
Abstract
Phaeodactylum tricornutum is a well-developed model diatom for both marine ecology and microalgal biotechnology, which has been enabled by the sequenced genome and the availability of gene delivery tools, such as biolistic transformation and E. coli-mediated conjugation. Till now, these tools have mainly relied on two selectable markers of bacterial origin which confer resistance to antibiotics Zeocin and nourseothricin. An alternative cost-effective and preferably endogenous selectable marker would facilitate gene stacking efforts through successive transformation or conjugation. We performed UV-mutagenesis of P. tricornutum to obtain mutations in the phytoene desaturase (PDS) gene, conferring resistance to the bleaching herbicide norflurazon. Two mutants displaying high tolerance to norflurazon and carrying unique mutations in PtPDS1 (PHATRDRAFT_45735) were selected. These mutants revealed novel point mutations at a conserved residue Gly290 to Ser/Arg. Homology-based structural modeling of mutated PDS1, over a resolved crystallographic model of rice PDS1 complexed with norflurazon, suggests steric hindrance by bulkier residue substitution may confer herbicide resistance. We report the characterization of PtPDS1 mutants and the development of the first endogenous selectable marker in diatoms suitable for industrial strain development, with the added benefit of biocontainment. The plasmid carrying the mutated PDS1 as a selection marker and eGFP as a reporter was created. An optimized biolistic transformation system is reported which allowed the isolation of positive transgenic events at the rate of 96.7%. Additionally, the ease of in vivo UV-mutagenesis may be employed as a strategy to create PDS-norflurazon-based selectable markers for other diatoms.
Similar content being viewed by others

Introduction
Diatoms are an extremely morphologically diverse group of photosynthetic microalgae that inhabit nearly all aquatic environments and contribute approximately 20 to 40% of atmospheric O21,2. In marine environments, photosynthetic diatoms display rapid growth by assimilating available nutrients, resulting in blooms. The capability of diatoms to use photosynthetic energy for rapid growth makes them excellent target organisms for microalgal biotechnology and genetic engineering3,4,5. As many diatoms are oleaginous species that are able to accumulate high amounts of storage lipids, they have attracted significant attention as potent biofuel organisms.
The pennate oleaginous diatom Phaeodactylum tricornutum has been engineered for enhanced fatty acid biosynthesis6,7 and storage lipid accumulation8,9,10 and improved LC-PUFA production11,12,13. This diatom is an emerging synthetic biology chassis, enabling synthesis of a variety of compounds, and a proven host for recombinant protein production. The production of triterpenoid compounds14, reconstruction of vanillin biosynthesis15, therapeutic peptide production16,17,18, and trophic conversion19,20 have been achieved in this industrially relevant diatom.
Numerous genetic engineering tools, such as gene delivery21,22,23,24, genome editing15,25,26,27,28,29 and gene silencing30, have been established to explore the genome information and empower the biotechnological exploitation of P. tricornutum. These tools depend on an efficient selectable marker for the selection of transgenic events. Limited availability of efficient and cost-effective selection schemes for nuclear transformation in P. tricornutum hinders the prospect of simultaneous or tandem delivery of multiple traits for gene stacking or engineering complex biosynthetic pathways. Furthermore, P. tricornutum was shown to be insensitive to a wide range of commonly used antibiotics and herbicides31. Currently, only two selectable markers sh-ble and nat are predominantly used to select transgenic events in P. tricornutum. Recently another selectable marker, blasticidin-S deaminase32 has been reported for P. tricornutum. However, these selectable markers are of bacterial origin and not suitable for generation of transgenic events for commercial use and are likely to face regulatory hurdles. Disruption of native genes leading to loss of function, such as PtUMPS and PtAPT, have been reported as counter selectable markers in P. tricornutum for genome editing applications29. However, their utility in generating transgenic events is limited due to the availability of a functional copy in the recipient cells which metabolizes the synthetic metabolite analog into a suicidal product leading to mortality of all cells. On the other hand, gain of function mutations in native genes may be used as endogenous selectable markers. Such endogenous selectable markers based on herbicide resistance offer an efficient and cost effective alternative to selectable markers of bacterial origin.
Herbicides, coupled with an endogenous or transgenic source of genetic resistance, have been routinely deployed as selectable markers for nuclear or plastid transformation in higher plants. The bleaching herbicide norflurazon competitively binds phytoene desaturase (PDS), the first enzyme in the carotenoid biosynthesis pathway, which catalyzes the introduction of two double bonds into phytoene, yielding ζ-carotene via the intermediate carotenoid phytofluene33. Several mutations in the PDS coding sequence conferring resistance to norflurazon have been reported34,35 and developed as selectable markers in higher plants and green microalgae, but none in microalgae with high biotechnological potential, which belong to Stramenopiles, such as diatoms and eustigmatophytes (e.g., Nannochloropsis spp.).
