
Abstract
The etiopathogenesis of type 1 diabetes (T1D), a common autoimmune disorder, is not completely understood. Recent studies suggested the gut microbiome plays a role in T1D. We have used public longitudinal microbiome data from T1D patients to analyze amyloid-producing bacterial composition and found a significant association between initially high amyloid-producing Escherichia coli abundance, subsequent E. coli depletion prior to seroconversion, and T1D development. In children who presented seroconversion or developed T1D, we observed an increase in the E. coli phage/E. coli ratio prior to E. coli depletion, suggesting that the decrease in E. coli was due to prophage activation. Evaluation of the role of phages in amyloid release from E. coli biofilms in vitro suggested an indirect role of the bacterial phages in the modulation of host immunity. This study for the first time suggests that amyloid-producing E. coli, their phages, and bacteria-derived amyloid might be involved in pro-diabetic pathway activation in children at risk for T1D.
Similar content being viewed by others


Introduction
Type 1 diabetes (T1D) is an autoimmune disorder driven by T cell-mediated destruction of the insulin-secreting β-cells of the pancreatic islets that often manifests during childhood1. There are a number of factors associated with the development of T1D2,3,4. The strongest susceptibility alleles for T1D encode certain human leukocyte antigens (HLAs), which play a central role in autoreactive T-cell activation in T1D pathogenesis5,6. Only a portion (3–10%) of children carrying HLA risk alleles will develop T1D, pointing out the role of environmental factors in disease initiation; however, the role of environmental factors in T1D etiology remains elusive3,7,8.
T1D is generally associated with a long pre-diabetic seroconversion period, during which autoantibodies to antigens of pancreatic β-cells or insulin are produced9. There are a few known factors that drive autoimmunity during infancy, such as so-called “natural apoptosis” or spontaneous cell death within the β-cell population, deposition of islet amyloid polypeptide (IAPP) aggregates, microbiota alterations, or viral infection that specifically targets pancreatic β-cells and leads to islet cell death, which contribute to the formation of β-cell antigens, activation of dendritic cells (DC), and antigen presentation1,10,11,12,13,14,15,16. However, the exact roles of these mechanisms in disease onset remain poorly understood1.
Pancreatotropic viruses, such as enterovirus, coxsackie B, mumps, rubella, and cytomegalovirus, have been evaluated as triggers of T1D since the discovery of antiviral antibodies in diabetic patients and because of the role of viral infection in elevated levels of type I interferon (IFN), which is known to play a role in T1D progression17,18,19. However, in many patients who develop T1D, there is no clear link with viral infections20. Moreover, the mimicry between β-cell autoimmune antibodies and viral epitopes of coxakieviruses, which are the most strongly associated with T1D, has recently been shown not to underlie T1D pathogenesis21,22. Natural apoptosis has been described only in certain patients and highlights the gaps in our knowledge about disease initiation1. Therefore, despite decades of research, the etiology of autoimmune response induction remains elusive22.
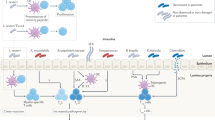
Previous studies have established a correlation between seroconversion and chronic inflammation resulting from impaired gut barrier function and increased intestinal permeability, which is suggested as a hallmark of T1D11. Given the overarching influence of gut bacteria on human health, including intestinal permeability and its regulatory role in autoimmunity, the role of the microbiota in T1D development has been recently explored12,23,24,25,26. The microbiota of the human intestinal tract is comprised of bacteria, fungi, and eukaryotic and bacterial viruses (bacteriophages). Bacteria in the human gut live within surface-associated microbial communities termed biofilms, which are characterized by the presence of self-produced extracellular matrix and a surface film that protects the microorganisms from the outer environment27,28,29. The extracellular matrix consists of different biomolecules, including extracellular nucleic acids, polysaccharides, and proteins. Several microorganisms within the human microbiome, predominantly members of Enterobacteriaceae, also produce amyloid proteins that can form so-called “curli fibers” which provide biofilms with unique mechanical properties30,31. Bacterial curli proteins of Enterobacteriaceae form highly ordered β-sheet rich amyloid composed of a major subunit, CsgA, and a minor subunit, CsgB32. Bacterial amyloids share certain similarity with human amyloids; however, whereas amyloid in bacteria is suggested to be physiological, in mammals, the formation of misfolded, insoluble, highly ordered prion-like cross-β structures is commonly associated with a variety of diseases10. In humans, pathological deposition of insoluble amyloid aggregates has been shown to be associated with the development of Creutzfeldt-Jakob disease, Alzheimer’s disease, Parkinson’s disease, and T1D, where an increased islet amyloid polypeptide (IAPP) concentration may constitute a risk factor for β-cell destruction33,34. Moreover, similarities between bacterial and eukaryotic amyloid β-sheets have been implicated in inter-kingdom communication through the toll-like receptor signaling pathway, leading to the deposition of misfolded protein in the brain and altered amyloidogenesis in Caenorhabditis elegans and rats following colonization with amyloid-producing E. coli35. Notably, in that work, the authors showed that bacterial curli fibers triggered T-cell immunity through the TLR-2-MyD88-NF-kB signaling pathway, which is also known to be associated with T1D progression; however, no microbiome studies have been conducted to reveal the association of amyloid-producing bacteria with diabetes36. Moreover, only a limited number of studies on the human microbiome have revealed alterations that may be associated with T1D. Kostic et al. and Zhao et al. found that an increased abundance of Blautia spp., Ruminococcus spp., and OTUs belonging to the genus Bacteroides positively correlated with T1D progression, suggesting that the primary pathological implication in autoimmunity might be through an altered metabolite profile associated with these bacteria, including triglyceride and branched-chain amino-acid production22,37. Notably, a previous microbiota study revealed microbiome alterations that emerged only after the appearance of autoantibodies, suggesting that such alterations are involved in clinical progression to T1D rather than in disease initiation12.
