (nos 1–9), which were derived from C57BL/6J (B6) fertilised eggs electroporated in a solution containing Rosa26-targeting gRNA and Cas9 protein (Table S2 and Supplementary Fig. S1b, left drawing). All tested samples were identified as positive (Supplementary Fig. S1b, upper right panel) as before. For the 2nd PCR, the 1st PCR products (0.5–1 ng) were subject to nested PCR using Bindel-PCR(Rosa26)-1S/-1A primer set (Tm, 60 °C; Supplementary Fig. S1b, left drawing and Table S3) under these conditions: 95 °C for 1 min, 40–45 cycles of 95 °C for 15 s, 68 °C for 10 s 72 °C for 40 s, and 72 °C for 40 s, and the reaction was performed in a PCR buffer [10 mM Tris (pH 8.3), 50 mM KCl, 200 μM dNTPs, and 1 mM MgCl2] containing rTaq DNA polymerase (0.5 U/25 μL). Samples numbered 6 and 9 were identified as negative (Supplementary Fig. S1b, lower right panel), suggesting that they are biallelic foetuses. This point was also confirmed by subsequent sequencing (Supplementary Fig. S2d).
Discussion
We developed a novel method, Bindel-PCR, that enables rapid identification of biallelic indel mutants among CRISPR/Cas9-mediated genome-edited animals having indels at a region within 5 bp 5′ upstream of the PAM (where 94% or more indels are known to generate). It was demonstrated that this new technique allows rapid identification of biallelic indel mutants among samples containing various types of indels and mosaic mutations with 100% accuracy. Notably, in proof-of-principle analyses of the Bindel-PCR method, we successfully identified biallelic KO individuals carrying indels in target loci such as Et1, Tyr, Ramp1, Ramp3 and Rosa26.
Bindel-PCR is characterised by the ability to detect biallelic KO samples alone among samples harbouring various types of indels through 2 PCR steps: 1st PCR, for generating the PCR product that indicates successful amplification of a target region; and 2nd PCR, for showing the lack of a PCR product, which indicates successful identification of biallelic mutation at a target region (Fig. 1a). This method requires only PCR primers, common enzymes for DNA amplification, and a PCR machine and agarose-gel electrophoresis system. These features distinguish our method from other detection systems that depend on the screening of mutations through entire samples by using complexed reagents, enzymatic reactions, and appropriate PCR conditions that require strict optimisation. For example, a sensitive screening technique based on conventional PCR, called ‘annealing at critical temperature PCR (ACT-PCR)’, for identifying mutants was recently reported20; ACT-PCR requires only a single PCR step followed by agarose-gel electrophoresis, but the critical annealing temperature should be carefully examined to avoid false-positive samples. Notably, in relation to this study, we observed that the PCR condition used under standard Mg2+ concentrations (1.5–2.5 mM) and an annealing step at higher temperatures close to or over Tm failed to identify biallelic KO individuals (see PCR with 1.5–2.5 mM in Fig. 2d, e for analysis of Et1 and lower right panels of Fig. 6a,b for analysis of Tyr, Ramp1, and lower right panels of Supplementary Fig. S1a,b for analysis of Ramp3 and Rosa26). Moreover, we tested various dNTP concentrations, 2-step PCR (shuttle PCR), and 3-step touchdown PCR (annealing at 72 °C to 61 °C/decreasing 1 °C per cycle), which are considered as a simple and rapid means to optimise PCR21, for potential establishment of Bindel-PCR, but failed (data not shown). From these analyses, we ascertained that the 3-step PCR (denature/annealing/extension) at low concentrations of MgCl2 is suitable for preferential selection of biallelic KO samples. PAGE has been considered useful for identifying organisms carrying indels22. However, detecting single-base changes such as exchange, deletion, or insertion of 1 nt remains challenging with the laboratory-standard PAGE (Fig. 3c). Furthermore, T7E1 assay is an excellent technique for identification of indels, but requires an additional in vitro assay (e.g., Cas9RNP cut assay) to detect biallelic KO mutations, as shown in Fig. 3b. Collectively, the present Bindel-PCR technique is more convenient than ACT-PCR, and more rapid than the pre-existing PAGE and T7E1 assay for the purpose of rapid identification of biallelic KO mutation, even though T7E1 assay appears to be superior for identifying the indels in the samples.
