
Abstract
The 25 human bitter taste receptors (hTAS2Rs) are responsible for detecting bitter molecules present in food, and they also play several physiological and pathological roles in extraoral compartments. Therefore, understanding their ligand specificity is important both for food research and for pharmacological applications. Here we provide a molecular insight into the exquisite molecular recognition of bitter β-glycopyranosides by one of the members of this receptor subclass, hTAS2R16. Most of its agonists have in common the presence of a β-glycopyranose unit along with an extremely structurally diverse aglycon moiety. This poses the question of how hTAS2R16 can recognize such a large number of “bitter sugars”. By means of hybrid molecular mechanics/coarse grained molecular dynamics simulations, here we show that the three hTAS2R16 agonists salicin, arbutin and phenyl-β-D-glucopyranoside interact with the receptor through a previously unrecognized dual binding mode. Such mechanism may offer a seamless way to fit different aglycons inside the binding cavity, while maintaining the sugar bound, similar to the strategy used by several carbohydrate-binding lectins. Our prediction is validated a posteriori by comparison with mutagenesis data and also rationalizes a wealth of structure-activity relationship data. Therefore, our findings not only provide a deeper molecular characterization of the binding determinants for the three ligands studied here, but also give insights applicable to other hTAS2R16 agonists. Together with our results for other hTAS2Rs, this study paves the way to improve our overall understanding of the structural determinants of ligand specificity in bitter taste receptors.
Similar content being viewed by others

Introduction
Human bitter taste or taste type 2 receptors (hTAS2Rs) are a group of 25 proteins (see Supplementary Table S1), belonging to the G-protein coupled receptor (GPCR) superfamily, the largest group of membrane proteins in humans1. Their classification within the GPCR superfamily has been the subject of debate. Based on evolutionary analysis, they were initially considered as part of class F, with which they share several common features, such as IFL in transmembrane helix (TM) 2, SFLL in TM5, and SxKTL in TM72. However, this classification was suggested with reservations, since more work may be required to clarify the evolution of bitter taste receptors. More recent studies suggest that hTAS2Rs belong to class A. Indeed, they share several conserved motifs (see references3,4,5). Nonetheless, the low sequence similarity with class A GPCRs also supports their classification as a distinct family (class T)6.
Originally, bitter taste receptors were identified in the taste buds of the tongue7,8, where they detect bitter molecules. These include several toxic compounds9, molecules with nutritional beneficial properties10,11,12,13 or that add a valued property to foods14,15,16,17. Nonetheless, in the last years hTAS2Rs have also been identified in extraoral tissues, including the brain18,19,20, the gut21,22 the upper and lower airways23,24 and the heart25. Depending on their location, they play different physiological roles and have been shown to be associated with different diseases22,26,27. Hence, hTAS2Rs constitute exciting novel targets for pharmaceutical intervention26,27,28,29. For instance, hTAS2R38 and hTAS2R14 are expressed in the upper airways and their activation is involved both in production of nitric oxide and in improvement of the cilia beat frequency, triggering antibacterial mechanisms and facilitating mucus excretion30. Another example is hTAS2R16, which is expressed in the brain and whose activation by salicin might modulate neurite outgrowth20.
In spite of their physiological and pathological importance, experimental structural information on hTAS2Rs is missing. Hence, molecular level understanding of ligand binding to hTAS2Rs currently relies on computational methods (namely homology modeling, docking and molecular dynamics (MD) simulations), combined with experimental mutagenesis and functional data4,31. Unfortunately, hTAS2Rs display a low sequence identity (<20%) with any of the available GPCR templates (62 unique GPCR structures, of which 52 correspond to class A, as of March 20196,32). As a consequence, the resulting homology models have low resolution33,34,35, regardless of the template used. The three dimensional structure, particularly the orientation of the side chains, is likely to be not correct, affecting the accuracy of the docking in predicting receptor/ligand interactions. Hence, MD simulations can be used to refine these low resolution models4,31, in particular, the hybrid molecular mechanics/coarse grained (MM/CG) approach. This was tested using the β2-adrenergic receptor (β2AR), a class A GPCR for which the crystal structure is available36. A model of the receptor was built based on a template with sequence identity comparable to that of hTAS2R/GPCR and the ligand was introduced in the structure using molecular docking. After a 0.8 μs MM/CG simulation, the obtained binding pose turned out to reproduce the receptor/ligand interactions present in the crystal structure of β2AR, attesting the validity of this computational protocol37. Furthermore, this methodology has also been used to study ligand binding in GPR3, a class A GPCR38. In the case of hTAS2Rs, MM/CG simulations have suggested receptor/ligand interactions in agreement with the binding residues inferred from the experiments37,39. Moreover, the simulations predicted new binding residues (not previously suggested by the experimental data) that were successfully confirmed a posteriori by carrying out additional experiments. Altogether, these integrated computational and experimental studies have provided important insights on agonist selectivity in bitter taste receptors37,39, as previously demonstrated for other GPCRs40.
While most of hTAS2Rs feature an intermediate or a narrow agonist spectrum, two outlier groups can be singled out: the broadly-tuned and the group-selective receptors41. The first group, which includes hTAS2R10, hTAS2R14 and hTAS2R46, can recognize almost half of the ~100 agonists tested against the whole set of 25 hTAS2Rs41,42,43,44. A possible rationale for such large agonist diversity has been put forward based on computer-aided structural predictions of hTAS2R46. This receptor shows a transient binding site – other than the canonical, orthosteric one – that might filter the receptor agonists out of the pool of bitter tastants39. This “access control”45 is also present in other class A GPCRs46,47,48,49,50.
The hTAS2R46 agonist diversity contrasts sharply with the ligand selectivity of the group-selective receptors hTAS2R38 and hTAS2R16. The first is the target predominantly of bitter compounds containing an isothiocyanate or thiourea group37,42,51. In line with its high specificity, computer-aided predictions on hTAS2R3837,51 have not identified (as yet) any transient binding site possibly serving as “access control”, differently from hTAS2R4631. The other group-selective receptor, hTAS2R16, mainly recognizes bitter β-D-glycopyranosides (hereafter, “bitter sugars”)52,53. These are composed by a sugar (usually β-glucose, but also β-mannose in a few cases52,53) attached to a hydrophobic aglycon moiety; the latter can be extremely diverse (see Fig. 1 and Supplementary Fig. S1). Indeed, studies focused on the ligand selectivity of hTAS2R16 discovered about 30 diverse β-glycopyranosides agonists53,54,55,56,57,58, but this number could be even larger42,59,60 (see Supplementary Text S1). This poses the question of how hTAS2R16 can accommodate so many highly diverse ligands belonging to the same chemical class.

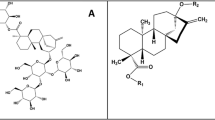
Chemical structures of the three agonists considered in this work: phenyl-β-D-glucopyranoside, arbutin and salicin (from top to bottom). Numbering of the glucopyranoside carbon atoms (and the corresponding oxygen atoms) is indicated; the phenyl substituent is numbered 7 for both arbutin and salicin for the sake of simplicity, despite the different position (para and ortho, respectively).
A structural characterization of the ligand binding determinants in hTAS2R16 would be crucial to understand the peculiar preference of hTAS2R16 for its agonists. Moreover, it may help design rationally potential novel drugs61, since several hTAS2R16 agonists are already known to affect human health62,63. These include arbutin, an inhibitor of bladder cancer proliferation64, and salicin, which has been long used for its analgesic, anti-inflammatory and antipyretic properties65. Salicin is a particularly interesting, yet challenging, lead compound, since it inhibits tumor growth and angiogenesis in endothelial cells66, but it can also have adverse effects by promoting neurite growth in neuroblastoma20.
Previous studies have attempted to characterize the structural determinants of agonist binding to hTAS2R16 (Supplementary Text S2). In an elegant series of experiments55,56, Sakurai and co-workers used site-directed mutagenesis to identify several residues that might be involved in agonist binding (Supplementary Table S2). They further showed that the mutations have similar effects across different ligands (such as salicin, arbutin and phenyl-β-D-glucopyranoside, see Fig. 1)55,56. This has led to the suggestion that all the receptor agonists share the same binding cavity55. In addition, Sakurai and coworkers proposed a structural model of the salicin binding pose55. However, this model does not predict ligand/receptor interactions with residues E262, L59 and V265, emerging from subsequent site-directed mutagenesis experiments53. This discrepancy may be consistent with the fact that bioinformatics-based models of human bitter taste receptors have only a limited predictive power31. Subsequently, Chen et al.67 presented a second model of the receptor in complex with salicin, using a computational approach similar to Sakurai, followed by molecular dynamics. Unfortunately, also this prediction seems not to be consistent with all the available mutagenesis data. In particular, residues F93, F236, F240, I243 and V265, putatively involved in ligand binding53,55 do not appear to interact with the ligand in their model. Therefore, none of the previously proposed models seems to be fully compatible with the available experimental data53,55 (see Supplementary Text S2 for details).
Classical MD simulations for low resolution models of chemosensory GPCR complexes appear to be not optimal because of TM stability issues. Indeed, the initial wrong orientation of the side chains may cause TM unfolding during the simulations. Hence, in an effort at shedding light on hTAS2R16/agonist interactions, here we use homology modeling and docking followed by extensive multiscale molecular mechanics/coarse grained (MM/CG) simulations68,69,70,71 on this receptor. The MM/CG approach has been shown to dramatically improve the predictive power of the computational models compared to using only bioinformatics methods31, by enhancing the sampling of the ligand binding cavity. Furthermore, the use of a Go-like model for the CG region helps to reduce the unfolding artifacts in the intracellular part of the receptor due to the low resolution of the initial model. Here we focus on three hTAS2R16 agonists: salicin, arbutin, and phenyl-β-D-glucopyranoside53,55,56. Our simulations (Supplementary Table S3) are validated a posteriori by comparison with available experimental site-directed mutagenesis data. Moreover they suggest that these three ligands display two possible binding modes for hTAS2R16, both consistent with the experimental data53,55,56. We hypothesize that this previously unrecognized dual binding mode mechanism might allow the receptor to accommodate hydrophobic aglycons of disparate sizes and with different substituents, thus helping hTAS2R16 to recognize a wider spectrum of bitter sugars. Based on these results, we also provide insights into the binding determinants of other hTAS2R16 agonists for which structure-activity relationship data are available.