There are multiple causes of gut bacterial alterations in T1D, and bacteriophages are considered to be one of the most influential, but are poorly studied in this regard38. Data from a phagobiota analysis of children with T1D are even more limited, and showed that alterations in Bacteroides phages are discriminative for disease status, but not clearly associated with disease progression38. Bacteriophages as possible agents that may negatively affect mammalian health have attracted scientific attention only recently39. We have previously shown that bacteriophages can alter the mammalian microbiota, leading to increased intestinal permeability and triggering chronic inflammation, which potentially participate in protein misfolding and might contribute to the onset of Parkinson’s disease40,41.
In this study, we conducted a detailed comparative metagenomic analysis of amyloid-producing gut bacteria and the associated phagobiome in a prospective, longitudinal cohort of HLA-matched infants up to 3 years of age. We retrieved a dataset of short sequence reads generated by Kostic et al. from the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/bioproject/382085) and analyzed amyloid-producing bacteria and bacteriophage diversity using MetaPhlAn and a custom method22,42.
This study for the first time revealed alterations in the abundance of intestinal amyloid-producing bacteria during early life due to prophage activation that that correlated with the development of T1D-associated autoimmunity. These data were supported by findings in vitro that revealed significant amyloid release from E. coli biofilms upon induction by prophages, suggesting a potential involvement of E. coli-derived curli fibers in pro-diabetic activation pathways in children at risk for T1D.
Results
Autoimmunity development is associated with an alteration in amyloid-producing bacterial abundance
To explore the potential link between the abundance of amyloid-producing bacteria and T1D-associated seroconversion, we used longitudinal shotgun metagenomics sequencing data of the fecal microbiome from 10 children who exhibited autoantibodies (six seroconverters and four children who developed T1D) and eight non-seroconverted HLA-matched control individuals. All patient characteristics were previously reported by Kostic et al. (Supplementary Table S1)22. HiSeq-2500 sequencing was used, producing an average of ~2.5 Gb per sample. Amyloid-producing bacteria were represented by E. coli, Staphylococcus aureus, and Salmonella spp29,30. Among these amyloid-producing bacteria, E. coli was the major group, while the other curli-producing bacteria were identified only in a single sample and at a few collection times and thus, were disregarded in subsequent analysis (Supplementary Table S2). We first compared the association between E. coli abundance, disease phenotype, and collection time using a two-tailed Mann–Whitney U test (Fig. 1; Supplementary Table S3). We clustered the samples into time-point bins of 300 days, and studied the dynamics of E. coli abundance over 1300 days within and among patient groups. E. coli abundances demonstrated a different dynamic relationship across time for case and control groups. E. coli tended to disappear in T1D and seroconverters over the time, whereas in controls, E. coli abundance tended to increase and did not change significantly over time. In both the seroconverter and T1D groups, we found a statistically significant decrease in E. coli abundance when comparing the first 0–300 day period and the following 300–600, 600–900, 900–1300 time bins (p < 0.05). Notably, the initial abundance of E. coli was significantly higher in cases than in controls at 0–300 days. The case groups responded differently over time, and importantly, data from the control group revealed that there was no variation in E. coli associated with age.

E. coli Abundance Over Time. Boxplot showing absolute E. coli abundances in the seroconverter, T1D, and control groups in the indicated periods. E. coli abundance across different time points was compared using two-tailed Mann–Whitney U test. Statistically significant variation between selected conditions (*p < 0.05, **p < 0.01 and ***p < 0.005).
To explore the relation between E. coli abundance and autoimmunity further, we next analyzed whether alterations in the abundance of E. coli could be associated with the development of autoantibodies in more detail, and whether or not E. coli abundance could distinguish the T1D disease state. To this end, we studied the appearance and disappearance of E. coli in each patient in dynamic association with the appearance of autoantibodies (Fig. 2). We found a positive correlation between the total disappearance or presumed disappearance (defined as a >50-fold decrease) in E. coli and autoantibody appearance in the T1D and seroconverter groups, but not in controls43. All T1D patients showed disappearance or an episode of disappearance of E. coli before the detection of autoantibodies and disappearance of E. coli prior to the diagnosis of diabetes (Supplementary Table S4). In seroconverters, E. coli disappeared in all patients, except E003989 and T013815. For patient E10629, data were difficult to interpret because of the long period between sample collections (556 days between the last sample taken before seroconversion and the first sample after seroconversion). In any case, E. coli was absent in the first sample following seroconversion; thus, it is reasonable to assume that it had disappeared before autoantibody appearance. Notably, in certain control patients, E. coli disappeared at some time points; however, this was followed by a restoration of the E. coli population in all cases, which was not regularly seen in case subjects.

Association Between the Dynamics of the Disappearance of E. coli and Seroconversion. Each row represents an individual, with each symbol indicating a single stool sample. The X-axis indicates age at sample collection. E. coli abundance is >0.01. Green indicates presence of E. coli, and red indicates disappearance or presumed disappearance (over 50-fold reduction when compared with initial abundance) of E. coli. Diamonds represent the time point at which autoantibodies were detected.
Next, we examined changes in the absolute abundance of E. coli in each patient individually among different periods, and we presented the data as a heatmap (Fig. 3). We detected alterations in E. coli abundance within children from the control group, followed by restoration of the E. coli population to the levels equal to or above the pre-seroconversion levels, whereas restoration of the E. coli population to preceding levels was noted in only a few children from the case groups. This more detailed analysis has also revealed some previously overlooked data, for patient T013815. Although in this subject, E. coli did not disappear prior to the seroconversion, the appearance of autoantibodies occurred in the period when E. coli started to significantly decrease from the highly elevated abundance compared to other patients. Another observation that attracted our attention was that the initial abundance of E. coli in T1D and seroconverter groups was significantly higher than that in the control group, which confirmed the data presented in Fig. 1. The result further supports that the initial higher abundance of E. coli and the decrease of amyloid-producing bacteria abundance in case group were signatures associated with autoimmunity and disease progression.