Interestingly, new techniques have emerged, using fluorescent-PCR or high-resolution melting-curve analysis, that facilitate rapid identification of single-nucleotide mutations23,24. However, the assays require expensive apparatus enabling high-resolution monitoring of the denaturation process of a double-stranded fluorescently labelled DNA fragment. The most promising approach for identifying these cells appears to be DNA sequencing of the target region showing indels by using the Sanger method or next-generation sequencing. Notably, the TIDE (Tracking of Indels by DEcomposition) method based on Sanger sequencing and its web-tool for analysis allow efficient evaluation of indel frequency25, but such strategies based on DNA sequencing are expensive and require several days for handling large numbers of samples.
In the present study, we estimated that the efficiency of success was >94% because the CRISPR/Cas9-induced indels spanned an area of 5 bp immediately before the PAM, and >94% of the genome-edited samples obtained thus far contained mutations in these regions (Fig. 1d); moreover, Bindel-PCR is focused on the identification of samples harbouring biallelic indels within an area of 5 bp upstream of the PAM. The other advantage of using Bindel-PCR is the simplicity of setting PCR conditions. For example, the forward PCR primer is designed to span a 20-bp sequence upstream of the PAM (NGG) or a 20-bp sequence [containing the N in the PAM (NGG)] upstream of the PAM (Figs 1a, 2a, 6 and Supplementary Fig. S1). Next, the reverse PCR primer is designed to correspond to the sequence ~200 bp downstream from the PAM. In this case, caution must be used to ensure that the Tm values of the reverse and forward primers are similar. Furthermore, the annealing temperature in the PCR performed ranged from about −3 °C to +10 °C of Tm value. Three PCR steps, featuring 40–50 cycles of 95 °C–68 °C–72 °C, were used, although Bindel-PCR directed towards Ramp1 was performed using only 2 steps of PCR, with 40–50 cycles of 95 °C–72 °C (Fig. 6b). Optimal conditions were determined by decreasing the MgCl2 concentration (starting from 1.5 and 2.5 mM for rTaq DNA polymerase and HiDi DNA polymerase, respectively). The optimal concentration of MgCl2 was determined under conditions wherein PCR products from the WT samples became invisible. For detection of the biallelic KO samples in which Et1 locus was targeted, the optimal concentration of MgCl2 was around 0.85 mM for rTaq DNA polymerase and around 2 mM for HiDi DNA polymerase. For detection of the biallelic KO samples in which Tyr, Ramp1, Ramp3, and Rosa26 loci had been targeted, the optimal concentration of MgCl2 was around 0.85, 0.85, 0.8, and 1 mM, respectively, for rTaq DNA polymerase (Fig. 6 and Fig. S1). The samples that were judged as negative after PCR was performed using these MgCl2 concentrations are highly likely to be biallelic KO mutants.
A challenge associated with Bindel-PCR might be that biallelic mutants must be selected as PCR-negative samples. To avoid potential false judgment, we performed 1st and 2nd PCR, with the 2nd PCR being tested in the presence of standard or low concentrations of MgCl2. The 1st PCR was set to allow amplification of fragments spanning 400–500 bp of sequence covering the gRNA-recognising region of a target gene (Figs 2a, 6, and Supplementary Fig. S1); this PCR should yield positive PCR products (Fig. 2c, upper right panels of Fig. 6, and Supplementary Fig. S1). Furthermore, the presence of samples containing long sequence deletions in a target gene can be readily detected, because the resultant PCR products differ in size or are occasionally absent (Fig. 6a). The 2nd PCR was set to allow amplification of the 1st PCR products under stringent conditions achieved by reducing the MgCl2 concentration to detect the absence of PCR products derived from biallelic KO samples (Figs 2d,e, 5, 6, and S1). The samples showing negative results when PCR was performed under the low concentration of MgCl2 were judged as biallelic mutants carrying indels of ≥1 bp at least within the 5-bp sequence upstream of the PAM or indels between at least 5 bp of sequence upstream or downstream of the PAM.