Methods
Homology modelling
The sequences of the 25 hTAS2Rs were retrieved from the Pfam database72. The multiple sequence alignment (MSA) was generated using PROMALS73 and its correctness was checked by ensuring the alignment of conserved X.50 positions32,74 and conserved structural motifs across hTAS2Rs5. This MSA was used as input for the GOMoDo webserver75. GOMoDo uses HHsearch 2.0.1676,77 to convert the input MSA into a Hidden Markov Model (HMM) and then aligns this HMM to the HMMs of all the possible GPCR templates available in the GOMoDo database. A two step procedure was used for template selection and model building. In the first step, the target-template HMM alignments were used to build only 10 models of hTAS2R16 (UniprotKB ID: Q9NYV7) for each of the templates stored in the GOMoDo webserver (https://gomodo.grs.kfa-juelich.de/) using the MODELLER program (version 9.10)78 included in GOMoDo. The selection of the most suitable template was performed using both sequence and structural parameters: (i) we discarded models based on inactive templates, as agonists bind to the active state of the receptor and no significantly higher sequence identity with inactive templates was detected79; (ii) we further discarded models generated using template-target alignments with gaps inside the TM regions longer than one residue; and (iii) we selected models with higher GA341 scores80 and lower DOPE scores81, i.e. higher overall structural quality. The selected template turned out to be the β2 adrenergic receptor (β2AR, PDB ID 4LDE, sequence identity 13%). The alignment between hTAS2R16 and β2AR was visually inspected to ensure that the conserved features shared between hTAS2Rs and class A GPCRs were well aligned5.
Once the β2AR template was selected, the second modeling step was performed. Using the aforementioned target-template alignment (Supplementary Fig. S2), 200 new models were generated using a standalone version of MODELLER (version 9.11). This second step is intended to have more solutions during the model building and optimization step at a reasonable computational cost, since the standalone version of MODELLER is faster than the implementation in the GOMoDo webserver. The most suitable β2AR-based model among the 200 was selected using the following structural criteria: (i) high quality scores, (ii) preservation of the secondary structure of the TM helices and (iii) lower number of residues in not allowed regions of the Ramachandran plot. The protonation state of the titratable residues was determined by the hydrogen placement algorithm in MolProbity, which optimizes hydrogen bonding networks and minimizes steric clashes82.
Ligand docking
The chemical structures of the ligands in Fig. 1 were downloaded from the PubChem database83. Their protonation states were identified using the pKa plugin in MarvinSketch (http://www.chemaxon.com/products/marvin/marvinsketch/). All three ligands have zero total charge.
Docking of the phenyl-β-D-glucopyranoside ligand to the hTAS2R16 model was performed with the HADDOCK webserver84,85. Fpocket86,87 was used to predict receptor residues putatively involved in ligand binding. After discarding residues belonging to the extracellular loops or pointing outside the receptor cavity, the remaining fpocket-predicted residues were used as active residues to define the so-called ambiguous interaction restraints (AIRs) for the data-driven HADDOCK procedure84,85. First, 1,000 initial structures were generated by rigid body docking and the 200 structures with lowest HADDOCK scores were submitted to further refinement using semi-flexible simulated annealing, followed by flexible explicit solvent refinement with a water layer. The resulting complexes were clustered using the algorithm in reference88 and a root mean square deviation (RMSD) cutoff of 2 Å. Clustering was used to compensate (at least in part) the known limitations of docking scores in ranking docking poses.
Indeed, the binding pose selected was the one with the lowest HADDOCK score belonging to the most populated cluster. In addition, another binding pose was also manually built using the tools of VMD89 (further details in the Results section). We would also like to note here the limitations of docking scores in discriminating the best docking poses for low resolution GPCR models90,91,92,93,94.
Since arbutin and salicin only differ by a small phenyl substituent, it is reasonable to assume that they are also accommodated in the same binding pocket and with the same orientation as phenyl-β-D-glucopyranoside. Consistently, mutations of residues presumably involved in binding affect similarly all three agonists53,55,56. Hence, a snapshot of the MD simulation of phenyl-β-D-glucopyranoside was taken at 12.5 ns (when the ligand is already located in the orthosteric binding site) and the phenyl aglycon was alchemically modified by adding the corresponding substituent (hydroxyl group in para for arbutin or hydroxymethyl group in ortho for salicin) with the Molefacture plugin in VMD89. Based on the second binding pose built for phenyl-β-D-glucopyranoside, a second binding pose was also manually built for arbutin and salicin using VMD89 (see Results).
Multiscale molecular dynamics simulations
In order to improve the bioinformatics predictions, the hTAS2R16 complexes were refined by performing molecular dynamics simulations, using the molecular mechanics/coarse grained (MM/CG) approach68,69,70. Figure 2 shows the simulation setup. The MM part consists of the extracellular part of the receptor, including the binding site with the docked ligand, along with the solvent (approx. 5,300 water molecules). The protein was described using the GROMOS96 43a1 force field95, whereas the SPC model was used for the water molecules96. The parameters for the ligands were built by combining GROMOS 56a6_CARBO forcefield97 parameters for the β-D-glucopyranose unit and PRODRG98 generated parameters for the aglycons. Following reference99, RESP charges100 were used for all ligands, calculated with Gaussian09101 at the B3LYP/6-31G(d,p) level of theory and fitted with Antechamber102. The intracellular part of the receptor, which is not involved in binding, was defined as the CG region and described using a Go-like potential103 between CG beads centered on the Cα atoms of the corresponding residues. We would like to note here that only the CG part is constrained (using the Go-like potential), whereas the MM part is fully flexible, both at the backbone and the side chain levels. In the MM/CG approach, the MM and the CG parts are connected by an interface region, which mediates the bonded and non-bonded interactions between the two levels of resolution. The membrane is represented in an implicit way, by introducing wall potentials (indicated with lines in Fig. 2). Further details can be found in recent reviews104,105,106.

Molecular mechanics/coarse grained (MM/CG) simulation setup. Water molecules, the extracellular part of the receptor and the ligand (in orange) constitute the all atom (MM) region, whereas the intracellular part of the receptor (in green) is the CG region. The interface (in violet) mediates the interaction between the MM and the CG regions. Carbon atoms in the atomistic and in the interface regions are coloured according to the region they belong to, whereas hydrogen atoms are in white, nitrogen in blue and oxygen in red. The potentials used to mimic the presence of the membrane and to cap the water molecules are represented as colored lines (blue, gray and yellow, respectively).
Six hTAS2R16 complexes were considered (three agonists, two binding poses for each). Each complex was first energy minimized with three runs of steepest descent followed by three runs of conjugate gradient minimization. In each minimization run performed with the same algorithm, the maximum minimization step size was progressively increased by one order of magnitude (from 0.001 to 0.01 to 0.1 nm). Each minimization was carried out until no further changes in potential energy were detected or the maximum force was smaller than the threshold value of 10 kJ mol−1 nm−1. Next, a simulated annealing protocol was carried out to increase the temperature of the complex from 0 to 300 K in 6 ns, using a velocity rescaling thermostat. During the heating, positional restraints were applied on the protein backbone using a force constant of 1000 kJ/mol*nm2 in each direction. After the simulated annealing phase, three further equilibration steps of 3 ns each were performed. In each step, the force constant applied on the protein backbone was progressively decreased (from 500 to 250 to 125 kJ/mol*nm2). After removing all restraints, two simulations were run for each ligand, starting from two different binding poses (Supplementary Table S3). All simulations were run for 0.8 μs using a timestep of 2 fs.
Interaction analysis
Hydrophobic interactions were calculated with the g_mindist tool of GROMACS107 and using a 5.5 Å distance cutoff (i.e., the sum of the van der Waals radii of two carbon atoms plus the diameter of a water molecule between them108,109,110). Hydrogen bonds (H-bonds) were identified with the “measure hbonds” tool in VMD89, using a 3.5 Å distance cutoff between donor and acceptor and a 30° deviation for the angle made by donor, hydrogen and acceptor atoms. The same criteria were used to define water-mediated H-bonds, in which the same water molecule is interacting simultaneously with both receptor and ligand. The persistency of a given interaction was calculated as the number of frames showing this interaction over the total number of frames, after excluding the first 100 ns of the simulation. Residues with hydrogen bond persistency higher than 10% or hydrophobic interaction persistency higher than 80% were considered as “computational binding” residues. We used two different persistency cutoffs for the two types of interactions to account for the higher fluctuations of the hydrogen bond network compared to the hydrophobic interactions. The rest were defined as “computational non-binding” residues. For the analysis of the static docking poses the computational binding and non binding residues were defined differently from the analysis of the MD trajectories. Indeed, they were identified based on the presence/absence of the interaction (defined as explained above) in the corresponding docked complex, as done in reference31. In other words, we excluded the persistency criterium because the interactions in the static binding poses are not time dependent, since no MD simulation had been run at this point.
A representative snapshot was selected for each simulation as follows. We first calculated the average structure with the GROMACS g_covar tool111 considering the whole system (except for the water molecules). Then, we computed the RMSD along the simulation using the GROMACS g_rms tool111, including only the transmembrane helices and the ligand. The snapshot with the lowest RMSD compared to the average structure was chosen as the representative structure of the corresponding ligand binding mode. The associated images were generated with VMD89.