Absolute Abundances of E. coli Across Samples. Fecal bacterial communities were analyzed by high-throughput Illumina Hiseq2500 sequencing. Threshold for E. coli abundance is >0.01. Each row represents an individual, with E. coli abundance in each stool sample (indicated with different symbols) being color-coded using a color gradient, ranging from red (low abundance) to purple (maximum abundance). Samples with no E. coli are marked with white circles. The X-axis indicates age at sample collection. Diamond sign represents the time of seroconversion detected.
In addition, we studied the potential correlation between the disappearance of E. coli and certain HLA alleles or the appearance of particular autoantibodies, IAA, GADA, IA2A, ZNT8A or ICA, in case groups. No correlations were observed (data not shown). We also detected no association between E. coli disappearance and breastfeeding duration, Bifidobacterium abundance, or antibiotic usage, which are known to influence Enterobacteriaceae (Supplementary Table S5, Supplementary Table S6)44.
E. coli bacteriophages as signatures associated with seroconversion
As we identified E. coli as T1D-discriminative bacteria, we next studied the relationships between this microorganism and bacteriophages of E. coli, as phages are known as main regulators of bacterial populations. First, we detected 63 E. coli phage species in case and control groups. We evaluated possible differences in E. coli phages, using α-diversity, in the pre-seroconversion period (Supplementary Table S7). For the samples from the control group, we set the medium time to seroconversion in case groups (540 days) as an artificial benchmark. Phage richness was statistically different between control and seroconversion individuals as indicated by ACE and Chao1 indexes (ACE: p = 0.0181; Chao1: p = 0.0137), but was not statistically significantly different between control and T1D, most likely because of the small T1D patient cohort (ACE: p = 0.6086; Chao1: p = 0.6359). E. coli phage diversity tended to be lower in the control subjects than in seroconverters (Shannon, p = 0.1526; Simpson, p = 0.5437), and was significantly lower in controls vs. T1D cases (Shannon, p = 0.0055; Simpson, p = 0.0248) (Supplementary Table S7).
We observed that nearly all E. coli phages were lysogenic, whereas strictly virulent lytic phages, such as Enterobacteria phage IME10 or Enterobacteria phage 9 g, were found only in a few samples (Supplementary Table S2). The predominance of lysogenic E. coli phages clarifies why α-diversity indices revealed trends of decreased evenness and diversity of phages in case groups compared to controls in pre-seroconversion samples, which most likely reflected lower E. coli abundance in controls (Supplementary Table S2).
Next, we analyzed the E. coli phage/E. coli bacterial cell ratio, which represents the “lytic potential” (Fig. 4; Supplementary Table S8; Supplementary Table S9)45. To this end, we first normalized the E. coli phage abundance to that of E. coli. In theory, the phage/bacteria ratio reflects whether or not a prophage is stably integrated within the host bacterial genome41. A low ratio indicates that prophage is most likely absent in the genomes of part of the bacterial host population, whereas a high ratio suggests active, productive phage-induced bacterial lysis. The phage/bacteria ratio increased in subjects from all groups prior to the decrease in E. coli abundance, indicating that productive phage infection was the cause of E. coli depletion. Notably, that observation reflected a statistically significant elevation in the phage/bacteria ratio before the appearance of autoantibodies in the T1D and seroconverter groups (Supplementary Table S10).

Association Between E. coli phage/E. coli Ratio Across Samples and Seroconversion. Each row represents an individual, with each symbol representing the phage/bacterial ratio, calculated as E. coli phage abundance normalized to that of the E. coli abundance, of a sample. Abundance is represented as a color gradient, ranging from purple (lowest ratio) to yellow (maximum ratio). Samples in which E. coli phages but no bacterial cells were present are marked with white circles. Samples with total E. coli disappearance or presumed disappearance with over 50-fold decrease in E. coli are indicated by red circles. The X-axis indicates age at sample collection. The diamond symbol represents the time point at which seroconversion was detected.
To evaluate the implication of the increase in the phage/bacteria ratio as a driving force behind E. coli depletion further, we analyzed which phages had a correlation with the depletion of E. coli abundance across most subjects. The most frequently found lysogenic phages with an inverse relationship with E. coli abundance are the representatives of the Peduovirinae subfamily and of the unclassified Lambdavirus subfamily within the Caudovirales order. The increase in number of the lysogenic E. coli phages along with the decrease in E. coli abundance indicated that there was a productive bacteriophage infection that led to bacterial host death and the release of phage progeny45. These data revealed previously overlooked particularities of the phagobiota in T1D, suggesting a primary role of induction of certain E. coli prophages in E. coli depletion, and an association with autoimmunity and T1D development.
Role of prophage induction in the release of amyloid from E. coli biofilms
We evaluated how the die-off of E. coli populations due to prophage induction could lead to amyloid release. We used 48-h old E. coli biofilms with confirmed curli expression (curli formation on 48-h colonies was visible with the naked eye on petri dishes) (Supplementary Fig. S1). Prophages were induced with mitomycin C. Lytic bacteriophage development was confirmed as an increase in plaque forming units (PFU) and a reduction in colony-forming units (CFU). The presence of phage in E. coli biofilm was confirmed by the production of plaques on a non-induced E. coli control culture between 4 h and 10 h. The effect of prophage induction in E. coli biofilm on CFU and PFU is summarized in Table 1. Phages started to appear in the biofilm as of 4 h after prophage induction, with maximum PFU at 8 h, which coincided with a decrease in CFU.
