
Abstract
Megalobrama terminalis distributed in Sino-Russian Heilong-Amur River basin has decreased dramatically in the last few decades. It has been listed in the Red Book of the Russian Federation as an endangered fish species. Here, the complete mitochondrial (mt) genome sequence of M. terminalis in the Heilong River (MTH) was first determined and characterized. Additionally, we identified a population-specific single nucleotide polymorphism (SNP) locus in MTH which could effectively separate MTH from the six other populations of the genus Megalobrama in the absence of hybridization. Moreover, phylogenetic analyses determined that the Xi River M. hoffmanni is located at the basal branch of the clade, and the rest of the group is divided into two assemblages, namely, one containing M. terminalis from Qiantang River and Jinsha River Reservoir/Longxi River M. Pellegrini/MTH and the other containing M. amblycephala from Liangzi Lake and Yi River. We clarify the intraspecies identity of MTH and construct a clearer phylogeny of the genus Megalobrama, which will contribute to the germplasm identification, protection and development of MTH in the future.
Similar content being viewed by others


Introduction
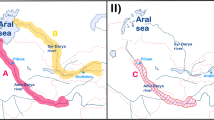
The genus Megalobrama belongs to Cultrinae in Cyprinidae, and is one of the most economically important fish genera in China. This genus contains four main species: M. amblycephala, M. pellegrini, M. hoffmanni and M. terminalis1,2. The most widely distributed species among these is M. terminalis. Historically, M. terminalis inhabited the middle and upper reaches of the Yangtze, Heilong River Basin, Min, Qiantang, Huai, Yellow and Liao Rivers1,3,4 (Fig. 1). Notably, there is only one wild population in the Jinsha River Reservoir of Hongan County in Hubei Province5, but its origin is unclear.

The historical distribution of M. terminalis. YAR, Yangtze River; MR, Min River; QTR, Qiantang River; JSR, Jinsha River Reservoir; HR, Huai River; YER, Yellow River; LR, Liao River; HLR, Heilong River (Fuyuan); AR, Amur River; SHR, Songhua River; NR, Nen River; JPL, Jingpo Lake; KL, Khanka Lake. A red dot represents that wild M. terminalis in the location has disappeared. A green dot indicates that wild M. terminalis in the location is still present.
To date, most of the wild populations of M. terminalis that lived in the original habitats have disappeared except for those in the Qiantang River, Heilong River and Jinsha River Reservoir (Fig. 1). The Heilong River is a Sino-Russian border river, and is known as the Amur River in Russia. M. terminalis in the Amur River has been listed as endangered in the Red Book of the Russian Federation6. In the Heilong River Basin, M. terminalis is called Faluo and lives in several water systems7, including the Heilong River, Songhua River, Nen River, Jingpo Lake, and Khanka Lake (Fig. 1)3,4. Faluo is a famous endemic fish species with several desirable qualities, such as a large body size, high intramuscular fat content and delicious taste. Since the 1960s, however, overfishing and pollution have caused the habitats of Faluo to decrease dramatically7. Currently, only dozens of individuals of Faluo can be captured per year near Fuyuan city (134°28′E, 48°37′N) (Fig. 1), which is located in Heilongjiang Province. Faluo is easy to catch due to its body shape and has late sexual maturity (more than five years)4,6. Therefore, its population growth is not fast enough to confer resilience, increasing the risk of complete extirpation resulting from environmental changes and anthropogenic factors.