Based on site-directed mutagenesis data and functional assays (agonist dose-response curves) data53,55, we also defined “experimental binding” and “experimental non binding” residues. In principle, changes in the half maximal effective concentration (EC50) values between the mutated and the wild-type (wt) receptor should be characteristic of residues involved in agonist binding, while an effect on the amplitude of the dose response curve upon mutation should be related with receptor activation112,113. However, in practice, it is difficult to ascertain whether a residue is involved only in binding, only in activation or in both. Actually, conformation-dependent effects114, changes indirectly related to the ligand affinity of the receptor (i.e. shaping of the binding cavity37,39) and second-shell effects115 can also affect the EC50 changes. Therefore, we used a second criterium based on the crystallographic structures of class A GPCR complexes79. Out of the residues whose mutation affects the EC50 value, only those located in the upper extracellular part of the receptor (i.e. the location of the canonical orthosteric binding site in class A GPCRs79) were considered as “experimental binding” residues, as done in reference31. Residues whose mutation does not change EC50 and/or that are located in the lower intracellular part of the receptor were considered as “experimental non-binding” residues.
Comparison of the computational and experimental residues allows us to define four different groups (Supplementary Text S3). “True positives” (TP) are amino acids identified as binding residues by both experiment and computation; “false positives” (FP) are amino acids identified as non-binding residues by experiment, but as binding residues in computation; “true negatives” (TN) are amino acids identified as non-binding residues by both experiment and computation; and “false negatives” (FN) are amino acids identified as binding residues by experiment, but not in computation. These were used to calculate the statistical parameters precision (PREC) and recall (REC):
in order to assess the agreement of the computational models with the experimental data, as done in our previous work31. These are two statistical parameters commonly used to evaluate method performance116,117,118,119. They are close to 1 when the computational predictions are consistent with the experimental data, and zero when they are not. Precision evaluates how many residues are correctly predicted as important for ligand-receptor interactions, whereas recall quantifies how many of the experimental binding residues are captured by the simulations. Precision and recall values were calculated for the initial docking poses and for a representative snapshot of each simulation. We did not consider other statistical parameters including TNs because mutagenesis experiments are designed in order to capture binding residues55 and hence the number of potential TNs is very low (e.g. one out of seven for hTAS2R16).
Results and Discussion
Phenyl-β-D-glucopyranoside
As in our previous MM/CG investigations of hTAS2Rs37,39, we started from the binding pose obtained by homology modelling and molecular docking. In this pose, the ligand is almost parallel to the receptor axis, with the aglycon moiety buried inside the receptor and the glucose unit pointing towards the extracellular side. The reliability of this initial docking pose was assessed by comparison with the available experimental mutagenesis data, in terms of precision and recall. The values of these two statistical parameters are equal to 0 (Table 1 and Supplementary Table S2), indicating a low agreement of the initial docking pose with the experimental data. Although the fpocket predicted residues used to drive the docking included some of the binding residues inferred from the mutagenesis data, the docking pose is not accurate enough, most likely due to the wrong orientation of the residue side chains in the low resolution homology model.
Hence, the docking pose was refined by performing MM/CG simulations. After a few ns, the phenyl-β-D-glucopyranoside ligand moved further inside the receptor and stabilized in this position for the rest (~0.8 μs) of the simulation (Supplementary Fig. S3). The identified binding cavity overlaps with the canonical orthosteric site observed in X-ray structures of class A GPCR/ligand complexes79 (Supplementary Fig. S4) and the receptor residues shown to be involved in binding are in agreement with the mutagenesis data (see below).
In this binding pose, the glucose unit of the ligand is in contact with several polar residues in the upper part of the binding pocket (see below), as well as solvated by water molecules, while the aglycon hydrophobic moiety is mostly surrounded by hydrophobic residues in the bottom part. Therefore, the residue distribution of the receptor binding site appears to match the chemical properties of the sugar derivative. The glucose ring is held in place by hydrogen bonds (H-bonds) formed mainly with four residues, located in two adjacent transmembrane helices. These are E863.33 and N893.36 in TM3, and E2627.39 and Y2667.43 in TM7 (the Ballesteros-Weinstein numbering74 commonly used for class A GPCRs is indicated as superscript). These two sets of residues are mirroring each other, so that E863.33 is in front of E2627.39, and similarly for the couple N893.36 and Y2667.43 (Fig. 3). On the other hand, the phenyl moiety is surrounded almost completely by hydrophobic residues (see Fig. 4).

Some of the residues interacting through H-bonds with phenyl-β-D-glucopyranoside. The ligand is shown with pink lines, whereas residues on TM3 are represented with green lines and the counterparts on TM7 with orange lines. Oxygen, hydrogen and nitrogen atoms are shown as red, white and blue spheres. The generalized Ballesteros-Weinstein numbering across class A GPCRs is reported below each residue (e.g. 3.33 for E86). (a) At the entrance of the binding cavity, residue E86 on TM3 mirrors E262 on TM7, as respectively N89 does with Y266 one helix turn below. They all form H-bonds with the ligand during the simulation, even if the H-bond pattern is dynamic. This binding mode was defined as TM3-facing, due to the orientation of the glucose hydroxymethyl group toward TM3. (b) Same as panel (a) but with the ligand rotated by 180 degrees along the receptor axis. The hydroxymethyl group is pointing toward the TM7, i.e. the TM7-facing binding mode.

Main amino acids interacting with the ligand aglycon. Residues shaping the bottom part of the binding cavity are represented as spheres using different colors. The only polar residue among this set of aminoacid is T92.
Given (i) the complementarity of the two sides of the binding cavity and (ii) the presence of hydroxyl groups on the ligand glucose moiety potentially able to form H-bonds with both sides of the previously described binding cavity, we asked ourselves whether a 180 degrees rotation of the phenyl-β-D-glucopyranoside along the receptor axis would still allow binding. The initial wrong orientation of the side chains and the limited exploration of the conformational space of the bioinformatics methodology may explain why this alternative binding pose was not identified by the docking algorithm. A further argument in support of the existence of a dual binding mode is the presence of a similar mechanism in several lectins120,121,122,123,124. In these sugar-binding proteins, the two binding modes involve the same residues, but use a different interaction pattern with the ligand. Therefore, we considered not only one but two possible binding modes: one in which the C6 substituent of the glucose ring is oriented towards TM3 (hereafter, TM3-facing mode, Fig. 3A) and the horizontally flipped alternative, in which the same substituent is pointing to TM7 (i.e. TM7-facing mode, Fig. 3B).
These two binding modes were investigated by running 0.8 microsecond MM/CG simulations (Supplementary Table S3 and Supplementary Fig. S3). In both cases, the ligand is stable inside the binding cavity (Supplementary Fig. S5). In addition, the receptor residues involved in interactions with the ligand are in agreement with the experimental mutagenesis data for both binding modes, as shown by the high values of recall and precision (see Table 1, Supplementary Table S2 and Supplementary Text S3). Hence, our simulations indicate that the phenyl-β-D-glucopyranoside agonist may have a dual binding mode.
In order to validate the quality of our predictions, an additional simulation was run, differing from the previously described in that the glucose unit is buried inside the receptor (instead of pointing toward the extracellular side) and the aglycon is pointing toward the extracellular rather than toward the inner part of the receptor (hereafter, glucose-in binding pose) (see Supplementary Text S2). This binding pose was discarded because of the instability of the ligand, which moves out of the receptor after a few nanoseconds (ca. 20) of MM/CG simulation (Supplementary Fig. S6). This behavior can be easily explained considering that initially the glucose unit is surrounded by hydrophobic residues, whereas the hydrophobic aglycon is surrounded by polar residues. The chemical mismatch between the glucose-in oriented ligand and the side chains constituting the binding site results in the glucose unit moving towards the extracellular side.
Arbutin and salicin
Next, we examined two derivatives of phenyl-β-D-glucopyranoside, arbutin and salicin (Fig. 1), which are also experimentally characterized agonists of hTAS2R1653,55. The former differs from phenyl-β-D-glucopyranoside by the presence of a hydroxyl group in para position, whereas in the latter the phenyl substituent is a hydroxymethyl group in ortho. The corresponding complexes were obtained by alchemical modification of phenyl-β-D-glucopyranoside, in either its TM3- or TM7-facing binding modes. During the 0.8 μs MM/CG simulations, both arbutin and salicin turned out to explore the same binding cavity as phenyl-β-D-glucopyranoside and to share similar protein-ligand interactions (Supplementary Tables S4-S7). This is consistent with the experimental observation that mutations of residues presumably involved in binding affect similarly all the ligands tested53,55,56. Moreover, the receptor-ligand interactions observed in the simulations are also compatible with experimental mutagenesis data53,55, as shown by the high values of precision (0.8–1.0) and recall (0.66–0.83) obtained for both binding modes (see Table 1 and Supplementary Table S2). Therefore, the dual binding mode proposed above for phenyl-β-D-glucopyranoside seems to apply also to arbutin and salicin. Indeed, six different MD trajectories (3 ligands, 2 binding poses for each) for a total of 4.8 μs are consistent with the existence of the dual binding mode for hTAS2R16 agonists.
In the next sections, we will discuss the specific receptor-ligand interactions observed in the simulations (Fig. 5 and Supplementary Tables S4–S7), as well as the comparison with the available experimental data carried out to validate the proposed models (Supplementary Table S2).

hTAS2R16 residues interacting with the glucose moiety of the ligands. Phenyl-β-D-glucopyranoside is shown as pink licorice (A,B), arbutin in yellow (C,D) and salicin in green (E,F). The top row (A,C,E) displays the TM3-binding mode of each ligand, while the TM7-binding mode is on the second row (B,D,F). The receptor interacting residues are in grey licorice and they are indicated with both their sequence and their Ballesteros-Weinstein numbering.