Next, we evaluated the effect of prophage induction on the amount of amyloid fibers released into the biofilm supernatant. To quantify the amount of aggregated amyloid, we evaluated its binding to the amyloid-diagnostic dye Congo red (CR)29. We observed a significant increase in CR absorbance upon binding to supernatant 8 h following prophage induction as compared with control samples (Fig. 5). Like for other curli fibers, heating of the E. coli supernatant induced a spectral shift of the CR solution, with a maximum difference in absorbance between CR alone and CR bound to amyloid fibers at ~541 nm46. These data suggested that the phage-mediated lysis of E. coli led to the release of amyloid aggregates into the biofilm.

Prophage Induction Promotes Amyloid Release from E. coli in vitro. CR absorbance upon binding to amyloid from intact E. coli supernatant (dashed line) and from E. coli supernatant after prophage induction (solid line). Both curves are approximately equal to zero at the isosbestic point (403 nm), and the maximum difference in absorbance was observed at 541 nm. Both spectra were normalized to the absorbance spectrum of free CR.
Discussion
This study revealed an association between the dynamics of intestinal amyloid-producing E. coli prior to seroconversion in HLA-matched children and the development of T1D-associated autoimmunity. Several lines of evidence support roles of E. coli, E. coli phages, and E. coli-derived amyloid in T1D.
First, in seroconverter and T1D patients, we observed a statistically significant elevation in E. coli abundance from day 0 to 300, followed by E. coli depletion. Notably, we observed the total disappearance or a pronounced depletion of E. coli prior to the appearance of autoantibodies in all children who progressed to T1D. A similar trend was noticed among most patients in the seroconverter group. Based on comparison with the control group, we confirmed that the observed changes in amyloid-producing bacteria were associated with disease state, not with age. Despite the relatively small patient cohort studied, the longitudinal analysis and the large total number of samples (120) analyzed provided sufficient statistical power to identify significant changes38. There are multiple factors that could lead to E. coli depletion within the gut microbiome, including interplay with other gut microorganisms, which constantly change during infancy. E. coli is a member of Enterobacteriaceae, which are known to positively correlate with Bifidobacteriaceae, which are affected by breastfeeding22,44. With that in mind, we evaluated a possible association between these families; however, we found no association between E. coli, duration of the breastfeeding, and Bifidobacteriaceae abundance. Another environmental factor that can affect the intestinal E. coli population is antibiotic usage. Although some children in this study had been exposed to antibiotics, no association between the timing of antibiotic usage and E. coli depletion was noticed47. However, the most profound regulators of bacterial populations are bacteriophages. Notably, we found a positive correlation between the marked decline in E. coli abundance and an increase in the E. coli phage/E. coli bacterial cell ratio. The strongest association was observed for lysogenic E. coli phages from the Peduovirinae subfamily in the family Myoviridae and the number of unclassified Lambdavirus, in the family Siphoviridae in the order Caudovirales suggesting these phages might play a role as a diabetogenic indirectly leading to the release of E. coli amyloid-DNA complexes. Most likely, prophage induction that led to E. coli depletion was triggered by an external stimulus39,40,48. Prophage induction signals vary among bacteriophages, but generally are activated in response to environmental and internal factors, such as bacterial SOS response molecules triggered by antimicrobial agents, oxidative stress, and DNA damage49,50. However, we found no relation between the timing of antibiotic usage and E. coli disappearance relevant to prophage induction.
Significant die-off of the host population by prophage activation results in microbial biofilm disruption, leading to the release of pathogen-associated molecular patterns (PAMPs)51. Accordingly, the E. coli die-off observed in this study might have led to the release of a variety of PAMPs, such as LPS, DNA, and amyloid; however, for the majority of PAMPs, their role in T1D remains elusive. Moreover, LPS derived from E. coli possess anti-diabetic activity, inhibit innate immune signaling, and enhance endotoxin tolerance in non-obese diabetic mice52. The predominant component expressed in E. coli biofilms that is unique to amyloid-producing Enterobacteria is curli amyloid fiber, which forms highly immunogenic complexes with DNA53. Amyloid-DNA composites are known to intensively stimulate TLR2 and TLR9, inducing immunogenic reactions, and they have been associated with the triggering of other autoimmune disorders, including systemic lupus erythematosus53,54. Therefore, it is reasonable to speculate that die-off of the intestinal E. coli population, particularly considering its initial high abundance among children from the case groups, might lead to the prominent release of curli fibers and DNA-amyloid complexes that in turn act as pro-diabetic protein and trigger the autoimmune cascade in susceptible hosts. In favor of the suggested diabetogenic role of E. coli amyloid, it has been previously implicated in autoimmunity associated with increased type I IFN-induced activation and protein misfolding55,56. Type I IFN is a signature cytokine in T1D patients and is known to be one of the injury agents during viral infection, which actually led to the hypothesis of the viral nature of pancreatic damage in T1D21. Type I IFN is implicated in early T1D stages by triggering β-cell apoptosis and the formation of β-cell autoantigens19. Notably, E. coli curli-DNA composites have the potency to stimulate the type I IFN response via two ways: through TLR stimulation of β-cells and of DCs54. Indeed one of the pathways underlying the potential diabetogenic action of E. coli amyloid-DNA complexes might be its direct triggering of pancreatic β-cell death by activating TLR2 and TLR9, which in turn leads to the production of type I IFN and the subsequent well-defined cascade of T1D autoimmune alterations19,55. Alternatively, E. coli-derived amyloid might interact with antigen-presenting cells. Recent studies on other autoimmune diseases, such systemic lupus erythematosus, have shown that microbial curli fibers can lead to the activation of TLR2 and TLR9 on DCs, thus stimulating immune responses, including increased type I IFN production55,56,57,58. Moreover, microbial amyloid has been shown to engage TLRs and trigger systemic inflammation through interaction with Peyer’s patches and lymph follicles59,60.