Here, we report the in vivo UV-mutagenesis of the native phytoene desaturase-1 (PHATRDRAFT_45735; P. tricornutum JGI database v2.0) coding sequence of the P. tricornutum strain UTEX 646 and the isolation of novel mutants conferring resistance to norflurazon. The PtPDS1 sequence from the most tolerant mutant was used to create the first endogenous selectable marker in P. tricornutum that is suitable for efficient selection of transgenic events and use in the production of industrial strains ensuring biocontainment.
Results
Determination of an optimal norflurazon selection concentration
For the development of an endogenous selection marker, suitable for genetic engineering in P. tricornutum, we focused on the resistance to the herbicide norflurazon. The sensitivity of WT P. tricornutum to increasing concentrations of norflurazon (0.25–5 μM) was tested by plating cells on RSSE-agar and incubation for 12 days followed by microscopy of Aniline Blue stained cells to assess cell damage as indicated by the cellular permeabilization of the stain (Fig. 1a,b). Uniform Aniline Blue permeabilization and staining of treated WT P. tricornutum cells was observed at 1.5 µM and higher norflurazon concentrations, which was associated with a breakdown of cellular membranes, a desintegration of the chloroplast and a loss of cell viability (Supplementary Fig. S1). Norflurazon-treated cells that were re-plated for recovery on the RSSE-agar medium free of norflurazon displayed 100% mortality at 2.5 µM and higher herbicide concentrations after 15 days of incubation (Fig. 1c). Based on these observations, selection was tested at 3 and 5 µM norflurazon concentrations in biolistic transformation experiments.

Determination of sensitivity of WT P. tricornutum UTEX 646 to norflurazon. (a) Schematic representation of experimental procedure to determine norflurazon sensitivity in WT P. tricornutum cells. 3 × 107 WT cells were plated on RSE-agar supplemented with norflurazon concentrations ranging from 0.25–5 µM. Cells were washed off plates after 12 days of incubation and stained with Aniline Blue for microscopic evaluation. (b) Percent Aniline Blue stained cells averaged over several non-overlapping fields. Error bars represent standard error. (c) Non-selective recovery of bleached WT cells to determine an optimal norflurazon selection concentration. Washed off RSE-agar + norflurazon cells were re-plated onto RSE-agar and grown under illumination of 60 μmol m−2 s−1 for 15 days to allow the recovery of any viable cells post-herbicide treatment. Numerical figures indicate treatment concentration of norflurazon before re-plating.
Isolation of P. tricornutum norflurazon-resistant PtPDS1 mutants and their characterization
Based on the results above and a preliminary assay of WT P. tricornutum sensitivity to norflurazon in liquid media (not shown), we chose 15 µM norflurazon concentration to enable the selection of highly resistant mutants and to limit escapes. Accordingly, UV-mutagenized colonies, obtained after exposure to different durations of UVB (see materials and methods), were selected on solid media containing 15 µM norflurazon. After 15 days, the two fastest growing norflurazon-resistant mutants (M1 and M2) were recovered from a 2-min UVB exposure.
A preliminary characterization of the M1 and M2 mutants was done in a 12-well plate, where mutants were exposed to a range of norflurazon concentrations between 10 and 40 µM. At the highest concentration of 40 µM norflurazon, M1 displayed chlorosis, whereas M2 was completely bleached at the end of day 20 of incubation (Fig. 2a). Both mutants displayed tolerance to norflurazon concentrations lower than 30 μM. Next, we compared the growth of mutants and WT in a column photobioreactor, with and without 5 and 10 µM norflurazon. Mid-log phase cultures were diluted to chlorophyll 2 mg L−1 at the start of experiment; chlorophyll and cell density were estimated by recording A680 and A720, respectively. By the end of experimental period, norflurazon-treated WT cells displayed greater Aniline Blue staining than their treated M1 and M2 counterparts (Fig. 2b), revealing greater WT sensitivity to herbicide and tolerance in the mutant lines. In the presence of 10 µM norflurazon, M1 and M2 mutants displayed a specific growth rate of 0.211 and 0.141 Abs-680 day−1, respectively, whereas chlorophyll production in WT cultures was completely arrested at 48 h, in the presence of 5 μM norflurazon (Fig. 2c). Untreated WT displayed slightly better chlorophyll accumulation than the M1 or M2 cultures in each experimental repetition. A similar trend was observed for cell density (A720) in norflurazon-treated and untreated cultures (Fig. 2d).

Tolerance of P. tricornutum mutants M1 and M2 to norflurazon. (a) Mutants M1 and M2 cultures in mid-log phase were diluted to 5 × 105 cells per mL and treated with norflurazon (10–40 µM) and cultured under illumination of 75 μmol m−2 s−1 for 20 days. Mutant M1 displayed greater tolerance to norflurazon. (b) WT and mutants M1 and M2 were growth in liquid cultures in a multi-cultivator for 93 h and examined for cell viability with Aniline Blue staining at the end of the experiment. (c,d) Growth characterization of mutants M1 and M2 in comparison with WT in the presence or absence of norflurazon (NRF).