Molecular basis of β-glucopyranoside binding to hTAS2R16
Common interactions between hTAS2R16 and the glucose unit of the ligands (W85 3.32, E86 3.33, N89 3.36, E262 7.39 )
Regardless of the ligand and the binding mode considered, the common glucose unit is mainly in contact with the same receptor residues (W853.32, E863.33, N893.36, Y2396.51 and E2627.39), thus supporting the presence of a dual binding mode. However, the pattern of the interactions and their persistence varies among different complexes, as described below.
W853.32 can form stacking or hydrophobic interactions with the glucose ring of the ligands (Fig. 5). This is in line with its mutation into R reducing the maximum response of the receptor towards salicin53, as well as the experimental data on this conserved tryptophan for other bitter taste receptors (reviewed in references31,39).
The two glutamates E863.33 and E2627.39 are located in the upper part of the binding cavity and, in our simulations, they form H-bonds with the glucose hydroxyl groups in both binding modes (Fig. 5). In the TM3-facing mode (Fig. 5A,C,E and Supplementary Table S4), E863.33 forms H-bonds mainly with O6, whereas in the TM7-facing mode (Fig. 5B,D,F and Supplementary Table S5), it does so with O2 and O3 (see oxygen numbering in Fig. 1). The persistency of these interactions along all the simulations suggests a fundamental role of E863.33 in ligand binding, in agreement with the experimental data. In particular, the isosteric E86Q mutation almost does not modify the EC50 value for any of the three ligands (mutant/wild-type ratio between 2.9 and 5.7), while the E86D mutation has a stronger impact (between 7.9 and 18.4), indicating that the length of the side chain at position 3.33 is crucial to establish H-bonds with the glucose unit55. On the other hand, E2627.39 is more flexible than E863.33, forming H-bonds with more than one glucose hydroxyl group and with variable persistency (Supplementary Tables S4 and S5). This is in line with the maximum receptor activity with salicin being only slightly decreased by the E262D mutation, compared to the stronger effect of E86D; only the E262A mutation completely abolishes receptor activation53. Moreover, mutations of the homologous E2657.39 in hTAS2R46 significantly altered the EC50 of this receptor for its cognate agonist45, thus further supporting the participation of position 7.39 in agonist binding in hTAS2Rs.
N893.36 can act as a H-bond donor to O1 and/or O2 in the TM7-facing binding mode or to their symmetric counterparts O5 and/or O6 in the TM3-facing binding pose (Fig. 5). The role in ligand binding of N893.36 is supported by the experimental data53,55, showing that its mutation abolishes the receptor activity for all three ligands.
Interestingly, residues W853.32 and N893.36 are highly conserved across the hTAS2R family (Supplementary Table S6) and mutagenesis data on other bitter taste receptors (see31,39 and references therein) also support their participation in the formation of the ligand binding cavity. In addition, residues at positions 3.32, 3.33, 3.36 and 7.39 discussed here, together with position 6.48 and 6.51 (see next sections), have been proposed to be part of the consensus ligand binding pocket across class A GPCRs79.
Common interactions between hTAS2R16 and the variable phenyl aglycon of the ligands (L59 2.53, F88 3.35, T92 3.39, F93 3.40, F236 6.48, V265 7.42, F268 7.45, I269 7.46 )
The variable phenyl aglycon is surrounded mostly by hydrophobic and aromatic residues, including L592.53, F883.35, F933.40, F2366.48, V2657.42, F2687.45 and I2697.46 (see Fig. 4); the only exception is the polar T923.39. Out of these residues, mutagenesis data on hTAS2R16 are available for L592.53, F2366.48 and V2657.42 (change in receptor response to salicin) and F933.40 (change in EC50 for all three ligands)53,55. For salicin, mutation of three aforementioned residues reduce the receptor activity (to 64%, 2% and 5% of the wild-type, see Supplementary Table S2), whereas mutation of the latter decreased EC50 by 19 to 55-fold, depending on the mutation. Consistently, in our simulations they form hydrophobic interactions with the phenyl moiety of all three ligands and are also involved in shaping the binding cavity (Supplementary Tables S6 and S7).
In addition, our simulations indicate that F2687.45 interacts with all the agonists. Although there are no experimental data for this residue in hTAS2R16, position 7.45 is conserved among hTAS2Rs (Supplementary Table S8) and there are mutagenesis data available for other bitter taste receptors31,39 that support its participation in the formation of the ligand binding cavity. T923.39 is also conserved across the whole bitter taste receptor family, but experimental data are missing. Nonetheless, position 3.39 is involved in sodium binding in class A GPCRs125 and thus we speculate that T923.39 may also play a functional role in hTAS2Rs (see next section).
Differences in binding among the three ligands (S63 2.57, N67 2.61, T92 3.39, Y266 7.43 )
As mentioned above, the common glucose unit establishes a stable, yet flexible, H-bond network with the receptor, which is very similar among the three ligands. The only significant differences involve salicin binding, most likely due to the bulkier hydroxymethyl substituent in para position of the phenyl aglycon. While the glucose moiety of arbutin and phenyl-β-D-glucopyranoside is mainly interacting with residues located on TM3 and TM7, salicin interacts with TM3 and TM2 residues. In other words, for salicin the TM2 helix helps TM7 to bind the ligand. In the TM3-facing binding mode, the O2 group of arbutin and phenyl-β-D-glucopyranoside is interacting with Y2667.43, while for salicin it does with S632.57 and N662.60 (Figs 4 and 5). In the TM7-facing mode, the O6 group of phenyl-β-D-glucopyranoside and arbutin also forms a H-bond with Y2667.43, whereas for salicin it is N672.61. Unfortunately, no mutagenesis studies have been performed on S632.57, N662.60 and N672.61, and thus their role in salicin binding remains to be verified. However, it is noteworthy that hTAS2R16 is the only bitter taste receptor having a H-bond donor/acceptor at position 2.57, whereas other hTAS2Rs have a hydrophobic residue (I or L, Supplementary Table S8). Therefore, it would be interesting to experimentally test the effect of mutations S63I and S63L on salicin binding.
T923.39 is another residue interacting differently with the three ligands. Although the bottom part of the binding site is lined with hydrophobic and aromatic residues and thus it is likely to be specialized in binding the phenyl moiety common to all three ligands, this region also contains a single polar amino acid, T923.39. This residue does not interact with phenyl-β-D-glucopyranoside, but its presence allows the formation of a H-bond with the ligands containing substituted phenyl aglycons, i.e. the phenolic oxygen of arbutin and the hydroxymethyl oxygen of salicin (O7 in Fig. 1). In the case of arbutin, a H-bond is present between the phenolic oxygen and T923.39 for both binding poses, because the O7 atom in para is optimally positioned to point toward T923.39. Instead, for salicin the H-bond pattern changes between the two binding modes, due to the O7 substituent being in ortho position. For the TM3-facing binding pose, the salicin hydroxymethyl group forms a H-bond with T923.39, whereas, for the TM7-facing mode, salicin O7 can either form a weak interaction with T923.39 or act as a H-bond acceptor to N893.36. In other words, the salicin O7 substituent is always pointing towards TM3, regardless of the binding mode. Based on the results of our simulations, we predict that mutation of T923.39 into a hydrophobic residue would affect the receptor response to salicin and arbutin, while the impact for phenyl-β-D-glucopyranoside might be weaker.
Moreover, position 3.39 is quite conserved across the hTAS2R family (see Supplementary Table S8), though there are no available mutagenesis data for any bitter taste receptor. Hence, we resorted to the information available for other class A GPCRs. In this class, it has been proposed that a conserved S/T at precisely this position forms part of the allosteric sodium binding site125. However, it should be noted that sodium binding in class A GPCRs requires a conserved D/E residue (located at position 2.50), and this acidic residue is replaced by a conserved R in hTAS2Rs. Therefore, one could surmise that the canonical sodium binding site might be missing in hTAS2Rs. Nonetheless, position 3.39 might still play a functional role in hTAS2Rs through a different mechanism, which remains to be tested with experiments.
Residues involved in shaping the binding cavity and in second shell effects (Q1775.39, H1815.43, Y2396.51, F2406.52, I2436.55)
The contact frequency analysis (Supplementary Tables S6 and S7) shows that, besides the aforementioned residues, H1815.43 is also close (i.e. within the 5.5 Å distance cutoff) for all three ligands. However, our simulations indicate that H181 is not directly interacting with the β-glucosides. Instead, it may have an indirect effect, since it forms a salt bridge with E863.33 in all the simulations. This glutamate directly interacts with the ligand and its interaction with H1815.43 might keep E863.33 in optimal position to act as H-bond acceptor for the glucose hydroxyl groups (Supplementary Fig. S7). In this regard, H1815.43 can be considered part of the second binding shell. Experimentally, the mutation H181T slightly reduces the EC50 with respect to wild-type hTAS2R16 for the three ligands (between 2 and 8.5 times), while H181L has a stronger impact on EC50 (no response for two out of the three ligands, see Supplementary Table S2)55. Therefore, the mutagenesis data supports the suggestion that a polar residue able to form H-bonds is needed at position 5.43.
Another second-shell residue is F2406.52. Although it is not directly interacting with many of the ligands, we noticed that it is part of an “aromatic cluster” which involves several π-stacked phenylalanines on TM6, TM7 and TM3. In particular, F2406.52 forms a T-stacking interaction with F2366.48, which in turn forms a parallel displaced stacking with F933.40; the latter interacts with F2687.45 through parallel displaced stacking (Supplementary Fig. S8). Considering that F933.40, F2366.48 and F2687.45 are involved in hydrophobic interactions with the ligand, F2406.52 may help to stabilize the other phenylalanines in the cluster and thus have a second shell effect in ligand binding. Mutation of F2406.52 to aromatic residues Y or W decreases EC50 only slightly (by 6.5–11-fold), whereas the L mutant shows no receptor activity53,55. Therefore, the experimental data supports that the stacking interactions of the aromatic cluster are likely to be important for ligand binding.