As Faluo is the only fish species of the genus Megalobrama that lives in the northern Yellow River, some morphological characters of Faluo in this region are different from those of populations of M. terminalis in southern China. Accordingly, some researchers have questioned the intraspecific taxonomic relationship between Faluo and southern M. terminalis7. So far, few data have been reported on the genetics and genomics of Faluo because of the difficulty in sampling. Evidence reflecting the molecular variations in Faluo and the phylogenetic relationships between Faluo and other fish species within the genus Megalobrama is scarce. On the other hand, regarding the interspecific relationships within the genus Megalobrama, most of the available data suggest close relationships among M. terminalis, M. pellegrini and M. amblycephala, and M. hoffmanni is more distantly related to the other species8,9,10,11,12,13,14. However, the relationships between M. terminalis, M. pellegrini and M. amblycephala have previously been debated. In a phylogenetic study of Eastern Asian Cyprinidae based on mitochondrial CYTB, M. terminalis and M. amblycephala are sister taxa that form a clade with M. pellegrini8. In contrast, in another study based on mitochondrial CYTB, Xie et al. proposed that M. terminalis was closely related to M. pellegrini but not to M. amblycephala9. In a subsequent study based on mitochondrial COI and ND2, Bai et al.10 supported the proposal of Xie et al.9. To some extent, the divergence in conclusions about the interspecific relationships within the genus Megalobrama interferes with our understanding of the phylogenetic position of Faluo within the genus Megalobrama.
Recently, increasing evidence has shown that the whole mt genome could reveal more accurate phylogenetic relationships with higher resolution than single genes or segments11,15,16,17,18. Here, we obtained the first complete mitochondrial (mt) genome sequence of Faluo in the Heilong River (MTH) and confirmed that the sequence (GenBank: AB626850) of M. terminalis (in the Amur River of Russia, Fig. 1) submitted by Imoto et al.19 was from Parabramis pekinensis strenosoma which had similar morphological features with M. terminalis10. Furthermore, we also isolated the complete mt genomes of M. terminalis from the Qiantang River (MTQ) and Jinsha River Reservoir (MTJ), and of M. amblycephala from Liangzi Lake (MAL) and Yi River (MAY). The single nucleotide variations, specific markers and phylogeny between MTH and six other representative geographic samples within the genus Megalobrama were analyzed from a mt genome perspective.
Materials and Methods
Sample collection and DNA extraction
A total of 210 fish from 7 populations (30 per population) of 4 species in the genus Megalobrama were sampled. Sampling locations are shown in Fig. 2. Total DNA was extracted from fin tissues from all samples for each population using the phenol/chloroform method20. All animal experiments were approved and conducted in accordance with the guidelines of the Animal Research and Ethics Committees of Heilongjiang River Fisheries Research Institute.

Sampling locations of fish in the genus Megalobrama. HLR, Heilong River; QTR, Qiantang River; JSR, Jinsha River Reservoir; YR, Yi River; LZL, Liangzi Lake; LXR, Longxi River; XR, Xi River.
Primer design, PCR amplification and sequencing
The complete mt genomic sequences from two species in the genus Megalobrama, namely, M. pellegrini from the Longxi River (MPL) and M. hoffmanni from the Xi River (MHX), were determined in a previous study11. Thus, one genomic sample from each population (MTH, MTQ, MTJ, MAL and MAY) was used as the template for whole mt genome sequencing. A pair of primers (mt F1 and mt R1) described by Huang et al.21 was used to amplify the D-loop region. Twenty-one other pairs of primers (Table 1) with putative overlapping amplification regions were designed using Primer 5.0 (Premier Biosoft International, Palo Alto, CA, USA) according to the complete mt genome sequences of species in the genus Megalobrama available in GenBank. The PCR conditions were as follows: 94 °C for 5 min, followed by 30 cycles at 94 °C 30 s, 59 °C for 15 s, and 72 °C for 45 s, and an extension at 72 °C for 5 min. PCR components contained 13.7 µL H2O, 2 µL 10 × PCR buffer, 1.6 µL 1.5 mM MgCl2, 0.6 µL of each primer (10 µM), 0.4 µL 10 mM dNTPs, 0.1 µL Taq DNA polymerase (5 U/µL) and 1 µL DNA template (50 ng/µL) in a total volume of 20 µL. PCR products were separated on 1.0% of agarose gels with 0.5 µg/mL of ethidium bromide. The DNA fragments were purified using an E. Z. N. A. Gel Extraction Kit (Omega Bio-Tek, Norcross, GA, USA) and subcloned into pMD18-T vectors (TaKaRa, Shiga, Japan). Recombinant clones containing each target fragment were sequenced on an ABI 3700 sequencer (Applied Biosystems, PerkinElmer, Foster City, CA, USA).