Phylogenetic and sequence analysis of PtPDS1 mutants
The plastid-targeted P. tricornutum PDS1 displayed the highest homology to P of other diatoms (Fig. S2). Within the phylogenetic tree for chloroplastic phytoene desaturases, P. tricornutum PDS1 clustered more closely to early divergent green microalgae (Prasynophytes), which form a clade separate from higher green algae (Chlorophytes) and land plants. Brown algal PDS peptide sequences were the most divergent and were used to root the phylogenetic tree.
Sanger sequencing of the cloned PtPDS1 CDS from mutant strains and the alignment of its translated peptide sequence to WT PtPDS1 showed substitutions at position Met28Ile (M1), Gly290Ala/Ser (M1/M2) (Supplementary Fig. S3a) and Arg494Cys (WT) of the amino acid sequence. Of these three substitutions, Gly290 was the only conserved residue (Supplementary Fig. S3b). Moreover, the Met28Ile substitution occurred in the chloroplast transit peptide (N1-MMFHYKTGSSWFLLLSASITTTLTTTTMTTTHAFAP-C36), which is predicted to be cleaved within the ASAFAP motif (Phe34)36,37,38. Mapping of known conserved residue substitutions conferring norflurazon resistance to the PtPDS1 peptide sequence confirms that Gly290Ala/Ser is a novel mutation (Fig. S3b).
Alignment of the PtPDS1 and OsPDS1 (GenBank accession number AF049356) peptide sequences showed a 59.51% sequence identity and a sequence coverage of 76%. Alignment between secondary structures of PtPDS1 and OsPDS1 showed highly similar topologies (Fig. 3a). The homology-based modeling of PtPDS1 over the OsPDS1 crystallographic structure (PDB: 5MOG.A, Fig. 3b) had a QMEAN value of −1.98 indicating high structural similarity and high accuracy of the model. Phe145 and Arg283 (Fig. 3c) residues are known to interact with norflurazon and allow its stable binding and inhibition of the phytoene desaturase enzyme. The PtPDS1 modeled structure overlaid on norflurazon-inhibited OsPDS1 indicated a clash between Gly290Ala (M1) and the backbone carbonyls of Arg283 and Glu287, which may interfere with residues stabilizing norflurazon in WT phytoene desaturase-1 enzyme (Fig. 3c), which may explain the basis of norflurazon resistance in mutants.

In-silico analysis of PtPDS1-M1 and M2 mutant sequences deduces a basis for norflurazon tolerance. (a) Representation of secondary structure alignments between PtPDS1 (target; SWISS-MODEL) and OsPDS1 (template; PDB: 5mogA) shows 59.51% sequence identity and sequence similarity score of 0.48. Sites of previously reported norflurazon conferring substitutions (green triangles) and PtPDS1-M1/M2 (red triangle) are indicated. (b) Representation of OsPDS1 (PDB: 5mogA) resolved structure complexed with Norflurazon and FAD. (c) Enlarged inset (shown in d) overlaid with modeled WT PtPDS1 displays structural homology at norflurazon biding site. (d) R283-Norflurazon (NRF) hydrogen bond (black dotted line) may be disrupted by Gly290Ala (M1) residue change due to side group clashes (brown dotted lines) leading to reduced affinity of PtPDS-M1 for norflurazon observed as norflurazon tolerance in M1.
Optimization of P. tricornutum transformation with the pPtPDS1-M1 construct
Next, we used the PDS1 sequence from mutant M1 to construct a pPtPDS1-M1vector (Fig. S4), harboring eGFP as a reporter (for details, please refer to Materials and Methods). An efficient norflurazon concentration, suitable for transformant selection, would maximize the selection of transgenic events and yet minimize the occurence of false positives. Transformation efficiency and the rate of false positive events were compared at 3 and 5 µM norflurazon selection post-biolistic delivery of pPtPDS1-M1 expression vector (Fig. 4).

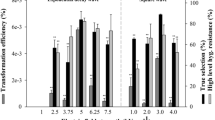
Optimization of selection efficiency with 3 or 5 µM norflurazon by biolistic transformation of pPtPDS1-M1 plasmid. Five microcarrier-DNA coating preparations were shot for each selection concentration of norflurazon. All putative transformants that appeared by day 12 of selection were screened by PCR for amplification of eGFP or PtPDS1-M1. (a) Percentage distribution out of total number colonies screened: 160 (3 µM norflurazon) and 181 (5 µM norflurazon). (b) Average number of colonies per microprojectile-DNA coating preparation of colonies screened by PCR. Bars marked with same alphabets indicate non-significant difference at α = 0.05. Error bars depict standard error values. FP = False positive.