Bindel-PCR allows highly sensitive detection of WT samples (Fig. 4a). This ability facilitates the detection of biallelic mutants among a group of genome-edited animals showing enrichment of mosaic mutations. Here, we examined whether Bindel-PCR can be used for effectively identifying the WT alleles included in samples enriched with mosaic mutations. For this purpose, we performed two experiments; one concerning the sensitivity of Bindel-PCR for identifying biallelic mutants among mixed samples containing various indels (Fig. 3), and the other concerning the sensitivity for detecting the presence of WT alleles in samples containing various ratios of mutant-type templates (Fig. 4). In the former, we found that Bindel-PCR could identify biallelic mutants among mixed samples containing various indels. In the latter, we prepared experimental samples, ‘WT:Indel Mix’ samples, in which genomic DNA carrying indels was mixed at various ratios with WT-mouse genomic DNA. Our results showed that Bindel-PCR performed using either rTaq or HiDi DNA polymerase successfully detected WT:Indel Mix = 1:127 (Fig. 4). Because mosaic mutations frequently occur at the 2-cell stage after transfection of zygotes with CRISPR/Cas9-related components, we consider Bindel-PCR to be highly effective in detecting biallelic mutants from offspring enriched with mosaic mutations. Furthermore, this high degree of sensitivity in detecting WT alleles should enable the identification of KO cells from a genome-edited polyploid cell line, such as 3T3.
Is rTaq or HiDi DNA polymerase optimal for Bindel-PCR? Our results support the view that both polymerases can be used for the detection of biallelic mutants. Notably, HiDi DNA polymerase was superior to rTaq DNA polymerase under stringent conditions: PCR product amounts were increased to a higher level with HiDi than rTaq DNA polymerase (Figs 2d,e and 4). Furthermore, HiDi DNA polymerase was also superior in terms of the ability to detect the WT samples among samples enriched with mosaic mutations (Fig. 4). In this context, it might be appropriate to select the DNA polymerase according to the complexity of the genome-edited samples to be examined.
In summary, Bindel-PCR is a T. aquaticus DNA polymerase-based PCR system that enables rapid detection of biallelic KO mutants in CRISPR/Cas9-mediated genome-edited organisms, and this PCR system requires no special reagents or apparatus. Given its simplicity and sensitivity, we expect Bindel-PCR to serve as an effective screening tool for identifying biallelic KO cells/individuals from large numbers of samples.
Methods
Ethics approval of mouse experiments
All experimental animal procedures were performed according to the guidelines of Shinshu University Committee on Recombinant DNA Security and were approved by the Animal Care and Experimentation Committee of Shinshu University (permit no. 300044).
Mice
C57BL/6J (hereafter ‘B6’) and ICR mice were purchased from CLEA Japan Inc. (Tokyo, Japan). Systemic Cas9-expressing Tg mice (‘sCAT’ mice) were produced in-house19.
Data source for nucleotide sites frequently showing CRISPR/Cas9-induced indels
To investigate the sites that are susceptible to CRISPR/Cas9-induced mutation, we used the data obtained in our recent study (Sakurai et al., unpublished data) for mouse Et1 and in other studies for rat Tyr, Rosa26, Sirpa, and Cyp2d15, and for mouse Fgf1011.
Reagents and PCR primers
Reagents were purchased from Wako Pure Chemical Industries, (Osaka, Japan), unless specified otherwise. The primers used for PCR are listed in Supplementary Table S3; the primers were ordered for synthesis as desalted oligonucleotides (Invitrogen Custom Primers Japan, Thermo Fisher Scientific K.K., Tokyo, Japan).
PCR, T7E1 assay, and gel image acquisition
We used 3 DNA polymerases in this study. For constructing a series of Et1 plasmids carrying indels, we used Tks Gflex DNA polymerase, a high-fidelity DNA polymerase derived from Thermococcus sp. (TaKaRa Bio. Inc., Shiga, Japan). For the establishment of Bindel-PCR and for its fidelity test, we used a cloned Taq (rTaq) DNA polymerase derived from T. aquaticus DNA polymerase (TaKaRa TaqTM #R001A, TaKaRa) and HiDi DNA polymerase (myPOLS Biotec #9001, myPOLS Biotec GmbH, Konstanz, Germany). We prepared MgCl2-free PCR reaction buffers for rTaq DNA polymerase [10×; 100 mM Tris (pH 8.3), 500 mM KCl, and 2 mM dNTPs] and HiDi DNA polymerase [10×; 500 mM Tris (pH 9.2), 160 mM (NH4)2SO4, 1% Tween-20, and 2 mM dNTPs], and before PCR, we added MgCl2 at appropriate concentrations to the PCR reaction buffer (1×). PCR was performed in a 25-μL volume, and 10-μL of the PCR products were electrophoresed using a 1.5% agarose-gel/TAE buffer system and a standard 5% polyacrylamide gel/TBE buffer system used in the laboratory. T7E1 assay was performed as described by Sakurai et al.12. The Gel images were acquired by 0.3 s exposure time with a CCD-gel imaging system (Nippon Genetics FAS-VI, Nippon Genetics, Tokyo, Japan) and cropped by Adobe Photoshop CS5 (12.0.4 × 64 version, Adobe Systems, CA, USA).