Mitochondrial genome assembly and sequence comparison
Sequencing data and the boundaries from each segment with overlapping domains were checked to ensure authenticity. The assembled sequences were annotated by comparisons with known complete mitochondrial genomes of closely related species, including MPL and MHX. The secondary structures of tRNAs were identified using the web-based tRNA-scan SE 2.0 program (http://lowelab.ucsc.edu/tRNAscan-SE/)22. Complete mt sequences from MTH, MTQ, MTJ, MAL and MAY have been deposited in GenBank with the accession numbers listed in Table 2. Furthermore, complete mt genome sequences from M. terminalis in the Amur River in Russia (MTA)19 and Parabramis pekinensis strenosoma in the Heilong River (PPH)23 were used in the sequence correction (Table 2). The mt genome comparisons between MTH and MTQ, MTJ, MPL, MAL, MAY, MHX, MTA and PPH were performed by BLAST using the CGView Comparison Tool (CCT)24.
Specific SNP loci identification
Single nucleotide polymorphism (SNP) loci of the complete mt genomes between MTH and other species from the genus Megalobrama were identified based on comparisons performed with the Clustal W method using the MegAlign program in DNASTAR Lasergene 7.1 software (DNASTAR Inc., Madison, WI, USA). Moreover, a pair of primers (forward: 5′-GCATCTGGCTTCA-ATCTCA-3′; reverse: 5′-GCGTAGGGGATTTGCTGA-3′) was designed to amplify a putative fragment of 380 bp containing the specific locus SNP locus C549T in the D-loop region in MTH based on the 349–728 bp domain of the MTH mt genome in GenBank (Accession No: MH289765). The PCR components were the same as those mentioned above, and the conditions of the reaction were as follows: 94 °C for 5 min, followed by 30 cycles at 94 °C 30 sec, 56 °C for 30 sec, and 72 °C for 30 sec, and an extension at 72 °C for 5 min. After PCR, 2 µL of RE × 10 buffer, 0.2 µL acetylated BSA and 5 U TaqI (Promega Co., USA) was added to each sample (~1 µg), and the reaction was incubated at 65 °C for 2 h. Then, all digested samples of 7 populations (30 per population) were separated on 4% agarose gels with 0.5 µg/mL ethidium bromide.
Phylogenetic analysis
All 7 complete mt genomes from the ingroup (MTH, MTQ, MTJ, MPL, MAL, MAY and MHX) and the outgroup consisting of genus Culter (Culter dabryi25 and Culter mongolicus18) (Table 2) were aligned using the Clustal W algorithm26 implemented in the MEGA 7 program27. Substitution saturation was evaluated by DAMBE 6.4.128. Phylogenetic analyses were conducted based on complete nucleotide sequences using maximum likelihood (ML), maximum parsimony (MP) and Bayesian inference (BI) methods. ML and MP calculations were completed using MEGA 7 with 1000 bootstraps27. The best model (HKY + G) for the ML analysis was estimated based on the Bayesian information criterion (BIC) in this program. Bayesian phylogenetic analysis was performed using MrBayes 3.229. The best model (GTR + I) was selected based on Akaike information criterion (AIC) in the Modeltest 3.7 program30. Two million generations with four chains were run, sampling per 500 generations and discarding the initial 25% of samples as a burn-in.