Besides, other residues possibly involved in shaping the binding cavity or in second shell effects according to our simulations are Q1775.39, Y2396.51 and I2436.55 (see Supplementary Tables S6 and S7). Q1775.39 is able to interact with the ligands through H-bonds (Supplementary Tables S4 and S5). However, the same hydroxyl group in contact with Q1775.39 can form more persistent H-bonds with other residues, suggesting that Q1775.39 is not essential for ligand binding or that can be easily replaced by other residues. Indeed, EC50 data shows that mutations to N, E or A do not affect the EC50 significantly (i.e. the EC50 change is only between 0.9 and 2.9 times, depending on the mutation and the ligand).
Y2396.51 can interact with the three ligands, but the low persistency of the corresponding H-bond (Supplementary Tables S4 and S5) is not enough to discriminate whether this residue is involved in ligand binding or in shaping the binding cavity. Indeed, in our simulations we observed that Y2396.51 can also establish a H-bond with N893.36 (Fig. 5A), thus contributing to the correct orientation of the latter residue inside the binding pocket. Future mutations Y239F and Y239T could help to clarify the role of this residue.
Finally, I2436.55 is located one helix turn above Y2396.51. We predict that the presence of a bulky residue in position 6.55 might help to keep the aromatic ring of Y2396.51 close to the ligand. In other words, I2436.55 may help Y2396.51 to adopt the right rotameric state. This is in line with mutations to bulky hydrophobic residues (I243L and I243V) reducing only slightly the EC50 (mutant/wild-type ratio between 2.0 and 5.2), while mutation to A (a smaller residue) shows no receptor response for any of the ligands studied here55,56.
Overall picture
In summary, our calculations suggest that the upper part of the hTAS2R16 binding cavity is mostly composed by polar residues that interact with the glucose unit. Interestingly, the residues capable of forming H-bonds are positioned mirroring each other on both sides of the cavity (E863.33 and N893.36 on TM3, as well as E2627.39 and Y2667.43 on TM7). The only exception is represented by salicin, which replaces some of the interactions with residues located on TM7 by polar residues on TM2 (S632.57, N662.60 and N672.61). Despite the slight differences for salicin binding, the picture remains the same for the three agonists studied here: the glucose ring is hold in place by two sets of polar residues, located in two spatially adjacent helices. In contrast, the bottom part of the hTAS2R16 binding cavity consists mostly of hydrophobic and aromatic residues that can interact with the hydrophobic aglycon (Fig. 4). Since these interacting residues are positioned all around the bottom part of the cavity and hydrophobic interactions are non-directional, the aglycon can be easily accommodated regardless of the ligand orientation (either TM3- or TM7-binding modes).
Binding mode of other hTAS2R16 agonists
We next provide qualitative insights into the binding determinants of other bitter sugars for which experimental data are available52,53,54. Assuming that these other hTAS2R16 agonists adopt a binding pose similar to that of the three ligands studied here (arbutin, phenyl-β-D-glucopyranoside and salicin), one can hypothesize a rationale for the changes in receptor activity caused by the variation in chemical structure among the different agonists. As shown below, this structure-activity relationship (SAR) data gives further support to our simulations.
In addition to β-D-glucopyranosides, hTAS2R16 can also detect some disaccharides, such as amygdalin. The O6 addition of another glucose unit in amygdalin (Supplementary Fig. S1) does not affect the receptor response significantly54. This can be explained, at least in part, assuming that amygdalin adopts a binding pose with the glucose moiety pointing toward the extracellular side of the receptor, as the one described here. The additional sugar will be placed towards the solvent and might be able to interact with other residues in the upper part of the binding cavity.
Replacement of the phenyl aglycon by smaller aliphatic groups (such as in methyl- or hexyl-β-D-glucopyranoside) decreases the receptor response53,54. These SAR data also supports the orientation of the ligand proposed here, since the aforementioned substitutions would result in loss of hydrophobic interactions between the aglycon and residues in the bottom part of the binding cavity, compared to phenyl-β-D-glucopyranoside. Moreover, the lack of hTAS2R16 response to the β-glucose monosaccharide52 may be due to the removal of the hydrophobic aglycon otherwise present in hTAS2R16 agonists (Supplementary Fig. S1), which eliminates the stabilizing hydrophobic interactions with the bottom part of the binding cavity. This is also in line with the higher hydrophobicity of bitter tastants compared to sweet compounds126.
Besides the aglycon substitutions, it is also interesting to compare with the SAR data for the sugar unit. The β configuration of the glycosidic oxygen and the equatorial orientation of the 4-OH group (Fig. 1) are known to be essential for hTAS2R16 recognition52,53,54. Consistently, the O1 and O4 atoms of the three ligands studied here are both involved in H-bonds with the receptor. The lack of receptor response to α-D-glucopyranoside and β-D-galactopyranoside52,54 can thus be ascribed to the change in orientation of the glycosidic oxygen or the 4-OH group relative to β-glucopyranoside, respectively (Supplementary Fig. S1). This in turn may severely affect the H-bond network that keeps the ligand bound. In addition, hTAS2R16 does not respond to phenyl-β-D-xylopyranoside53, in which the glucose C6 hydroxymethyl group present in β-glucopyranoside is absent (Supplementary Fig. S1). This may be interpreted based on the loss of H-bond interactions (with E863.33 and Y2396.51 in TM3-binding pose or with E2627.39 and Y2667.43 for TM7-binding pose).
In contrast, β-D-mannopyranoside (Supplementary Fig. S1) can still be detected by hTAS2R1652,53. This may be possible, at least in part, by an exchange of H-bonding residues upon epimerization of the 2-OH group from equatorial (glucose) to axial (mannose). In the TM3-facing mode, the O2 atom would go from forming H-bonds with E2627.39 and Y2667.43 for glucose to be near Y2396.51 for mannose. Complementarily, in the TM7-facing mode, W853.32 could take the place of E863.33 or N893.36.
Conclusions
We have presented here the binding determinants of hTAS2R16, a group-selective bitter taste receptor that preferentially binds β-glycopyranosides with a hydrophobic aglycon (i.e. bitter sugars). Our simulations suggest receptor-ligand interactions that are validated a posteriori by comparison with a plethora of experimental data. Moreover, they predict new putative binding residues not yet experimentally characterized, in particular S632.57 and T923.39. Additional mutagenesis and functional experiments, complemented with MD simulations of the mutated complexes, will help to clarify the putative role of these residues in binding39,127,128.
Although hTAS2R16 is highly specialized in detecting bitter β-glucopyranosides, its ligands display a large diversity of aglycons (Fig. 1 and Supplementary Fig. S1), varying in size and hydrophobicity53,54,55,56. This poses the question of how the binding cavity of hTAS2R16 is capable to adapt to such a broad range of ligands. Here, using MM/CG simulations on the complexes with the hTAS2R16 agonists arbutin, phenyl-β-D-glucopyranoside and salicin (Fig. 1), we suggest that the solution to this apparent dichotomy is the existence of a previously unrecognized dual binding mode. Unlike previously published models (see Supplementary Text S2 55,67, our findings can provide a molecular explanation to all the available experimental data, even if the mutagenesis data were not used to drive the docking or bias the simulations. Moreover, they provide a rationale for the SAR data53,54,55,56 and thus give insights applicable to other hTAS2R16 agonists, besides the three studied here.
Our work suggests, for the first time, the presence of a dual binding mode mechanism in bitter taste receptors. Nonetheless, this is not the only protein exhibiting a dual binding mode. Several other protein-ligand complexes have been reported129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146, including several lectins120,121,122,123,124, i.e. carbohydrate-binding proteins that are able to recognize sugars with high specificity. In these lectin-sugar complexes, the two observed binding modes involve interactions between the same protein residues and the same sugar hydroxyl groups, but with a different H-bond pattern. This is essentially the same strategy used by hTAS2R16 to bind the glucose unit of the bitter sugar using both the TM3- and TM7- binding modes. In addition, for lectins it was shown that the two binding modes coexist in equilibrium, as observed in the crystal structures120,121, and can exchange at room temperature122,123. Although our simulations are not long enough to explore this exchange, we speculate that this might be also the case for the two binding modes identified here for the hTAS2R16 complexes.
The physico-chemical characteristics of the hTAS2R16/glycopyranoside complex are in agreement with the features suggested to be important to enable multiple binding modes in other protein-ligand complexes136,138. The first one is ligand flexibility, which allows the ligand to adopt several possible conformations with a low energy cost. For the bitter sugars considered here, the flexibility is ensured by the rotatable glycosidic bond, which permits to position the aglycon in different orientations. The second one is the presence of a mostly hydrophobic binding site, which can provide rather unspecific and non-directional interactions. In our case, the intracellular part of the hTAS2R16 binding cavity is surrounded by hydrophobic residues that can stabilize the hydrophobic aglycon of the bitter sugars regardless of the orientation. However, the extracellular part of the binding cavity is mostly composed by polar residues, in order to accommodate the glucose unit. hTAS2R16 overcomes this issue by placing two sets of interacting residues mirroring each other in adjacent helices (TM3 and TM7). This peculiar residue distribution is still compatible with the dual binding mode because it allows the formation of H-bonds with the sugar in either of the two 180 degree-rotated binding modes (see Supplementary Text S4).