A total of 160 (3 µM norflurazon) and 181 (5 µM norflurazon) putative transgenic events were PCR screened for the presence of eGFP or PtPDS1-M1 (Fig. 4a,b). The mean number of colonies per microcarrier coating reaction in each selection regime were about 32–36 and statistically non-significant (p = 0.54) (Fig. 4b). However, selection at 5 µM norflurazon produced significantly more Pt-PDS1 positive lines (p = 0.003) than 3 µM norflurazon: 96.1% vs. 45.6%, respectively (Fig. 4a). A similar trend was seen for eGFP positive lines: 96.7% vs. 36.9% (p = 0.002). Selection at 5 µM norflurazon effectively reduced non-transgenic escapes to ~4%, compared to 50% of the total colonies screened at 3 µM (p = 0.014) (Fig. 4a,b).
Validation of transgene integration and expression
Putative transgenic events screened by PCR, for the integration of PtPDS1-M1 and eGFP (Supplementary Fig. S5) in the nuclear genome of the transformants T1, T2, T3, and T4, displayed positive amplification. Furthermore, Southern blotting of KpnI-digested genomic DNA of transgenic events and WT with eGFP and PtPDS1 probes, indicated in Fig. 5a, validated transgene integration (Fig. 5b). Cytoplasmic eGFP protein corresponding to 37.2 kDa was detected in a western blot of transgenic events (Fig. 5c). In vivo expression of cytoplasmic eGFP was observed by fluorescent microscopy in transgenic lines (Fig. 5e). Spotted 104–102 cells of transgenic lines on RSE-agar + 5 µM norflurazon plates displayed no growth inhibition, confirming PtPDS1-M1 expression (Fig. 5d).

Characterization of transgenic lines. (a) Schematic representation of pPtPDS1-M1 vector depicting PCR primer binding sites and restriction sites used for validation of transgene insertion in to P. tricornutum nuclear genome. (b) Southern blot of genomic DNA digested with KpnI restriction enzyme was hybridized with a PDS1 probe (581 bp), stripped and re-hybridzed with eGFP fragment (606 bp) to confirm transgene integration in nuclear genome of P. tricornutum. (c) Western blot of crude protein extracts from transgenic events was probed with anti-GFP antibody to confirm eGFP expression. (d) Norflurazon tolerance assay of transgenic events on RSE-agar + 5 µM Norflurazon to confirm expression of PtPDS-M1 from integrated cisgene. (e) Cytoplasmic eGFP expression was confirmed in transgenic lines through fluorescent microscopy (see Materials and Methods). DIC - differential interference contrast; eGFP -enhanced green fluorescent protein; PAF - plastidial auto-fluorescence.
Evaluation of long-term expression stability of transgenic lines
Transgenic lines (T1, T2 and T3), maintained without selection for about 48 months, representing a range of transgene copy number integration were characterized for norflurazon sensitivity and eGFP expression. Transgenic lines cultured in column photobioreactor, with or without 5 µM norflurazon displayed robust growth over three repetitions of the experiment (Fig. 6a,b). In the presence of 5 µM norflurazon, transgenic lines displayed minimal inhibition of growth when compared to untreated controls, including wild-type. Furthermore, transgenic lines displayed tolerance over a range of norflurazon concentrations (10–120 µM) and remained uninhibited in a plate-based assay of norflurazon sensitivity (Fig. 6c). Integration of eGFP and PtPDS1-M1 was validated by PCR (Supplementary Fig. S5a). Stable eGFP expression in all three transgenic lines was validated by fluorescence microscopy (Supplementary Fig. S6b).

Evaluation of long-term expression stability of transgenic lines. Transgenic lines T1, T2 and T3 (from Fig. 5b), displaying different transgene copy number and maintained in nonselective medium for nearly 48 months were evaluated for stable expression of selectable marker. (a,b) Growth characterization of transgenic lines T1, T2 and T3 compared with WT in the presence or absence of 5 µM norflurazon (NRF). (c) Transgenic lines T1, T2 and T3 grown to mid-log phase were diluted to 5 × 105 cells per mL and treated with norflurazon (10–120 µM) and cultured under illumination of 75 μmol m−2 s−1 for 20 days.
Discussion
The diatom P. tricornutum has developed into the primary model microalga for biotechnological exploitation, specifically for lipids, LC-PUFA and recombinant protein production, and a wide range of molecular tools used in developing this diatom into a valuable synthetic biology chassis have been reported15,39. UV-mutagenesis is a simple method for the introduction of dominant genic mutations in a wide range of microorganisms. In P. tricornutum, UV-mutagenesis has been successfully employed to enhance EPA content up to 44%40. Here, we report a novel dominant endogenous selection marker in diatoms for the transformation of P. tricornutum, based on a novel amino acid substitution in PtPDS1 (PHATRDRAFT_45735; P. tricornutum JGI database v2.0) conferring resistance to the bleaching herbicide norflurazon.