Results
Characteristics of the MTH mt genome
We determined the complete mt genome of MTH. The assembled mt genome of MTH had a typical circular structure with a length of 16,621 bp, which is similar (16,621–16,623 bp) to that of its closely related species in the genus Megalobrama (Table 3). The whole genome contains 13 protein-coding genes, 22 tRNA genes, 2 rRNA (12SrRNA and 16SrRNA), and a putative control region (Table 3). The heavy (H) strand encodes 28 genes, while the light (L) strand encodes 9 genes (Fig. 3, Table 3). Based on prediction by tRNAscan-SE, 21 of 22 tRNAs fold into the typical cloverleaf secondary structure, while tRNASer(AGN) lacks the dihydrouridine (DHU) stem. ATG is the major start codon of the protein-coding genes, and only one start codon (GTG) is used in the COXI gene. In 13 protein-coding genes, seven (ND1, COX1, ATP8, ATP6, ND4L, ND5, and ND6) have a TAA stop codon, while the other six have incomplete stop codons of TA (COX3 and ND4) and T (ND2, COX2, ND3, and CYTB) (Table 3). The overall GC content of the MTH mt genome is 44.1%, while that of the D-loop region is only 35.9% (Fig. 3 and Table 4).

Circular map of the mitochondrial genome of M. terminalis in the Heilong River (MTH). Genes encoded by heavy and light strands with inverse arrow directions are shown outside and inside the circle, respectively. The black ring indicates the GC content (outward and inward peaks showing above or below average GC content, respectively). The purple-green ring indicates the GC skew [(G − C)/(G + C), purple (between 0 and 1), green (between −1 and 0)].
Comparison with closely related species of Megalobrama and Parabramis
We compared the percent identity and pairwise distances of mitogenomes among MTH, MTQ, MTJ, MAL, MAY, MTA and PPH. The number of the percent identity results were showed in Table 5, and the sequences identity comparison results were reflected in the CCT BLAST map (Fig. 4). High identity was found between MTH and MTQ, MTJ, and MPL (99.6–99.7%). The identity was low between MTH and MHX (96.1%), and the lowest identities were observed between MTH and MTA and between MTH and PPH (95.2% each). However, MTA had high identity with PPH (99.7%). Seventeen specific SNPs were detected between MTH and other fish species in the genus Megalobrama. The numbers of specific SNPs of MTQ and MTJ were 8 and 11, respectively (Table 6). Thirteen MTH-specific SNPs were found in coding regions (ND1, 2; ND2, 1; Cox1, 1; CoxII, 1; CoxIII, 1; ND3, 1; ND4, 2; ND5, 3; and ND6, 1). Only three nonsynonymous SNPs are found, and they were located in ND1 (nucleotide location 4,542 in the MTH assembly and with a codon change from GTCVal to GCCAla), COXIII (nucleotide location 10,180 in the MTH assembly and with a codon change from AAGLys to ACGThr) and ND4 (nucleotide location 11,400 in the MTH assembly and with a codon change from GCCAla to ACCThr) (Table 7). There were four MTH-specific SNPs in noncoding regions (D-loop, 1; 12SrRNA, 1; and 16SrRNA, 2) (Table 7).

Graphical map of the nucleotide percent identity of mitochondrial genomes determined by BLAST between Megalobrama terminalis in the Heilong River (MTH) and other fish of the genus Megalobrama and between MTH and the closely related Parabramis pekinensis strenosoma. The rings labeled 1 to 8 represent the BLAST results of sequences from MTH against those from M. terminalis in the Qiantang River (MTQ), M. terminalis in the Jinsha River Reservoir (MTJ), M. pellegrini in the Longxi River (MPL), M. amblycephala in the Yi River (MAY), M. amblycephala in the Liangzi Lake (MAL), M. hoffmanni in the Xi River (MHX), P. pekinensis strenosoma in the Heilong River (PPH) and M. terminalis in the Amur River (MTA).
Population-specific marker identification
The SNP C549T in the D-loop region formed one TaqI restriction site (T/CGA or TTGA) in the MTH population with one genotype (180/200 bp, T/CGA) (Fig. 5). In the six other populations, the base at the 549 site was T. However, only in the populations MTQ, MPL and MHX, there was one genotype (380 bp, TTGA) (Fig. 5). In the populations MTJ, MAL and MAY, there was one TaqI restriction site (T/CGA or TCAG) resulting from SNP G573A and SNP A574G, which is different from the position in MTH. In the MTJ population, there were three genotypes (157/223 bp; 157/223/380 bp; 380 bp). In populations MAL and MAY, there were two genotypes (157/223 bp or 380 bp) (Fig. 5).