The three hTAS2R16 agonists studied here (arbutin, phenyl-β-D-glucopyranoside and salicin) can bind to the receptor using either of the two binding modes. Nonetheless, given the large diversity of aglycons recognized by hTAS2R16, some might exhibit only one binding mode (specially those with non-symmetric aglycons and bulky substituents). Indeed, the changes in salicin binding compared to unsubstituted phenyl-β-D-glucopyranoside (Fig. 1) suggest that the dual binding mode of hTAS2R16 agonists might be modulated by the aglycon substituents. This is in line with a previous analysis of protein-ligand complexes exhibiting dual binding mode136 showing that two types of mechanisms can be observed. Ligands belonging to the same chemical class can bind either with two different orientations or with only one depending on their substituents.
In summary, our simulations suggest that the particular residue distribution of the hTAS2R16 binding cavity enables the existence of the dual binding mode mechanism, which in turn allows to accommodate a wide variety of ligands.
Data Availability
Data generated or analyzed during the current study are available upon request.
References
Lagerstrom, M. C. & Schioth, H. B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7, 339–357, https://doi.org/10.1038/nrd2518 (2008).
Fredriksson, R., Lagerstrom, M. C., Lundin, L. G. & Schioth, H. B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63, 1256–1272, https://doi.org/10.1124/mol.63.6.1256 (2003).
Nordström, K. J., Sällman Almén, M., Edstam, M. M., Fredriksson, R. & Schiöth, H. B. Independent HHsearch, Needleman–Wunsch-based, and motif analyses reveal the overall hierarchy for most of the G protein-coupled receptor families. Mol Biol Evol 28, 2471–2480 (2011).
Di Pizio, A. & Niv, M. Y. Computational Studies of Smell and Taste Receptors. Isr J Chem 54, 1205–1218, https://doi.org/10.1002/ijch.201400027 (2014).
Di Pizio, A. et al. Comparing Class A GPCRs to bitter taste receptors: Structural motifs, ligand interactions and agonist-to-antagonist ratios. Methods in cell biology 132, 401–427 (2016).
Horn, F. et al. GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res 31, 294–297 (2003).
Adler, E. et al. A novel family of mammalian taste receptors. Cell 100, 693–702 (2000).
Chandrashekar, J. et al. T2Rs function as bitter taste receptors. Cell 100, 703–711 (2000).
Mueller, K. L. et al. The receptors and coding logic for bitter taste. Nature 434, 225 (2005).
Liu, R. H. Potential synergy of phytochemicals in cancer prevention: mechanism of action. J Nutr 134, 3479S–3485S, https://doi.org/10.1093/jn/134.12.3479S (2004).
Weisburger, J. H. Eat to live, not live to eat. Nutrition 16, 767–773 (2000).
Bravo, L. Polyphenols: chemistry, dietary sources, metabolism, and nutritional significance. Nutr Rev 56, 317–333 (1998).
Mennella, J. A., Spector, A. C., Reed, D. R. & Coldwell, S. E. The bad taste of medicines: overview of basic research on bitter taste. Clin Ther 35, 1225–1246, https://doi.org/10.1016/j.clinthera.2013.06.007 (2013).
Anliker, J. A., Bartoshuk, L., Ferris, A. M. & Hooks, L. D. Children’s food preferences and genetic sensitivity to the bitter taste of 6-n-propylthiouracil (PROP). Am J Clin Nutr 54, 316–320, https://doi.org/10.1093/ajcn/54.2.316 (1991).
Drewnowski, A. The science and complexity of bitter taste. Nutr Rev 59, 163–169 (2001).
Duffy, V. B. & Bartoshuk, L. M. Food acceptance and genetic variation in taste. J Am Diet Assoc 100, 647–655, https://doi.org/10.1016/S0002-8223(00)00191-7 (2000).
Beckett, E. L. et al. Bitter taste genetics–the relationship to tasting, liking, consumption and health. Food Funct 5, 3040–3054, https://doi.org/10.1039/c4fo00539b (2014).
Ansoleaga, B. et al. Dysregulation of brain olfactory and taste receptors in AD, PSP and CJD, and AD-related model. Neuroscience 248, 369–382, https://doi.org/10.1016/j.neuroscience.2013.06.034 (2013).
Pydi, S. P. et al. Amino Acid Derivatives as Bitter Taste Receptor (T2R) Blockers. J Biol Chem 289, 25054–25066, https://doi.org/10.1074/jbc.M114.576975 (2014).
Wolfle, U. et al. Salicin from Willow Bark can Modulate Neurite Outgrowth in Human Neuroblastoma SH-SY5Y Cells. Phytother Res 29, 1494–1500, https://doi.org/10.1002/ptr.5400 (2015).
Rozengurt, E. Taste receptors in the gastrointestinal tract. I. Bitter taste receptors and α-gustducin in the mammalian gut. Am J Physiol-Gastr L 291, G171–G177 (2006).
Behrens, M. & Meyerhof, W. Gustatory and extragustatory functions of mammalian taste receptors. Physiol Behav 105, 4–13, https://doi.org/10.1016/j.physbeh.2011.02.010 (2011).
Deshpande, D. A. et al. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med 16, 1299–1304, https://doi.org/10.1038/nm.2237 (2010).
Lee, R. J. & Cohen, N. A. Bitter and sweet taste receptors in the respiratory epithelium in health and disease. J Mol Med 92, 1235–1244, https://doi.org/10.1007/s00109-014-1222-6 (2014).
Foster, S. R. et al. Expression, regulation and putative nutrient-sensing function of taste GPCRs in the heart. PLoS One 8, e64579, https://doi.org/10.1371/journal.pone.0064579 (2013).
Shaik, F. A. et al. Bitter taste receptors: Extraoral roles in pathophysiology. Int J Biochem Cell Biol 77, 197–204, https://doi.org/10.1016/j.biocel.2016.03.011 (2016).
Lu, P., Zhang, C. H., Lifshitz, L. M. & ZhuGe, R. Extraoral bitter taste receptors in health and disease. J Gen Physiol 149, 181–197, https://doi.org/10.1085/jgp.201611637 (2017).
Foster, S. R., Roura, E. & Thomas, W. G. Extrasensory perception: odorant and taste receptors beyond the nose and mouth. Pharmacol Ther 142, 41–61, https://doi.org/10.1016/j.pharmthera.2013.11.004 (2014).
Ferrer, I. et al. Olfactory Receptors in Non-Chemosensory Organs: The Nervous System in Health and Disease. Front Aging Neurosci 8, 163, https://doi.org/10.3389/fnagi.2016.00163 (2016).
Lee, S. J., Depoortere, I. & Hatt, H. Therapeutic potential of ectopic olfactory and taste receptors. Nat Rev Drug Discov 18, 116–138, https://doi.org/10.1038/s41573-018-0002-3 (2019).
Fierro, F. et al. Agonist binding to chemosensory receptors: a systematic bioinformatics analysis. Frontiers in molecular biosciences 4, 63 (2017).
Isberg, V. et al. GPCRdb: an information system for G protein-coupled receptors. Nucleic Acids Res 44, D356–D364, https://doi.org/10.1093/nar/gkv1178 (2016).
Chothia, C. & Lesk, A. M. The relation between the divergence of sequence and structure in proteins. EMBO J 5, 823–826 (1986).
Piccoli, S., Suku, E., Garonzi, M. & Giorgetti, A. Genome-wide Membrane Protein Structure Prediction. Curr Genomics 14, 324–329, https://doi.org/10.2174/13892029113149990009 (2013).
Olivella, M., Gonzalez, A., Pardo, L. & Deupi, X. Relation between sequence and structure in membrane proteins. Bioinformatics 29, 1589–1592 (2013).
Cherezov, V. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265, https://doi.org/10.1126/science.1150577 (2007).
Marchiori, A. et al. Coarse-Grained/Molecular Mechanics of the TAS2R38 Bitter Taste Receptor: Experimentally-Validated Detailed Structural Prediction of Agonist Binding. Plos One 8, https://doi.org/10.1371/journal.pone.0064675 (2013).
Capaldi, S. et al. Allosteric sodium binding cavity in GPR3: a novel player in modulation of Abeta production. Sci Rep 8, 11102, https://doi.org/10.1038/s41598-018-29475-7 (2018).
Sandal, M. et al. Evidence for a Transient Additional Ligand Binding Site in the TAS2R46 Bitter Taste Receptor. J Chem Theory Comput 11, 4439–4449, https://doi.org/10.1021/acs.jctc.5b00472 (2015).
Munk, C., Harpsoe, K., Hauser, A. S., Isberg, V. & Gloriam, D. E. Integrating structural and mutagenesis data to elucidate GPCR ligand binding. Curr Opin Pharmacol 30, 51–58, https://doi.org/10.1016/j.coph.2016.07.003 (2016).
Behrens, M. & Meyerhof, W. Vertebrate Bitter Taste Receptors: Keys for Survival in Changing Environments. J Agric Food Chem, https://doi.org/10.1021/acs.jafc.6b04835 (2017).
Meyerhof, W. et al. The molecular receptive ranges of human TAS2R bitter taste receptors. Chem Senses 35, 157–170, https://doi.org/10.1093/chemse/bjp092 (2010).
Sainz, E. et al. Functional characterization of human bitter taste receptors. Biochem J 403, 537–543, https://doi.org/10.1042/BJ20061744 (2007).
Wiener, A., Shudler, M., Levit, A. & Niv, M. Y. BitterDB: a database of bitter compounds. Nucleic Acids Res 40, D413–D419, https://doi.org/10.1093/nar/gkr755 (2012).
Brockhoff, A., Behrens, M., Niv, M. Y. & Meyerhof, W. Structural requirements of bitter taste receptor activation. Proc Natl Acad Sci USA 107, 11110–11115, https://doi.org/10.1073/pnas.0913862107 (2010).
Kruse, A. C. et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556, https://doi.org/10.1038/nature10867 (2012).