We can speculate that finally, bacterial amyloid might lead to β-cell destruction and β-antigen release by stimulating IAPP aggregation and deposition in the pancreas59. IAPP plays multiple roles in T1D progression: on the one hand, it leads to β-cell destruction, and on the other hand, it reportedly is an autoantigen for T1D triggering60,61,62. How bacterial amyloid can contribute to IAPP accumulation remains unclear; however, recent studies have revealed a trans-kingdom interplay between E. coli curli fibers and alpha-synuclein aggregation in the gut and brain of Parkinson’s disease models, and in triggering amyloidosis in systemic lupus erythematosus following Salmonella typhimurium curli-DNA treatment35,54.
Next, we evaluated the diabetogenic potential of E. coli prophages by the fact that their induction that modulated an elevated E. coli phage/E. coli ratio from microbiome analysis) could contribute to the release of bacterial amyloid48,63. We have determined that in vitro activation of E. coli prophages with mitomycin C results in the pronounced amyloid release from preformed microbial biofilms, which according to data from metagenomics analysis, suggests that the same process occurs in the gut of those children who further develop autoimmunity and T1D64.
For E. coli-derived gut amyloid to activate either one of the above signaling pathways triggering autoimmune cascade and T1D, it has to reach antigen-presenting cells or pancreatic cells59. This can happen in the case of increased intestinal permeability, leading to the translocation of microbial byproducts to the lamina propria, or through interaction with Peyer’s patches59,65. Although we did not analyze intestinal permeability, altered gut barrier function is well described in T1D, with certain studies pointing it out as an important pathogenic factor for T1D11,66. Therefore, the model we propose might shed another light on the role of increased intestinal permeability–the mechanistic impact of which on T1D pathogenesis was unclear to date– suggesting that leaky gut allows E. coli amyloid-DNA complexes to pass to the lamina propria and trigger autoimmunity and T1D progression66.
We cannot conclusively explain the disappearance of E. coli in some control patients (who also had HLA risk for T1D); the lower abundance of E. coli prior to the decrease in these subjects compared to the T1D and seroconverters groups attracted our attention (Fig. 4). It is possible that the lower amount of E. coli resulted in insufficient amyloid release to trigger autoimmunity. Another reasons for the differential effects of E. coli depletion across patients could be alteration of permeable state for intestinal barrier and the presence of E. coli strains with different levels of curli expression or amyloid synthesis under the different growth conditions; however, these possibilities need further study.
Finally, it is reasonable to speculate that the released E. coli phages themselves might have played a particular role in triggering T1D autoimmunity. The role of bacteriophages in the interplay with the immune system is poorly described; however, recent studies have suggested that bacterial viruses interact with eukaryotic cells, including immune cells67,68.
Our analysis revealed previously overlooked correlations between an initially high level of amyloid-producing E. coli in the intestine, followed by their depletion, most likely due to prophage induction, and the initiation of autoimmunity and T1D progression. The diabetogenic role of E. coli prophages was supported by the fact that the activation of E. coli prophages with mitomycin C resulted in pronounced amyloid release from preformed microbial biofilms in vitro. Together with the data from metagenomics analysis, these findings suggested the same process might occur in the gut of children who developed autoimmunity and T1D. Combing with existing data on the immunogenic role of enterobacterial amyloid, our findings suggest that curli released by E. coli might trigger autoimmunity in susceptible children, highlighting the need to pay specific attention to the relationships between amyloid-producing bacteria and their bacteriophages in genetically susceptible hosts, although additional studies using an in vivo animal models are required to determine the role of phages in amyloid-release. Another important issue that has to be addressed in the follow-up studies is the identification of the stimuli that might induce E. coli prophage induction, and one such possible stimuli might arise from bacterial and fungal components of microbiota that are known to release a variety of molecules responsible for prophage induction. Determining the exact role of E. coli-derived amyloid in the progression of T1D may lead to novel diagnostics and interventional approaches; however, obviously, further, detailed studies are required.
Materials and Methods
Study population
We used data from a prospective, longitudinal cohort study by Kostic et al. of 16 HLA-matched infants followed from birth until 3 years of age22. The children, from Finland and Estonia, were recruited to the study between September 2008 and August 2010 (http://www.diabimmune.org/). The study generated shotgun metagenomics sequencing data of the fecal microbiome from 10 children who exhibited autoantibodies (six who developed serum autoantibodies with no progression to T1D, and four who were seroconverted and developed T1D) and eight non-seroconverted control individuals22. Inclusion criteria were: presence of HLA DR-DQ alleles associated with T1D development. Data on diabetes-associated autoantibodies are presented in the original study. Demographic parameters of the study participants and data collection details are presented in Supplementary Table S1.
Microbiota sequencing and processing
High-throughput shotgun sequencing was performed on the Illumina HiSeq2500 platform, generating ~2.5 Gb of sequence per sample with 101-bp paired-end reads. Human contamination was removed with the BMTagger (ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/). Bacterial and phage contents were quantified separately using the SRA shotgun metagenomic sequencing data. Bacterial content was quantified by taxa directly from SRA reads using Metaphalan (v. 2.0), which maps sequence reads to a database of predefined clade-specific marker genes42. All bacterial taxa with relative abundances <0.01 in all samples were excluded from statistical analysis. Phage content was assessed using a custom method. First, reads from each SRA file were assembled de novo into contigs with metaSPAdes (v. 3.11.1)69. Then, contigs >200 bp were aligned to the EBI collection of phage genomes (https://www.ebi.ac.uk/genomes/phage.html) by BLAST, with a threshold e-value <1e-5 and alignment length >50% of contig length. All of the original reads were then re-mapped with Bowtie2 (v. 2.3.4.1) to the contigs with good phage BLAST matches in order to increase sensitivity and to more accurately count the abundance of reads from each type of phage70. Phage read counts per contig were combined per phage genome (taxa) and normalized to relative abundance. A detection threshold of two reads per sample (>90% identity to the phage genome) was used, based on a previous report71.