Differentiation between Megalobrama terminalis in the Heilongjiang (MTH) and other populations of the genus Megalobrama determined using polymerase chain reaction-restriction fragment length polymorphism of a D-loop region fragment with TaqI digestion. M = DL 1,000 DNA ladder, 1–3 = MTH, 4–6 = M. pellegrini in the Longxi River (MPL), 7–9 = M. amblycephala in the Liangzi Lake (MAL), 10–12 = M. amblycephala in the Yi River (MAY), 13–15 = M. terminalis in the Qiantang River (MTQ), 16–18 = M. terminalis in the Jinsha River Reservoir (MTJ), 19–21 = M. hoffmanni in the Xi River (MHX), 22 = Negative control.
Phylogenetic analysis
The substitution saturation of all nucleotide sequences was not tested. The maximum parsimony, maximum likelihood, and Bayesian methods produced identical tree topologies with strong bootstrap and posterior probability values, and the maximum likelihood tree is shown here (Fig. 6). M. terminalis (MTH, MTQ and MTJ), M. pellegrini (MPL), M. amblycephala (MAL and MAY) and M. hoffmanni (MHX) composed a monophyletic clade. MHX was located at the basal branch of this clade. MTH, MTQ, MTJ and MPL formed one subclade, and MAL and MAY formed the other subclade. MTA, MTQ and MPL constituted a monophyletic clade. MTH has a sister relationship with a clade comprising MTQ, MTJ, and MPL.

Phylogenetic tree of the genus Megalobrama (ingroup) inferred by using the maximum likelihood (ML) method based on the complete mitochondrial genome data. Values shown at each node of the tree correspond to the ML bootstrap values, maximum parsimony bootstrap values and Bayesian posterior probabilities given in percentages. Culter dabryi and Culter mongolicus were used as the outgroup. GenBank accession numbers: MTH, Megalobrama terminalis in the Heilong River (MH289765); MTQ, M. terminalis in the Qiantang Rive (MH289767); MTJ, M. terminalis in the Jinsha River Reservoir (MH289766); MPL, M. pellegrini in the Longxi River (JX242529); MAL, M. amblycephala in the Liangzi Lake (MH289764); MAY, M. amblycephala in the Yi River (MH289763); MHX, M. hoffmanni in the Xi River (JX242530); C. dabryi (NC_021418); C. mongolicus (AP009060).
Discussion
Imota et al. submitted the full mtDNA sequence of M. terminalis in the Amur River (MTA) in Russia19. Based on a comparative analysis, however, we found that the lengths of the complete mt genomes of MTA, MHX, and PPH were identical (16,623 bp), and this sequence of MTA showed higher identity with that of PPH (99.7%) than with that of MTH or other M. terminalis (95.2%). In contrast, the length of the mt genome sequence (MTH) obtained in the present study was identical to those of MTQ, MTJ and MPL (16,621 bp), and showed a high identity with them (99.6–99.8%). These results suggest that the M. terminalis sequence provided by Imoto et al. was from PPH. M. terminalis and PPH have the same distribution in the Heilongjiang-Amur River basin and share some morphological and biological characteristics31,32. Thus, the confusion between the two is very easy to happen.