Dror, R. O. et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–+, https://doi.org/10.1038/nature12595 (2013).
Granier, S. & Kobilka, B. A new era of GPCR structural and chemical biology. Nat Chem Biol 8, 670–673, https://doi.org/10.1038/nchembio.1025 (2012).
Manglik, A. et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326, https://doi.org/10.1038/nature10954 (2012).
Wheatley, M. et al. Lifting the lid on GPCRs: the role of extracellular loops. Br J Pharmacol 165, 1688–1703, https://doi.org/10.1111/j.1476-5381.2011.01629.x (2012).
Biarnes, X. et al. Insights into the binding of Phenyltiocarbamide (PTC) agonist to its target human TAS2R38 bitter receptor. PLoS One 5, e12394, https://doi.org/10.1371/journal.pone.0012394 (2010).
Bufe, B., Schöley-Pohl, E., Krautwurst, D., Hofmann, T. & Meyerhof, W. Identification of human bitter taste receptors. Am Chem Soc Symp S 867, 45–59 (2003).
Thomas, A. et al. The bitter taste receptor TAS2R16 achieves high specificity and accommodates diverse glycoside ligands by using a two-faced binding pocket. Sci Rep-Uk 7, 7753 (2017).
Bufe, B., Hofmann, T., Krautwurst, D., Raguse, J. D. & Meyerhof, W. The human TAS2R16 receptor mediates bitter taste in response to beta-glucopyranosides. Nat Genet 32, 397–401, https://doi.org/10.1038/ng1014 (2002).
Sakurai, T. et al. Characterization of the beta-D-glucopyranoside binding site of the human bitter taste receptor hTAS2R16. J Biol Chem 285, 28373–28378, https://doi.org/10.1074/jbc.M110.144444 (2010).
Sakurai, T. et al. The human bitter taste receptor, hTAS2R16, discriminates slight differences in the configuration of disaccharides. Biochem Biophys Res Commun 402, 595–601, https://doi.org/10.1016/j.bbrc.2010.10.059 (2010).
Soranzo, N. et al. Positive selection on a high-sensitivity allele of the human bitter-taste receptor TAS2R16. Curr Biol 15, 1257–1265, https://doi.org/10.1016/j.cub.2005.06.042 (2005).
Hinrichs, A. L. et al. Functional variant in a bitter-taste receptor (hTAS2R16) influences risk of alcohol dependence. The American Journal of Human Genetics 78, 103–111 (2006).
Kingsbury, J. M. Poisonous plants of the United States and Canada. Soil Science 98, 349 (1964).
Meyerhof, W. Elucidation of mammalian bitter taste. Rev Physiol Bioch P 154, 37–72, https://doi.org/10.1007/s10254-005-0041-0 (2005).
Chan, H. S. et al. Exploring a new ligand binding site of G protein-coupled receptors. Chemical Science 9, 6480–6489 (2018).
Campa, D. et al. Bitter taste receptor polymorphisms and human aging. PLoS One 7, e45232, https://doi.org/10.1371/journal.pone.0045232 (2012).
Risso, D. S. et al. A bio-cultural approach to the study of food choice: The contribution of taste genetics, population and culture. Appetite 114, 240–247, https://doi.org/10.1016/j.appet.2017.03.046 (2017).
Li, H., Jeong, Y.-M., Kim, S. Y., Kim, M.-K. & Kim, D.-S. Arbutin inhibits TCCSUP human bladder cancer cell proliferation via up-regulation of p21. Die Pharmazie-An International Journal of Pharmaceutical Sciences 66, 306–309 (2011).
Jones, R. Nonsteroidal anti-inflammatory drug prescribing: past, present, and future. The American Journal of Medicine 110, S4–S7 (2001).
Kong, C. S. et al. Salicin, an Extract from White Willow Bark, Inhibits Angiogenesis by Blocking the ROS-ERK Pathways. Phytotherapy research 28, 1246–1251 (2014).
Chen, Z. et al. Insights into the binding of agonist and antagonist to TAS2R16 receptor: a molecular simulation study. Molecular Simulation 44, 322–329 (2018).
Neri, M., Anselmi, C., Cascella, M., Maritan, A. & Carloni, P. Coarse-grained model of proteins incorporating atomistic detail of the active site. Phys Rev Lett 95, https://doi.org/10.1103/PhysRevLett.95.218102 (2005).
Neri, M. et al. Microseconds dynamics simulations of the outer-membrane protease T. Biophys J 94, 71–78, https://doi.org/10.1529/biophysj.107.116301 (2008).
Leguebe, M. et al. Hybrid molecular mechanics/coarse-grained simulations for structural prediction of G-protein coupled receptor/ligand complexes. PLoS One 7, e47332, https://doi.org/10.1371/journal.pone.0047332 (2012).
Tarenzi, T., Calandrini, V., Potestio, R., Giorgetti, A. & Carloni, P. Open boundary simulations of proteins and their hydration shells by Hamiltonian adaptive resolution scheme. J Chem Theory Comput 13, 5647–5657 (2017).
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res 32, D138–141, https://doi.org/10.1093/nar/gkh121 (2004).
Pei, J. & Grishin, N. V. PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 23, 802–808, https://doi.org/10.1093/bioinformatics/btm017 (2007).
Ballesteros, J. A. & Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in neurosciences 25, 366–428 (1995).
Sandal, M. et al. GOMoDo: A GPCRs Online Modeling and Docking Webserver. Plos One 8, https://doi.org/10.1371/journal.pone.0074092 (2013).
Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960, https://doi.org/10.1093/bioinformatics/bti125 (2005).
Soding, J., Biegert, A. & Lupas, A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33, W244–248, https://doi.org/10.1093/nar/gki408 (2005).
Eswar, N., Eramian, D., Webb, B., Shen, M. Y. & Sali, A. Protein structure modeling with MODELLER. Methods Mol Biol 426, 145–159, https://doi.org/10.1007/978-1-60327-058-8_8 (2008).
Venkatakrishnan, A. J. et al. Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194, https://doi.org/10.1038/nature11896 (2013).
Melo, F., Sanchez, R. & Sali, A. Statistical potentials for fold assessment. Protein Sci 11, 430–448, https://doi.org/10.1002/pro.110430 (2002).
Shen, M. Y. & Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci 15, 2507–2524, https://doi.org/10.1110/ps.062416606 (2006).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography 66, 12–21 (2010).
Wang, Y. et al. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res 37, W623–W633 (2009).
Dominguez, C., Boelens, R. & Bonvin, A. M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125, 1731–1737, https://doi.org/10.1021/ja026939x (2003).
De Vries, S. J., van Dijk, M. & Bonvin, A. M. J. J. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc 5, 883–897, https://doi.org/10.1038/nprot.2010.32 (2010).
Le Guilloux, V., Schmidtke, P. & Tuffery, P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 10, 168, https://doi.org/10.1186/1471-2105-10-168 (2009).
Schmidtke, P., Le Guilloux, V., Maupetit, J. & Tuffery, P. fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res 38, W582–W589, https://doi.org/10.1093/nar/gkq383 (2010).
Daura, X. et al. Peptide folding: when simulation meets experiment. Angewandte Chemie International Edition 38, 236–240 (1999).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. Journal of molecular graphics 14, 33–38 (1996).
Michino, M. et al. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov 8, 455–463, https://doi.org/10.1038/nrd2877 (2009).
Katritch, V., Rueda, M., Lam, P. C., Yeager, M. & Abagyan, R. GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins 78, 197–211, https://doi.org/10.1002/prot.22507 (2010).
Kufareva, I., Katritch, V., Participants of, G. D., Stevens, R. C. & Abagyan, R. Advances in GPCR modeling evaluated by the GPCR Dock 2013 assessment: meeting new challenges. Structure 22, 1120–1139, https://doi.org/10.1016/j.str.2014.06.012 (2014).
Kufareva, I. et al. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 19, 1108–1126, https://doi.org/10.1016/j.str.2011.05.012 (2011).
Beuming, T. & Sherman, W. Current assessment of docking into GPCR crystal structures and homology models: successes, challenges, and guidelines. J Chem Inf Model 52, 3263–3277, https://doi.org/10.1021/ci300411b (2012).
Scott, W. R. P. et al. The GROMOS biomolecular simulation program package. J Phys Chem A 103, 3596–3607, https://doi.org/10.1021/jp984217f (1999).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The Missing Term in Effective Pair Potentials. J Phys Chem-Us 91, 6269–6271, https://doi.org/10.1021/j100308a038 (1987).
Plazinski, W., Lonardi, A. & Hünenberger, P. H. Revision of the GROMOS 56A6CARBO force field: Improving the description of ring-conformational equilibria in hexopyranose-based carbohydrates chains. Journal of computational chemistry 37, 354–365 (2016).
Schuttelkopf, A. W. & van Aalten, D. M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr 60, 1355–1363, https://doi.org/10.1107/S0907444904011679 (2004).
Lemkul, J. A., Allen, W. J. & Bevan, D. R. Practical considerations for building GROMOS-compatible small-molecule topologies. Journal of chemical information and modeling 50, 2221–2235 (2010).
Bayly, C. I., Cieplak, P., Cornell, W. & Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. The Journal of Physical Chemistry 97, 10269–10280 (1993).
Frisch, M. J. et al. Gaussian 09, revision A. 1. Gaussian Inc. Wallingford CT 27, 34 (2009).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Antechamber: an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc 222, U403 (2001).
Go, N. & Abe, H. Non-Interacting Local-Structure Model of Folding and Unfolding Transition in Globular-Proteins .1. Formulation. Biopolymers 20, 991–1011, https://doi.org/10.1002/bip.1981.360200511 (1981).
Giorgetti, A. & Carloni, P. In Protein Modelling 165–174 (Springer International Publishing, 2014).