Statistical analysis of microbial community composition and differential abundance
The QIIME pipeline (v1.9.1) was used for quality filtering of bacterial and bacteriophage DNA sequences, chimera removal (with USEARCH software), taxonomic assignment, and calculation of α-diversity, as previously described40,72. Downstream data analysis and calculation of diversity metrics were conducted in R v3.5.1, using ggplot2 and phyloseq libraries; DESeq. 2 was used to calculate logarithm of fold change. Bacterial and bacteriophage communities at the genus, family, and species levels were characterized based on α- and β-diversities. α-Diversity indices (ACE, Chao 1 richness estimator, Shannon and Simpson indexes) were calculated using the phyloseq R library73. Differences in α-diversities between datasets were examined by the Mann-Whitney test; p < 0.05 was considered statistically significant.
Differences among groups where two variables exist (phenotype and time point) were analyzed by two-way ANOVA and Tukey’s multiple comparison test. When one variable was compared, an unpaired two-tailed t-test was used. Data were visualized using multidimensional scaling (MDS).
Correlations between E. coli disappearance, Bifidobacterium abundance, breastfeeding and antibiotic usage were assessed pairwise using the Jaccard similarity index74. Differences were considered statistically significant at p < 0.05.
Bacteria and bacteriophages
E. coli strain MG1655 was used as a host for prophage induction experiments75. Bacteria were subcultured from freezer stocks onto Mueller–Hinton agar plates (Oxoid) and incubated at 37 °C overnight. All subsequent liquid subcultures were derived from colonies isolated from these plates and were grown in Mueller–Hinton broth (Oxoid).
The E. coli strain, MG1655 was grown on YESCA plates (0.5 g/L yeast extract; 10 g/L casamino acids, 20 g agar per liter) in the presence or absence of 25 μg/mL CR for 48 h at 26 °C.
Bacteriophage λ from our collection was employed. Bacteriophage suspensions were routinely stored in TM buffer (10 mM Tris-HCl, 10 mM MgSO4, pH 7.2) at 4 °C. E. coli lysogenic strains were obtained by infection of E. coli MG1655 with the phage and titration of cells on Mueller–Hinton agar plates as previously described76.
Prophage induction
Mitomycin C (Sigma-Aldrich) was prepared in 0.1 M phosphate buffer (pH 7.2), filtered through a 0.22-μm-pore filter (Millipore Corp., Bedford, MA), and stored at 4 °C77. A modified protocol according to Reyrolle et al. designed for prophage induction on 96-well plates was used63,78. Twenty microliters of an overnight E. coli MG1655 culture, with an absorbance at 600 nm of 0.2, was dispensed in each well of a 96-well flat-bottom polystyrene tissue culture microtiter plate (Sarstedt, Numbrecht, Germany), after which 180 μl of MHB (Oxoid) was added. The plates were incubated at 37 °C for 48 h. Induction of prophages in the E. coli biofilms was provoked by the addition of mitomycin C (Sigma) to a final concentration 1 μg/ml to each well. The plates were further incubated at 37 °C and samples were taken at 4, 6, 8, and 10 h following prophage induction.
Determination of CFU and PFU
To estimate CFU, biofilms were thoroughly scraped28. Well contents were aspirated, placed in 1 ml of isotonic phosphate buffer (0.15 M, pH 7.2), and the total CFU number was determined by serial dilution and plating on Mueller–Hinton agar.
The number of phage virions produced after induction was estimated by phage titration (using strain MG1655 as a host) and phage plaque assay. Following induction, at the indicated times, 200-μl samples of biofilms were collected. Then, 30 μl of chloroform was added to each sample, the mixture was vortexed and centrifuged at 3000 × g for 5 min in a microcentrifuge (Eppendorf 5415D). A supernatant fraction of the bacterial lysate was further used. The phage titer (number of phages per ml) was determined using the double agar overlay assay method as described previously16. To this end, 2.5 μl of each titration point of phage lysate was spotted on Mueller−Hinton agar (Oxoid). Then, a mixture of 1 ml of E. coli MG1655 culture and 2 ml of 0.7% nutrient agar (heated to 45 °C) supplemented with MgSO4 and CaCl2 (to a final concentration of 10 mM each) was poured over the plate. Plates were incubated at 37 °C overnight. Each experiment was repeated in triplicate.
Supernatant CR depletion assay
The amount of bacterial amyloid following prophage induction was measured in the supernatant of E. coli MG1655 biofilms. Biofilms were obtained as described above. Biofilm supernatant was taken at 4, 6, 8, and 10 h following prophage induction. To isolate aggregated amyloid fibers from the supernatant, the supernatant was centrifuged at 10,000 × g, filtered through a 0.22 µm filter to separate bacterial cells, and treated with proteinase K to eliminate contaminating proteins. CR (Sigma-Aldrich) was added to a final concentration of 10 μg/ml from a filtered stock solution of 1 mg/ml29. After 5 min of equilibration, absorbance spectra were recorded from 400 to 600 nm (SmartSpec 3000, Bio-Rad). Each trace represents the average of 5 accumulated spectra. For all samples, spectra of corresponding nutrient MHB solutions with CR were used as blanks.
Data Availability
The other sequencing datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Lehuen, A., Diana, J., Zaccone, P. & Cooke, A. Immune cell crosstalk in type 1 diabetes. Nature Reviews Immunology 10, 501–513 (2010).
Atkinson, M. & Eisenbarth, G. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. The Lancet 358, 221–229 (2001).
Rewers, M. & Ludvigsson, J. Environmental risk factors for type 1 diabetes. The Lancet 387, 2340–2348 (2016).
Soltesz, G., Patterson, C. & Dahlquist, G. Worldwide childhood type 1 diabetes incidence? what can we learn from epidemiology? Pediatric Diabetes 8, 6–14 (2007).
Barrett, J. et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nature Genetics 41, 703–707 (2009).
Ikegami, H., Noso, S., Babaya, N. & Kawabata, Y. Genetics and pathogenesis of type 1 diabetes: prospects for prevention and intervention. Journal of Diabetes Investigation 2, 415–420 (2011).