Through random or targeted mutagenesis of the PDS enzyme, norflurazon resistance has been successfully developed as an endogenous selectable marker in several algae41,42,43,44,45,46,47 and plant species48,49. With respect to the PtPDS1 sequence, five amino acid substitutions at highly conserved residues in the PDS enzyme that are known to confer resistance to norflurazon have been reported so far: Phe145Val43, Arg283Pro/Ser/Thr/Cys49,50,51,52, Leu403Pro34, Val505Gly34,53,54, and Leu550Arg/Phe41,42,44,45,46,47. These residue substitutions were all shown to occur in the vicinity of the reaction center comprising the FAD-binding site and the plastoquinone/norflurazon binding pocket in the Rice PDS (OsPDS1) crystal structure33,55. In vivo UV mutagenesis and stringent selection allowed isolation of norflurazon-resistant mutants of P. tricurnutum with novel substitutions at a highly conserved residue, Gly290. Homology modeling of the PtPDS1 peptide over the OsPDS1-elucidated structure, suggests that Gly290Ala/Ser substitutions introduce bulky residue that clash with the backbone carbonyls of Arg283 and Glu287, which may destabilize norflurazon interaction with phytoene desaturase enzyme and likely result in norflurazon resistance.
Nuclear transformation in P. tricornutum is routinely achieved by two selectable markers of bacterial origin, Sh-ble- Zeocin™ and nat- nourseothricin. Apart from Sh-ble and nat, nptII-geneticin and cat-chloramphenicol are the only other selectable markers that have been used with less success in transformation of P. tricornutum and therefore not used routinely. Use of alternate selectable markers such as nptII and nat are fraught with difficulties, such as being effective only at higher concentrations and at reduced salt strength in the selection medium. Moreover, an array of commonly used antibiotic selection agents, such as kanamycin, streptomycin, spectinomycin, and hygromycin are ineffective as selection agents in P. tricornutum22,56. Commercial herbicides and their corresponding resistance genes provide a cost-effective and efficient selection scheme when genetically encoded antibiotic resistance markers are unavailable. However, widely used herbicides, such as glyphosate, bialaphos, phosphinothricin, sulfometuron-methyl, chlorosulfuron, and imazapyr, have proven to be ineffective in killing P. tricornutum22. Due to this dearth of efficient selectable markers, PDS-norflurazon resistance based selection system was explored in P. tricornutum. In contrast to selectable markers of bacterial origin, endogenous PDS1 gene, resistant to norflurazon provides additional benefits of efficient selection, biocontainment and cost-effectiveness. When compared on a cost per liter basis for producing one liter of selective medium, norflurazon, Zeocin™ and nourseothricin, respectively, are ca. USD 2 (Merck, USA), 20 (InVivoGen, San Diego, USA) and 3000 (Merck, USA).
In order to develop PtPDS1-M1 into an efficient selectable marker, optimization of the selection pressure on biolistic-transformed cells was carried out at 3 and 5 µM norflurazon concentrations. Selection at 5 µM norflurazon allowed significant improvement in selection of positive transgenic lines at the rate of 96.7% of total colonies obtained. Similarly, non-transgenic escapes were limited to 3.8% of the total colonies screened. With 5 µM norflurazon selection, on an average each microprojectile coating preparation produced 35 transformants as compared to ~100 with sh ble-Zeocin™; this may be attributed to the PtPDS1-M1 CDS length of 1875bp, which is five times that of sh-ble CDS.
The Phaeodactylum tricornutum genome contains two homologs of the phytoene desaturase enzyme viz. PtPDS1 (PHATRDRAFT_45735) and PtPDS2 (PHATRDRAFT_55102), of which only the former is functional57. A phylogenetic analysis of the PtPDS1 peptide sequence revealed close similarity to other diatom PDS sequences, which is in agreement with Dambek et al.57. Based on the endosymbiotic history of diatom chloroplasts, it would be expected that plastid-targeted nuclear-encoded proteins would share greater homology with their red algal endosymbiont ancestor; however, a recent study of the genes involved in the carotenoid biosynthesis of the chromist algae revealed that about two-thirds of these genes are closely related to the prasinophytes58. Retention of sequences from a prasinophyte source has been implicated in the evolution of fitness in diatoms and their capability to dominate the marine environment59. PtPDS1 clustered to a clade comprising diatoms, cryptomonads, and prasinophytes, which displayed divergence from the clade comprising the green algae, and the lower and higher plants, in agreement with Frommolt et al.58.