Furthermore, to understand the characteristics of the mt genome of MTH, we detected 17 SNP sites of specific to MTH, which was more than the number obtained for conspecific MTQ (8) and MTJ (11). In China, all fish species in the genus Megalobrama inhabit the southern Yellow River except Faluo, which lives in the Heilongjiang basin1,32, where the effective growing season of fish is only ~4 months, and the overwintering period beneath ice reaches ~5 months33. Therefore, unique habitats and long-term geographical isolation may have led to MTH having more polymorphic sites in its mt DNA than intraspecific MTQ and MTJ. Three nonsynonymous SNPs in ND1, COX3 and ND4 discovered between MTH and other fish species in the genus Megalobrama remain to be elucidated. Based on the comparative data of specific SNPs, we identified one population-specific marker in the D-loop region (C 549 T), which effectively separated MTH from the six other fish species in the genus Megalobrama in the absence of hybridization. All individuals of MTH used in the present study were homozygous at the specific marker, suggesting that MTH still has a relatively pure gene pool. The genus Megalobrama emerged relatively recently in Cyprinidae fish species8,34. M. terminalis, M. amblycephala and M. pellegrini are very similar in morphology before sexual maturity, and can hybridize with each other by artificial fertilization35,36,37. Recently, a culture variety of M. amblycephala bred by Li et al. began to appear in the aquaculture waters of northeast China38, and posed a potential threat to precious M. terminalis resources. Thus, further work is required to develop population-specific markers at the nuclear gene level for germplasm purity identification of MTH during sample collection and artificial breeding.
The species taxonomy of Faluo has yet to be fully accepted7. One of the major reasons is that Faluo shows some distinct morphological characters (number of vertebrae, length of the intestine/body length, body length/eye diameter, and hypopharyngeal teeth type) from southern M. terminalis7. Additionally, Faluo was not used as a representative specimen of M. terminalis in the early classification of the genus Megalobrama1. In our present phylogenetic analysis, however, MTH, MTJ and MTQ were in a well-supported monophyletic group. Considering that MTH showed the highest percent identity with MTJ and MTQ, we speculate that the present species classification of Faluo is undisputable.
To the best of our knowledge, this is the first phylogenetic analysis including samples from three different geographical populations of M. terminalis with clear origins. MTH in particular represents a population distribution in northernmost China. In previous studies, however, samples were only taken from one population of M. terminalis other than MTH8,9,10,11,12,13,14. Notably, MTQ, MTJ and MPL constituted a monophyletic clade, shared a common ancestor with MTH, and further clustered with MAL and MAY in the present study. This result clearly indicates that MPL rather than MTH has a close phylogenetic relationship with MTJ and MTQ. The present results give strong evidence by utilizing complete mt genome data, supporting M. pellegrini as one of the populations of M. terminalis.
The sequence of a complete mt genome from MTH was obtained for the first time. We expect the analysis of specific markers of MTH and identification at the population level will contribute to germplasm purity detection in future sampling and breeding practices. Furthermore, we confirmed that MTH belongs to the species M. terminalis by sequence comparisons and phylogenetic tree reconstruction. The introduction of MTH provided clearer phylogenetic relationships among species in the genus Megalobrama at the mitochondrial genome level, which will also be helpful in the protection and development of this endangered species in the future.
Data Availability
The mitochondrial genome data of Megalobrama terminalis in the Heilong River (MH289765), Qiantang River (MH289767) and Jinsha River Reservoir (MH289766), and Megalobrama amblycephala in the Liangzi Lake (MH289764) and Yi River (MH289763) have been submitted to GenBank. The authors confirm that the data supporting the findings of this study are available within the article. However, any datasets generated and/or analysed during the current study are available from the corresponding author on reasonable requests.
References
Chen, Y. Y. Fauna Sinica: Osteichthyes Cypriniformes II. (Science Press, Beijing, 1998).
Luo, Y. L. A revision of fishes of the cyprinid genus Megalobrama. Acta Hydrobiol Sin 14, 160–165 (1990).
Ren, M. L. Ichthyofauna of the Heilongjiang River. Chin J Fish 7, 1–14 (1994).
Zhang, J. M. Fishes of the Heilongjiang Province (Heilongjiang Science and Technology Press, Harbin, 1995).
Liang, K. X. & Li, M. B. Developing now breeds and large-scale culturing Megalobrama terminalis. J Hydroecol 6(37–38), 41 (1994).