Noble, J. & Valdes, A. Genetics of the HLA Region in the Prediction of Type 1 Diabetes. Current Diabetes Reports 11, 533–542 (2011).
Achenbach, P., Bonifacio, E. & Ziegler, A. Predicting type 1 diabetes. Current Diabetes Reports 5, 98–103 (2005).
Vehik, K. et al. Islet Autoantibody Seroconversion in the DPT-1 Study: Justification for repeat screening throughout childhood. Diabetes Care 34, 358–362 (2011).
Paulsson, J. et al. High Plasma Levels of Islet Amyloid Polypeptide in Young with New-Onset of Type 1 Diabetes Mellitus. PLos One 9, e93053 (2014).
Vaarala, O., Atkinson, M. & Neu, J. The “Perfect Storm” for Type 1 Diabetes: The Complex Interplay Between Intestinal Microbiota, Gut Permeability, and Mucosal Immunity. Diabetes 57, 2555–2562 (2008).
Knip, M. & Siljander, H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nature Reviews. Endocrinology 12, 154–167 (2016).
Eizirik, D., Colli, M. & Ortis, F. The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nature Reviews Endocrinology 5, 219–226 (2009).
Drescher, K., Kono, K., Bopegamage, S., Carson, S. & Tracy, S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology 329, 381–394 (2004).
Hyöty, H. Viruses in type 1 diabetes. Pediatric Diabetes 17, 56–64 (2016).
Todd, J. Etiology of Type 1 Diabetes. Immunity 4, 457–467 (2010).
Flodström, M. et al. Target cell defense prevents the development of diabetes after viral infection. Nature Immunology 3, 373–382 (2002).
Op de Beeck, A. & Eizirik, D. Viral infections in type 1 diabetes mellitus — why the β cells? Nature Reviews Endocrinology 12, 263–273 (2016).
Filippi, C. & von Herrath, M. Viral Trigger for Type 1 Diabetes: Pros and Cons. Diabetes 57, 2863–2871 (2008).
Schloot, N. et al. Molecular mimicry in type 1 diabetes mellitus revisited: T-cell clones to GAD65 peptides with sequence homology to Coxsackie or proinsulin peptides do not crossreact with homologous counterpart. Human Immunology 62, 299–309 (2001).
Harrison, L. Type 1 diabetes. Molecular, cellular, and clinical immunology. Medical Journal of Australia 169, 272–272 (1998).
Kostic, A. et al. The Dynamics of the Human Infant Gut Microbiome in Development and in Progression toward Type 1 Diabetes. Cell Host & Microbe 20, 121 (2016).
Paun, A., Yau, C. & Danska, J. The Influence of the Microbiome on Type 1 Diabetes. The Journal of Immunology 198, 590–595 (2017).
Vatanen, T. et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 562, 589–594 (2018).
Stewart, C. et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588 (2018).
Flemming, H. & Wingender, J. The biofilm matrix. Nature Reviews Microbiology 8, 623–633 (2010).
Tetz, G., Artemenko, N. & Tetz, V. Effect of DNase and Antibiotics on Biofilm Characteristics. Antimicrobial Agents and Chemotherapy 53, 1204–1209 (2008).
Costerton, J. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 284, 1318–1322 (1999).
Chapman, M. Role of Escherichia coli Curli Operons in Directing Amyloid Fiber Formation. Science 295, 851–855 (2002).
Barnhart, M. & Chapman, M. Curli Biogenesis and Function. Annual Review of Microbiology 60, 131–147 (2006).
Schwartz, K. & Boles, B. Microbial amyloids – functions and interactions within the host. Current Opinion in Microbiology 16, 93–99 (2013).
Fowler, D., Koulov, A., Balch, W. & Kelly, J. Functional amyloid – from bacteria to humans. Trends in Biochemical Sciences 32, 217–224 (2007).
Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 539, 217–226 (2016).
Chen, S. et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Scientific Reports 6 (2016).
Tükel, Ç. et al. Toll-like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms. Cellular Microbiology 12, 1495–1505 (2010).
Davis-Richardson, A. et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Frontiers in Microbiology 5 (2014).
Correction for Zhao. et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proceedings of the National Academy of Sciences 115, E11426–E11426 (2018).
Tetz, G. & Tetz, V. Bacteriophages as New Human Viral Pathogens. Microorganisms 6, 54 (2018).
Tetz, G. et al. Bacteriophages as potential new mammalian pathogens. Scientific Reports 7 (2017).
Tetz, G., Brown, S., Hao, Y. & Tetz, V. Parkinson’s disease and bacteriophages as its overlooked contributors. Scientific Reports 8 (2018).
Segata, N. et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature Methods 9, 811–814 (2012).
Walterspiel, J., Morrow, A., Cleary, T. & Ashkenazi, S. Effect of subinhibitory concentrations of antibiotics on extracellular shiga-like toxin I. Infection 20, 25–29 (1992).
Candela, M. et al. Interaction of probiotic Lactobacillus and Bifidobacterium strains with human intestinal epithelial cells: Adhesion properties, competition against enteropathogens and modulation of IL-8 production. International Journal of Food Microbiology 125, 286–292 (2008).
Waller, A. et al. Classification and quantification of bacteriophage taxa in human gut metagenomes. The ISME Journal 8, 1391–1402 (2014).
Reichhardt, C. et al. Congo Red Interactions with Curli-Producing E. coli and Native Curli Amyloid Fibers. Plos One 10, e0140388 (2015).
Mills, S. et al. Movers and shakers. Gut Microbes 4, 4–16 (2013).
Penadés, J., Chen, J., Quiles-Puchalt, N., Carpena, N. & Novick, R. Bacteriophage-mediated spread of bacterial virulence genes. Current Opinion in Microbiology 23, 171–178 (2015).
Nanda, A., Thormann, K. & Frunzke, J. Impact of Spontaneous Prophage Induction on the Fitness of Bacterial Populations and Host-Microbe Interactions. Journal of Bacteriology 197, 410–419 (2014).