Norflurazon-resistant PDS mutants with instances of significant impairment of in vivo carotenogenic activity upon PDS mutagenesis have been reported34,53,54. In vitro characterization of mutations Leu550Phe/Arg and Phe145Val in rice PDS, under normal conditions, showed less than 5% activity compared to WT rice PDS except for Arg283Ser, which retained about 15% activity55. It has been proposed that such mutants may compensate for reduced carotenogenic activity by transcriptional up-regulation via retrograde signaling to the nucleus. On the other hand, norflurazon-resistant mutants without major retardation of carotene biosynthetic capability have been isolated51. Such mutants are preferable for exploitation as selectable markers as they would neither significantly alter cellular transcription nor adversely affect transgene expression. The two norflurazon-mutants, M1 and M2, displayed growth rates, comparable to WT under untreated conditions. The specific growth rates of WT, M1 and M2 lines displayed significant retardation in the presence of a killing concentration of norflurazon (10 µM), to 0%, 65% and 36%, respectively, when compared to their untreated controls.
Consequently, we used the PDS1 sequence from mutant M1 to develop a novel selectable marker for P. tricornutum. The established transformation vector pPha-T121 expressing eGFP was modified by replacement of the ShBle selectable marker with PtPDS-M1 to create a test vector for nuclear transformation, pPtPDS1-M1 (Fig. 4a). After confirmation of successful transformation, a new transformation vector, pBS-PtPDS1-M1 (Fig. 4b), was created by ligation of an excised PtPDS1-M1 selectable marker cassette into the SpeI restriction site of the pBlueScript SK II (+) vector with extensive multiple cloning sites for insertion of additional transcriptional units.
Stable integration and expression of transgenes is essential for a transformation system to be considered reliable. To this end, three transgenic lines maintained over 48 months without selection were subjected to norflurazon sensitivity assay in plate and growth characterization in column photobioreactor. These transformants displayed enhanced tolerance range with slight inhibition at 120 µM norflurazon which is nearly three times the upper limit for mutant M1. Similar results were observed in the column photobioreactor, with minimal growth inhibition for transgenic lines in the presence of the herbicide. These results indicate that the PtPDS1-Gly290Ala is a functional enzyme, which displays resistance to norflurazon.
Identification of a novel endogenous dominant selection marker for the model species P. tricornutum is of great significance towards advancing synthetic biology approaches in this versatile and extensively researched species. Availability of an endogenous selection marker will not only facilitate the selection of successful transformants but will also allow the creation of cisgenic P. tricornutum events provided that only native sequences are repurposed for genetic engineering, so that regulatory hurdles for the cultivation and marketing of such products may be overcome.
Conclusion
We demonstrate a simple in-vivo UV-mutagenesis strategy to isolate dominant mutants, viz. norflurazon resistance in the model diatom P. tricornutum. These novel mutants were characterized for herbicide resistive strength, and the coding sequence of PtPDS1 from M1 mutant with greater resistance was used to create the first successful endogenous selectable marker in P. tricornutum which is cost-effective, efficient and free of biocontainment concerns. In the future, domestication of type-IIS restriction sites within the selectable marker would make it a preferred selectable marker for use in large-scale transcriptional assembly methods such as Golden braid60 or Mobius assembly61 for engineering synthetic pathways in P. tricornutum.
Materials and Methods
Algal strain and culture conditions
An axenic culture of P. tricornutum UTEX 646 was maintained on Red Sea Salt Enriched (RSE)-agar (Red Sea, Israel)62 plates under illumination of 50 μmol photons m−2 s−1, at 22 °C. Liquid cultures (100 mL culture in a 250-mL Erlenmeyer flask) were routinely diluted and maintained in an exponential growth phase in an incubator shaker at 18 °C and 120 rpm, with the continuous illumination of 75 μmol photons m−2 s−1 under a CO2-enriched atmosphere (200 mL min−1).
Determination of an effective norflurazon concentration for selection on solid medium
Agar plates, containing half-strength RSE salt supplemented with different concentrations of norflurazon (Millipore-Sigma, West Chester, USA) dissolved in DMSO (0.25, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0 and 5.0 μM), were plated with 3 × 107 cells from a mid-log phase wild type (WT) P. tricornutum and incubated at 22 °C under constant illumination of 60 μmol photons m−2 s−1. After 12 days, cells were washed off the plates and re-plated on an RSE-agar medium without norflurazon and incubated under constant illumination of 60 μmol photons m−2 s−1 for 12 days at 22 °C to test for recovery of viable cells. In addition, cells were stained with 10 µL of Aniline Blue (2.5% in 2% acetic acid; Millipore-Sigma, St. Louis, USA) and observed under a fluorescent microscope to ascertain damage to the cells63.
In vivo UV-mutagenesis of WT P. tricornutum and selection of norflurazon-resistant mutants
The UV-mutagenesis of P. tricornutum cells was performed on a RSE-agar medium. Cells (3 × 107 cells) from an exponentially growing culture were spread uniformly onto a 90-mm diameter Petri dish and allowed to dry. The lid was removed, and cells were mutagenized by direct irradiation of the agar surface with UVB rays from a UV-transilluminator (312 nm; Wilber Lourmant, France) for 15 s, 30 s, 1 min, 1.5 min and 2 min. UV-irradiated plates were left for 24 h recovery at an illumination of 30 μmol photons m−2 s−1 at 22 °C before re-plating the cells on a RSE-agar plate supplemented with 15 μM norflurazon. After 15 days, obtained colonies were re-streaked onto a fresh RSE-agar plate supplemented with 15 μM norflurazon, and the two fastest growing norflurazon-resistant lines were selected for further characterization.