Novomodny, G., Sharov, P. & Zolotukhin, S. Amur Fish: Wealth and Crisis (WWF RFE, Russia, 2004).
Wang, Z. Y. et al. The preliminary study on Megalobrama skolkovii Dybowsky from Heilong River. Fish Heilongjiang. Suppl, 10–13 (1993).
He, S. P. et al. Molecular phylogenetic relationships of Eastern Asian Cyprinidae (Pisces: Cypriniformes) inferred from cytochrome b sequences. Sci China Ser C 47, 130–138 (2004).
Xie, N., Liu, X. Y., Feng, X. Y. & Guo, S. R. Sequences analysis on mitochondrial Cytochrome b gene fragment of Megalobram spp. Mod Agric Sci Technol 1, 290–292 (2012).
Bai, X. H. et al. Species identification and evolutionary inference of the genera Megalobrama and Parabramis (Cyprinidae: Cultrinae) in China. Mitochondrial DNA 26, 357–366 (2015).
Lai, R. F. et al. Comparison of mitochondrial genomes of the genus Megalobrama and their phylogenetic analysis. J Fish China 38, 1–14 (2014).
Li, S. F., Zhu, Z. W., Zou, S. M., Zhao, J. L. & Cai, W. Q. Interspecific phylogenesis and intraspecific genetic differences of genus Megalobrama: Bluntnose black bream (M. Amblycephala), Guangdong black bream (M. Hoffmanni) and black bream (M. Terminalis). Acta Zoologica Sinica 48, 339–345 (2002).
Song, W., Wang, Y. Z., Zhu, D. H., Ren, L. & Wang, W. M. Morphological variations among the genus Megalobrama. Freshwater Fish 43, 21–27 (2013).
Chen, J., Li, F. G., Huang, C. X., Jiang, X. Y. & Zou, S. M. Morphological variaitons of genera Parabramis and Megalobrama teleost populations. J Shanghai Ocean Uni 23, 388–394 (2014).
Cao, Y., Adachi, J., Janke, A., Paabo, S. & Hasegawa, M. Phylogenetic relationships among eutherian orders estimated from inferred sequences of mitochondrial proteins: instability of a tree based on a single gene. J Mol Evol 39, 519–527 (1994).
Miya, M., Kawaguchi, A. & Nishida, M. Mitogenomic exploration of higher teleostean phylogenies: a case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol Biol Evol 18, 1993–2009 (2001).
Yu, L., Li, Y. W., Ryder, O. A. & Zhang, Y. P. Analysis of complete mitochondrial genome sequences increases phylogenetic resolution of bears (Ursidae), a mammalian family that experienced rapid speciation. BMC Evol Biol 7, 198 (2007).
Saitoh, K. et al. Mitogenomic evolution and interrelationships of the Cypriniformes (Actinopterygii: Ostariophysi): The first evidence toward resolution of high-level relationships of the world’s largest freshwater fish clade based on 59 whold mitogenome sequences. J Mol Evol 63, 826–841 (2006).
Imoto, J. M. et al. Phylogeny and biogeography of highly diverged freshwater fish species (Leuciscinae, Cyprinidae, Teleostei) inferred from mitochondrial genome analysis. Gene 514, 112–124 (2013).
Butler, J. M. Advanced Topics in Forensic DNA Typing: Methodology. (Academic Press, San Diego, 2012).
Huang, Z. J. et al. Application and primer design of mitochondrial DNA D-loop of freshwater fishes. Acta Sci Natur Univ Sunyatseni 48, 84–88 (2009).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: Search and contextual analysis of transfer RNA genes. Nul Acids Res 44, W54–57 (2016).
Duan, X. K. et al. The complete mitochondrial genome sequence of Parabramis pekinensis streosoma (Cypriniformes: Cyprinidae). Mitochondrial DNA Part A 27, 86–87 (2016).
Grant, J. R., Arantes, A. S. & Stothard, P. Comparing thousands of circular genomes using the CGView comparison tool. BMC Genomics 13, 202 (2012).