Suku, E., Fierro, F., Giorgetti, A., Alfonso-Prieto, M. & Carloni, P. Multi-scale simulations of membrane proteins: the case of bitter taste receptors. Journal of Science: Advanced Materials and Devices (2017).
Schneider, J. et al. Predicting ligand binding poses for low-resolution membrane protein models: Perspectives from multiscale simulations. Biochemical and biophysical research communications 498, 366–374 (2018).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J Comput Chem 26, 1701–1718, https://doi.org/10.1002/jcc.20291 (2005).
Lee, B. & Richards, F. M. The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55, 379-IN374 (1971).
Bohacek, R. S. & McMartin, C. Definition and display of steric, hydrophobic, and hydrogen-bonding properties of ligand binding sites in proteins using Lee and Richards accessible surface: validation of a high-resolution graphical tool for drug design. J Med Chem 35, 1671–1684 (1992).
Graziano, G. On the size dependence of hydrophobic hydration. Journal of the Chemical Society, Faraday Transactions 94, 3345–3352 (1998).
Lindahl, E., Hess, B. & Van Der Spoel, D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. Molecular modeling annual 7, 306–317 (2001).
Colquhoun, D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125, 924–947 (1998).
Strange, P. G. Agonist binding, agonist affinity and agonist efficacy at G protein-coupled receptors. Br J Pharmacol 153, 1353–1363, https://doi.org/10.1038/sj.bjp.0707672 (2008).
Kenakin, T. Drug efficacy at G protein-coupled receptors. Annu Rev Pharmacol Toxicol 42, 349–379, https://doi.org/10.1146/annurev.pharmtox.42.091401.113012 (2002).
Singh, N., Pydi, S. P., Upadhyaya, J. & Chelikani, P. Structural basis of activation of bitter taste receptor T2R1 and comparison with Class A G-protein-coupled receptors (GPCRs). J Biol Chem 286, 36032–36041, https://doi.org/10.1074/jbc.M111.246983 (2011).
Raghavan, V., Bollmann, P. & Jung, G. S. A critical investigation of recall and precision as measures of retrieval system performance. ACM Transactions on Information Systems (TOIS) 7, 205–229 (1989).
Manning, C. D. & Schütze, H. Foundations of statistical natural language processing. Vol. 999 (MIT Press, 1999).
Davis, J. & Goadrich, M. In Proceedings of the 23rd international conference on Machine learning. 233–240 (ACM).
Saito, T. & Rehmsmeier, M. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One 10, e0118432, https://doi.org/10.1371/journal.pone.0118432 (2015).
Loris, R., Maes, D., Poortmans, F., Wyns, L. & Bouckaert, J. A structure of the complex between concanavalin A and methyl-3, 6-di-O-(α-D-mannopyranosyl)-α-D-mannopyranoside reveals two binding modes. J Biol Chem 271, 30614–30618 (1996).
Feinberg, H., Castelli, R., Drickamer, K., Seeberger, P. H. & Weis, W. I. Multiple modes of binding enhance the affinity of DC-SIGN for high mannose N-linked glycans found on viral glycoproteins. J Biol Chem 282, 4202–4209 (2007).
Angulo, J. et al. Saturation Transfer Difference (STD) NMR Spectroscopy Characterization of Dual Binding Mode of a Mannose Disaccharide to DC-SIGN. ChemBioChem 9, 2225–2227 (2008).
Marcelo, F. et al. Delineating Binding Modes of Gal/GalNAc and Structural Elements of the Molecular Recognition of Tumor-Associated Mucin Glycopeptides by the Human Macrophage Galactose-Type Lectin. Chemistry–A European Journal 20, 16147–16155 (2014).
Bourne, Y. et al. Crosslinking of mammalian lectin (galectin-1) by complex biantennary saccharides. Nature Structural and Molecular Biology 1, 863 (1994).
Katritch, V. et al. Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39, 233–244, https://doi.org/10.1016/j.tibs.2014.03.002 (2014).
Di Pizio, A., Shoshan-Galeczki, Y. B., Hayes, J. E. & Niv, M. Y. Bitter and sweet tasting molecules: it’s complicated. Neuroscience letters (2018).
Burney, P. R., Nordwald, E. M., Hickman, K., Kaar, J. L. & Pfaendtner, J. Molecular dynamics investigation of the ionic liquid/enzyme interface: Application to engineering enzyme surface charge. Proteins: Structure, Function, and Bioinformatics 83, 670–680 (2015).
Sprenger, K., Plaks, J., Kaar, J. & Pfaendtner, J. Elucidating sequence and solvent specific design targets to protect and stabilize enzymes for biocatalysis in ionic liquids. Phys Chem Chem Phys 19, 17426–17433 (2017).
Poulos, T. L. & Howard, A. J. Crystal structures of metyrapone- and phenylimidazole-inhibited complexes of cytochrome P-450cam. Biochemistry 26, 8165–8174 (1987).
Badger, J. et al. Structural analysis of a series of antiviral agents complexed with human rhinovirus 14. Proc Natl Acad Sci USA 85, 3304–3308 (1988).
Holtz, K. M. et al. Alternate modes of binding in two crystal structures of alkaline phosphatase-inhibitor complexes. Protein Science 9, 907–915 (2000).
Dohnálek, J. et al. A distinct binding mode of a hydroxyethylamine isostere inhibitor of HIV-1 protease. Acta Crystallographica Section D: Biological Crystallography 57, 472–476 (2001).
Wojtczak, A., Cody, V., Luft, J. R. & Pangborn, W. Structure of rat transthyretin (rTTR) complex with thyroxine at 2.5 Å resolution: first non-biased insight into thyroxine binding reveals different hormone orientation in two binding sites. Acta Crystallographica Section D: Biological Crystallography 57, 1061–1070 (2001).
Uytterhoeven, K., Sponer, J. & Van Meervelt, L. Two 1: 1 binding modes for distamycin in the minor groove of d(GGCCAATTGG). Eur J Biochem 269, 2868–2877 (2002).
Rosenfeld, R. J. et al. Conformational changes in nitric oxide synthases induced by chlorzoxazone and nitroindazoles: crystallographic and computational analyses of inhibitor potency. Biochemistry 41, 13915–13925 (2002).
Lewis, P. J. et al. On the detection of multiple-binding modes of ligands to proteins, from biological, structural, and modeling data. J Comput Aid Mol Des 17, 129–134 (2003).
Nazaré, M. et al. Probing the subpockets of factor Xa reveals two binding modes for inhibitors based on a 2-carboxyindole scaffold: a study combining structure-activity relationship and X-ray crystallography. Journal of medicinal chemistry 48, 4511–4525 (2005).
Baumgartner, R. et al. Dual binding mode of a novel series of DHODH inhibitors. Journal of medicinal chemistry 49, 1239–1247 (2006).
Vogtherr, M. et al. NMR characterization of kinase p38 dynamics in free and ligand-bound forms. Angewandte Chemie International Edition 45, 993–997 (2006).
Kim, M. et al. Crystal structure of papain-E64-c complex. Binding diversity of E64-c to papain S2 and S3 subsites. Biochemical Journal 287, 797–803 (1992).
Arnatt, C. K. & Zhang, Y. G. Protein-Coupled Estrogen Receptor (GPER) Agonist Dual Binding Mode Analyses Toward Understanding of Its Activation Mechanism: A Comparative Homology Modeling Approach. Molecular informatics 32, 647–658 (2013).
Nash, M. A., Smith, S. P., Fontes, C. M. & Bayer, E. A. Single versus dual-binding conformations in cellulosomal cohesin–dockerin complexes. Curr Opin Struc Biol 40, 89–96 (2016).
Zhu, Z. et al. Discovery of Novel Hydroxamates as Highly Potent Tumor Necrosis Factor-α Converting Enzyme Inhibitors: Part I. Discovery of Two Binding Modes. Journal of medicinal chemistry 51, 725–736 (2008).
Pilger, J. et al. A Combination of Spin Diffusion Methods for the Determination of Protein–Ligand Complex Structural Ensembles. Angewandte Chemie International Edition 54, 6511–6515 (2015).
Singh, R. P. et al. Dual binding mode of antithyroid drug methimazole to mammalian heme peroxidases–structural determination of the lactoperoxidase–methimazole complex at 1.97 Å resolution. FEBS open bio 6, 640–650 (2016).
Jobst, M. A. et al. Resolving dual binding conformations of cellulosome cohesin-dockerin complexes using single-molecule force spectroscopy. Elife 4, e10319 (2015).
Acknowledgements
The authors gratefully acknowledge the computing time granted by the JARA-HPC on the supercomputer CLAIX at RWTH Aachen Rechenzentrum (project ID jara0165). We thank the financial support of the “Ernesto Illy Foundation” (Trieste, Italy). We are also grateful to Jakob Schneider for his help with the molecular dynamics trajectories analysis and for careful reading of the manuscript.
Author information
Authors and Affiliations
Contributions
F.F. performed molecular modeling and simulations, analyzed the data and wrote the manuscript. A.G., P.C. and M.A.P. supervised, reviewed, discussed the project and wrote the manuscript. W.M. conceptualized data and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fierro, F., Giorgetti, A., Carloni, P. et al. Dual binding mode of “bitter sugars” to their human bitter taste receptor target. Sci Rep 9, 8437 (2019). https://doi.org/10.1038/s41598-019-44805-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44805-z
This article is cited by
-
VirtuousPocketome: a computational tool for screening protein–ligand complexes to identify similar binding sites
Scientific Reports (2024)
-
BitterMatch: recommendation systems for matching molecules with bitter taste receptors
Journal of Cheminformatics (2022)
-
Bitter Taste and Olfactory Receptors: Beyond Chemical Sensing in the Tongue and the Nose
The Journal of Membrane Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