Preliminary determination of herbicide tolerance of norflurazon-resistant mutants
Mutants, cultured in liquid RSE medium until the mid-log phase, were diluted to 5 × 105 cells mL−1 and transferred to a 24-well plate and challenged with norflurazon in the range of 10–40 μM. These cultures were incubated in an incubator shaker (120 rpm) at 18 °C and illumination of 75 μmol photons m−2 s−1, in an atmosphere enriched with CO2 (200 mL min−1). After 20 days incubation, tolerance to norflurazon was determined by visual observation. Preliminary experiments under the similar setup showed that these norflurazon concentrations are lethal for WT P. tricornutum.
Comparative growth characterization of WT and mutants with and without norflurazon
WT and mutant (M1 and M2) cultures, grown to mid-log phase, were diluted to a chlorophyll concentration of 2 mg L−1 and cultivated in cylindrical glass columns in a Multi-Cultivator MC 1000-OD (Photon Systems Instruments, Czech Republic) with different norflurazon concentrations (0, 5 and 10 μM) at 22 °C, a light intensity of 50 μmol photons m−2 s−1, and bubbling with filtered air enriched with 2% CO2. Chlorophyll and cell density were recorded in situ, every 10 minutes for 93 h, via optical density measurements, at 680 nm and 720 nm, respectively. The experiment was repeated three times, and the average values are reported. At the end of the experiment, cells were observed microscopically under a bright field and were also stained with Aniline Blue to assess cellular damage.
Phylogeny of the P. tricornutum phytoene desaturase-1 enzyme
Homologs of the P. tricornutum phytoene desaturase (PtPDS1; EEC48362.1) were searched against the NCBI database using the BLASTp algorithm64 and chloroplastic phytoene desaturase/dehydrogenase peptide sequences, representing a diverse phylogenetic group. Alignments were made using EMBL-EBI Clustal Omega65. Phylogenetic trees were built and tested using the ATGC: PhyML 3.0 server66 with Smart Model Selection67. Within PhyML 3.0, BIONJ68 was used to build a bootstrap tree with Subtree-Pruning-Regrafting69; tree improvements and branch support bootstrapping were set to 1000. Species with bootstrap values below 500 were iteratively dropped to arrive at the final phylogenetic tree.
Cloning of the PtPDS1 gene
P. tricornutum genomic DNA was isolated using the Quick-DNA Plant/Seed Kit (Zymo Research, Irvine, USA) following the manufacturer’s instructions. The PtPDS1 CDS (PHATRDRAFT_45735; JGI database P. tricornutum v2.0) was PCR amplified from the genomic DNA of two mutants, designated M1 and M2, and WT as a reference, by Phusion HotStart II DNA polymerase (ThermoFisher Scientific, USA) with primers PtPDS1-F (5′-ATGATGTTTCACTATAAGACAGGGTCGTCATG-3′) and PtPDS1-R (5′-CTAGGCTTCCACGAATTGACTAGGATCAAC-3′). The PCR product was gel-eluted (Zymoclean™ Gel DNA Recovery Kit, Irvine, USA), ligated into a pJET1.2 vector (ThermoFisher Scientific, USA) and transformed into E. coli. Plasmids from six individual colonies were Sanger sequenced using pJET1.2-F (5′-CGACTCACTATAGGGAGAGCGGC-3′), pJET1.2-R (5′-AAGAACATCGATTTTCCATGGCAG-3′) and PtPDS1-Seq-F (5′ TATTCTCATGAACCAGAAAATGCTCACTTTGC-3′) primers. Sequencing chromatograms were aligned to the PHATRDRAFT_45735 coding sequence in Benchling (https://benchling.com) to identify mutations.
Secondary and tertiary structure analysis of PtPDS1 peptide sequence
The PtPDS1 tertiary structure was modeled on Oryza sativa phytoene desaturase-1 (OsPDS1; PDB: 5MOG.A) with SWISS-MODEL70. Secondary structure alignments rendered above the amino acid sequence alignment, using the ESPript 3.0 server71. The modeled tertiary structure of PtPDS1 was superimposed on the OsPDS1 chain A complexed with norflurazon, and the effect of amino acid substitution leading to norflurazon tolerance was analyzed using Coot72.