Et1 plasmids carrying a series of indels
We generated a series of Et1 plasmids carrying various types of indels (Fig. 2b). First, we constructed an Et1 plasmid carrying WT Et1 cDNA. Genomic DNA isolated from a B6 mouse was PCR-amplified using an Et1ch-2S/-2A primer set to obtain a 450-bp PCR product, which was then subcloned into pBluescript II (Agilent Technologies Japan, Tokyo, Japan) to obtain pBs/WT Et1. The resultant plasmid was sequenced to confirm that the inserted fragment was the authentic cDNA. Next, we generated a series of Et1 plasmids harbouring indels through inverse-PCR performed using pBs/WT Et1 as template, and the resultant Et1 plasmids were sequenced to confirm that they contained the appropriate target sequences.
Preparation of model samples used for fidelity test of Bindel-PCR
For testing the fidelity of Bindel-PCR, we used samples carrying indels in the loci Et1 (Fig. 5), Tyr, Ramp1, Ramp3 and Rosa26 (Figs 6, S1, S2, and Supplementary Tables S1 and S2). For generating mice harbouring indels, we employed the in vitro zygote electroporation method11 using CUY21Edit II (BEX, Tokyo, Japan). The gRNAs targeting Tyr, Ramp1, Ramp3, and Rosa26 (Fig. 6 and Supplementary Fig. S1) were prepared as described by Sakurai et al.12, and the zygotes were prepared according to the methods described in our previous study19. For producing mice harbouring indels in Tyr, Ramp3 and Rosa26 loci, we used B6-derived zygotes and a Tyr, Ramp3 and Rosa26-targeting gRNA complexed with Cas9 protein (IDT #1074181, IDT Inc., IL, USA). For generating mice carrying indels in Ramp1 locus, we used sCAT zygotes19 and a Ramp1-targeting gRNA alone. Zygotes electroporated in vitro in the presence of CRISPR/Cas9 components were transferred to the oviducts of pseudopregnant ICR females, which then delivered the pups and 12.5 dpc foetuses. For indel analysis, genomic DNA was isolated from a piece of ear dissected from the pups that were born, and 12.5 dpc foetuses, and the region recognised by the gRNAs was PCR-amplified. Subsequently, the resultant PCR products were subcloned into a TA cloning vector (TaKaRa). Sequencing was performed using BigDye Terminator Cycle Sequencing Kit ver3.1 and an ABI Genetic Analyzer 3130 (Applied Biosystems, Life Technologies Japan, Tokyo, Japan). Data were analysed using Genetyx-Mac ver.13.0.3 (Software Development, Tokyo, Japan) and ClustalW server (http://www.genome.jp/tools-bin/clustalw).
References
Fernandez, A., Josa, S. & Montoliu, L. A history of genome editing in mammals. Mamm Genome 28, 237–246 (2017).
Hsu, P. D., Lander, E. S. & Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014).
Knott, G. J. & Doudna, J. A. CRISPR-Cas guides the future of genetic engineering. Science (New York, N.Y 361, 866–869 (2018).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science (New York, N.Y 339, 819–823 (2013).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (New York, N.Y 337, 816–821 (2012).
Cortizas, E. M. et al. Alternative end-joining and classical nonhomologous end-joining pathways repair different types of double-strand breaks during class-switch recombination. J Immunol 191, 5751–5763 (2013).
McVey, M. & Lee, S. E. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet 24, 529–538 (2008).