Zhang, X. J., Ma, Z. H., Chen, D. X., Xu, W. J. & Yang, R. B. The complete mitochondrial genome of Culter dabryi (Cyprinidae: Cultrinae). Miochondral DNA Part A 25, 98–99 (2014).
Thompson, J. D., Higgins, D. J. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680 (1994).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33, 1870–1874 (2016).
Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics and molecular evolution. J Hered 108, 431–437 (2017).
Ronquist, F. et al. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Systematic Biol 61, 539–542 (2012).
Posada, D. & Crandall, K. A. MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Cai, M. J., Zhang, M. Y., Zeng, Q. L. & Liu, H. Z. A study on morphometrics of the genus Megalobrama. Acta Hydrobiol Sin 25, 631–635 (2001).
Wu, X. W. The cyprinid fishes of China (Vol. 1). (Shanghai Scientific and Technical Publishers, Shanghai, 1982).
Hu, X. S. et al. Inheritance of growth traits in Songpu mirror carp (Cyprinus carpio L.) cultured in Northeast China. Aquaculture 477, 1–5 (2017).
Wang, X. Z., Gan, X. N., Li, J. B., Mayden, R. L. & He, S. P. Cyprinid phylogeny based on Bayesian and maximum likelihood analysis of partitioned data: implications for Cyprinidae systematics. Sci China Life Sci 55, 761–773 (2012).
Zhang, D. L. et al. The interspecific hybridization in four freshwater bream Megalobrama Sp. J Dalian Fish Univ 29, 121–125 (2014).
Yang, H. Y., Li, S. F. & Zou, S. M. A primary study on inheritance of morphological traits from Megalobrama amblycephala, Megalobrama terminalis to their reciprocal hybrids (F1). J Shanghai Ocean Uni 11, 305–309 (2002).
Guan, W. Z. et al. Comparative analysis of growth and morphological variations among Megalobrama amblycephala, M. terminalis, Parabramis pekinensis and their hybrids. J Fish Sci China 24, 31–39 (2017).
Li, S. F. New breed of Megalobrama amblycephala: Pujiang No.1. Nongcun Baishi Tong 10, 34 (2016).
Acknowledgements
We thank Prof. Xiaoyu Feng, Prof. Weimin Wang, Prof. Zuogang Peng, Prof. Zhixun Li and Mr. Changyi Qu for their help during the sample collection. The research was funded by the Central Public-interest Scientific Institution Basal Research Fund, CAFS (Grant No. 2018HY-XKQ02-04), Central Public-interest Scientific Institution Basal Research Fund, HRFRI (Grant No. HSY201807M), and Key Laboratory of Freshwater Aquatic Biotechnology and Breeding, Ministry of Agriculture and Rural Affairs (Grant No. FBB2017-01).
Author information
Authors and Affiliations
Contributions
X.H., Z.M., J.T. and L.S. conceived and designed the experiments. X.H., C.C., C.L., Z.J., M.S. and S.W. performed sample collection and experiments. X.H., P.L. and Y.G. analyzed the data. X.H. wrote the manuscript. All authors have reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, X., Luan, P., Cao, C. et al. Characterization of the mitochondrial genome of Megalobrama terminalis in the Heilong River and a clearer phylogeny of the genus Megalobrama. Sci Rep 9, 8509 (2019). https://doi.org/10.1038/s41598-019-44721-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44721-2
This article is cited by
-
Exploring cross-species genetic diversity: unveiling new insights in Megalobrama through whole genome-wide simple sequence repeats
Conservation Genetics (2023)
-
Extensive mitogenomic heteroplasmy and its implications in the phylogeny of the fish genus Megalobrama
3 Biotech (2023)
-
Next-generation sequencing reveals the mitogenomic heteroplasmy in the topmouth culter (Culter alburnus Basilewsky, 1855)
Molecular Biology Reports (2022)
-
Genetic diversity and genetic differentiation of Megalobrama populations inferred by mitochondrial markers
Genes & Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