De Paepe, M., Leclerc, M., Tinsley, C. & Petit, M. Bacteriophages: an underestimated role in human and animal health?. Frontiers in Cellular and Infection Microbiology 4 (2014).
Kelley, W. Lex marks the spot: the virulent side of SOS and a closer look at the LexA regulon. Molecular Microbiology 62, 1228–1238 (2006).
Vatanen, T. et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 165, 842–853 (2016).
Tursi, S. et al. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLOS Pathogens 13, e1006315 (2017).
Gallo, P. et al. Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity 42, 1171–1184 (2015).
Honda, K. & Taniguchi, T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nature Reviews Immunology 6, 644–658 (2006).
Di Domizio, J. et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proceedings of the National Academy of Sciences 109, 14550–14555 (2012).
Gallucci, S. et al. Bacterial amyloids promote type I interferon production and accelerate autoimmunity, (BA6P. 124) (2006).
Spaulding, C., Dodson, K., Chapman, M. & Hultgren, S. Fueling the Fire with Fibers: Bacterial Amyloids Promote Inflammatory Disorders. Cell Host & Microbe 18, 1–2 (2015).
Friedland, R. & Chapman, M. The role of microbial amyloid in neurodegeneration. Plos Pathogens 13, e1006654 (2017).
Teng, F. et al. Gut Microbiota Drive Autoimmune Arthritis by Promoting Differentiation and Migration of Peyer’s Patch T Follicular Helper Cells. Immunity 44, 875–888 (2016).
Westermark, G., Krogvold, L., Dahl-Jørgensen, K. & Ludvigsson, J. Islet amyloid in recent-onset type 1 diabetes—the DiViD study. Upsala Journal of Medical Sciences 122, 201–203 (2017).
Delong, T. et al. Islet Amyloid Polypeptide Is a Target Antigen for Diabetogenic CD4 + T Cells. Diabetes 60, 2325–2330 (2011).
Denroche, H. & Verchere, C. IAPP and type 1 diabetes: implications for immunity, metabolism and islet transplants. Journal of Molecular Endocrinology 60, R57–R75 (2018).
Reyrolle, J., Chopin, M. C., Letellier, F. & Novel, G. Lysogenic strains of lactic acid streptococci and lytic spectra of their temperate bacteriophages. Applied and environmental microbiology 43, 349–356 (1982).
Hartmann, P., Haimerl, M., Mazagova, M., Brenner, D. A. & Schnabl, B. Toll-Like Receptor 2–Mediated Intestinal Injury and Enteric Tumor Necrosis Factor Receptor I Contribute to Liver Fibrosis in Mice. Gastroenterology 143, 1330–1340 (2012).
Carratù, R. et al. Altered intestinal permeability to mannitol in diabetes mellitus type I. Journal of pediatric gastroenterology and nutrition 28, 264–269 (1999).
Górski, A. et al. Phages and immunomodulation. Future microbiology 12, 905–914 (2017).
Nguyen, S. et al. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. MBio 8, e01874–17 (2017).
Hao, Y. et al. HPViewer: sensitive and specific genotyping of human papillomavirus in metagenomic DNA. Bioinformatics 34, 1986–1995 (2017).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335 (2010).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P.A. metaSPAdes: a new versatile metagenomic assembler. Genome research gr-213959 (2017).
McMurdie, P. J. & Holmes, S. phyloseq: An R Package For Reproducible Interactive 519 Analysis And Graphics Of Microbiome Census Data. PloS one 8, e61217 (2013).
Jaccard, P. The distribution of flora in the alpine zone. New Phytologist 11, 37–50 (1912).
Jensen, K. F. The Escherichia coli K-12” wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. Journal of bacteriology 175, 3401–3407 (1993).
Maniatis, T., Fritsch, E.F. & Sambrook, J. Molecular cloning: a laboratory manual, 545, Cold Spring Harbor, NY: Cold spring harbor laboratory (1982).
Georgopoulos, M., Vass, C., Vatanparast, Z., Wolfsberger, A. & Georgopoulos, A. Activity of dissolved mitomycin C after different methods of long-term storage. Journal of glaucoma 11, 17–20 (2002).
McDonald, J. E., Smith, D. L., Fogg, P. C., McCarthy, A. J. & Allison, H. E. High-throughput method for rapid induction of prophages from lysogens and its application in the study of Shiga toxin-encoding Escherichia coli strains. Applied and environmental microbiology 76, 2360–2365 (2010).
Yuan, Y., Gao, M., Wu, D., Liu, P. & Wu, Y. Genome characteristics of a novel phage from Bacillus thuringiensis showing high similarity with phage from Bacillus cereus. PLoS One 7, e37557 (2012).
Author information
Authors and Affiliations
Contributions
G.T. designed the analysis. S.B., Y.H., G.T. conducted the analysis. G.T. and V.T. supervised data analysis and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tetz, G., Brown, S.M., Hao, Y. et al. Type 1 Diabetes: an Association Between Autoimmunity, the Dynamics of Gut Amyloid-producing E. coli and Their Phages. Sci Rep 9, 9685 (2019). https://doi.org/10.1038/s41598-019-46087-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46087-x
This article is cited by
-
Exploring the Human Virome: Composition, Dynamics, and Implications for Health and Disease
Current Microbiology (2024)
-
Mutant and curli-producing E. coli enhance the disease phenotype in a hSOD1-G93A mouse model of ALS
Scientific Reports (2023)
-
Hyperglycemia is associated with duodenal dysbiosis and altered duodenal microenvironment
Scientific Reports (2023)
-
Characterization of the human gut virome in metabolic and autoimmune diseases
Inflammation and Regeneration (2022)
-
Islet autoantibody seroconversion in type-1 diabetes is associated with metagenome-assembled genomes in infant gut microbiomes
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