Sfeir, A. & Symington, L. S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends in biochemical sciences 40, 701–714 (2015).
Lino, C. A., Harper, J. C., Carney, J. P. & Timlin, J. A. Delivering CRISPR: a review of the challenges and approaches. Drug delivery 25, 1234–1257 (2018).
Sato, M., Ohtsuka, M., Watanabe, S. & Gurumurthy, C. B. Nucleic acids delivery methods for genome editing in zygotes and embryos: the old, the new, and the old-new. Biology direct 11, 16 (2016).
Hashimoto, M. & Takemoto, T. Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Scientific reports 5, 11315 (2015).
Sakurai, T., Watanabe, S., Kamiyoshi, A., Sato, M. & Shindo, T. A single blastocyst assay optimized for detecting CRISPR/Cas9 system-induced indel mutations in mice. BMC biotechnology 14, 69 (2014).
Vouillot, L., Thelie, A. & Pollet, N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda, Md 5, 407–415 (2015).
Watanabe, S., Sakurai, T., Nakamura, S., Miyoshi, K. & Sato, M. The Combinational Use of CRISPR/Cas9 and Targeted Toxin Technology Enables Efficient Isolation of Bi-Allelic Knockout Non-Human Mammalian Clones. International journal of molecular sciences 19 (2018).
Yoshimi, K. et al. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nature communications 7, 10431 (2016).
Drum, M., Kranaster, R., Ewald, C., Blasczyk, R. & Marx, A. Variants of a Thermus aquaticus DNA polymerase with increased selectivity for applications in allele- and methylation-specific amplification. PloS one 9, e96640 (2014).
Yen, S. T. et al. Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Developmental biology 393, 3–9 (2014).
Hashimoto, M., Yamashita, Y. & Takemoto, T. Electroporation of Cas9 protein/sgRNA into early pronuclear zygotes generates non-mosaic mutants in the mouse. Developmental biology 418, 1–9 (2016).
Sakurai, T. et al. A non-inheritable maternal Cas9-based multiple-gene editing system in mice. Scientific reports 6, 20011 (2016).
Hua, Y., Wang, C., Huang, J. & Wang, K. A simple and efficient method for CRISPR/Cas9-induced mutant screening. Journal of genetics and genomics 44, 207–213 (2017).
Don, R. H., Cox, P. T., Wainwright, B. J., Baker, K. & Mattick, J. S. ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic acids research 19, 4008 (1991).
Zhu, X. et al. An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Scientific reports 4, 6420 (2014).
Dahlem, T. J. et al. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS genetics 8, e1002861 (2012).
Palais, R. A., Liew, M. A. & Wittwer, C. T. Quantitative heteroduplex analysis for single nucleotide polymorphism genotyping. Analytical biochemistry 346, 167–175 (2005).
Brinkman, E. K., Chen, T., Amendola, M. & van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic acids research 42, e168 (2014).
Steitz, T. A. A mechanism for all polymerases. Nature 391, 231–232 (1998).
Acknowledgements
T.Sa. received a JSPS KAKENHI Grant (16K15233 and 19K07477) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology. This study was also supported by the Core Research for Revolutionary Science and Technology (CREST), Japan Science and Technology Agency (JST), and Japan Agency for Medical Research and Development (AMED) Core Research for Revolutionary Medical Science and Technology (AMED-CREST).
Author information
Authors and Affiliations
Contributions
T.Sa. designed the experiments, performed experiments, and drafted the manuscript. T.Sa. and M.S. analysed the data and edited the manuscript. S.W. helped with PCR experiments. N.T., A.K., and T.Sh. assisted in conducting the experiments.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sakurai, T., Kamiyoshi, A., Takei, N. et al. Bindel-PCR: a novel and convenient method for identifying CRISPR/Cas9-induced biallelic mutants through modified PCR using Thermus aquaticus DNA polymerase. Sci Rep 9, 9923 (2019). https://doi.org/10.1038/s41598-019-46357-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46357-8
This article is cited by
-
Identification of CRISPR-Induced Mutations in Plants: with a Focus on the Next-Generation Sequencing Assay
Journal of Plant Biology (2022)
-
Nucleotid-genaue PCR: Von der Mutationsdetektion zur Genom-Editierung
BIOspektrum (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