Vector construction of pPtPDS1-M1 and pBS-PtPDS1-M1
A pPtPDS1-M1 (GenBank accession MK645852) vector was generated by replacing sh-ble CDS in pPha-T1::eGFP (a kind gift from Prof. Assaf Vardi) with PtPDS1-M1 CDS. The pPha-T1::eGFP vector digested with XhoI (3967 bp) served as a vector backbone to assemble fcpB-Prom (fcpB-Prom-F 5′-cacaggatgagttttctcgaACTAGTACATACCTTCAGCGTCGTCTTCACTG-3′ + fcpB-Prom-R 5′-gtcttatagtgaaacatcatCTTGACATCTGGCAACCGTGA-3′), PtPDS1-M1 (inf-PtPDS1-F 5′-cacggttgccagatgtcaagATGATGTTTCACTATAAGACAGGG-3′ + inf-PtPDS1-R attaaatttttaaggaaggtCTAGGCTTCCACGAATTGAC) and fcpA-Term (fcpA-Term-F 5′-gtcaattcgtggaagcctagACCTTCCTTAAAAATTTAATTTTCATTAGTTGCA-3′ + fcpA-Term-R 5′-tatagggagaccggcctcgaACTAGTCTCGAGAAAACTCATCCTGTGCCTTC-3′) fragments in a one-pot InFusion cloning reaction (Takara-Clontech, USA). Underlined sequences within a primer pair were used for annealing and amplification of the desired template with overlapping complementary overhangs (indicated in lower case). SpeI restriction sites (5′-ACTAGT-3′) were incorporated in primers flanking the PtPDS1-M1 expression cassette for easy sub-cloning into the vector of choice.
The pBS-PtPDS1-M1 (GenBank Accession MK645853) vector was generated by SpeI excision of the PtPDS1-M1 expression cassette from the pPtPDS1-M1 vector and ligation into the respective site in pBlueScript II SK (+) plasmid (Agilent, Cat#212205). The cloning strategy was conceptualized in Benchling (https://benchling.com), and vector maps were generated in SnapGene Viewer 4.0.4 (http://www.snapgene.com).
Biolistic transformation of P. tricornutum
The WT P. tricornutum strain UTEX 646 culture in mid-log phase was diluted with an equal volume of RSE medium and grown for another three days. On the day of transformation, cells were pelleted by centrifugation (2200 × g for 5 min), and 3 × 107 cells were spread on each RSE-agar plate. The plates were left to dry in the laminar hood for 1.5 h before particle bombardment. Tungsten-M17 particles (Bio-Rad) were coated with 5 µg of DNA, as per the manufacturer’s instructions. Each DNA-coated micro-projectile preparation was spent in two shots. Biolistic gene delivery was performed using the PDS-1000/He™ gene gun (Bio-Rad, Hercules, USA); shots were fired at 1350 psi under a vacuum of 27 in. Hg at a shelf distance of 6 cm22. Bombarded cells were incubated at 22 °C under constant illumination of 30 μmol photons m−2 s−1 for 24 h to recover. Post-recovery, cells were re-plated on ½ × RSE-agar + 3 or 5 μM norflurazon and illuminated at 60 μmol photons m−2 s−1 at 22 °C for 12 days to allow the selection of transgenic events.
Calculation of transformation efficiency with pPtPDS1-M1 and statistical analysis of transformation optimization
All putative transgenic events from five plasmid-microcarrier coating reactions each, shot and selected on 3 or 5 µM norflurazon plates, were screened by PCR amplification for eGFP and PtPDS1-M1 integration.
Transformation efficiency was calculated as follows:
Transformation optimization experiments were compared by t-test at α = 0.05 in Microsoft excel using Data AnalysisToolPak.
Molecular characterization and validation of transgene expression
On-colony PCR screening of putative transgenic P. tricornutum cells was performed as described by Nymark et al.26. Insertion of PtPDS-M1 in P. tricornutum genomic DNA was confirmed using the primer pair seq-fcpB-P-F (5′-ACATACCTTCAGCGTCGTCTTCACTG-3′) + PtPDS1-R (5′-CTAGGCTTCCACGAATTGACTAGGATCAAC-3′); similarly, eGFP was confirmed using eGFP-F (5′-CGTAAACGGCCACAAGTTCAG-3′) + eGFP-R (5′-AACTCCAGCAGGACCATGTG-3′).
Southern hybridization of purified genomic DNA from transgenic lines was performed to confirm integration of pPtPDS1-M1 DNA in the P. tricornutum genome. A wet pellet weighing 3 g was lyophilized and subsequently used for high quality genomic DNA purification following the Dellaporta protocol73. KpnI-digested genomic DNA (9 µg) was resolved on a 1% agarose gel and neutral blotted to a HyBond-N + nylon membrane (GE Healthcare, Little Chalfont, UK) for Southern blotting using a DIG High Prime DNA Labelling and Detection Starter Kit I (Roche, Penzberg, Germany). Templates for DIG labeling were PCR amplified and gel-eluted. A template for the eGFP probe (606 bp) was amplified with eGFP-F and eGFP-R primers. Similarly, a template for the PtPDS1 probe (581 bp) was amplified with PtPDS1-F and PtPDS1-R primers, followed by the addition of XhoI restriction enzyme (New England BioLabs, Ipswich, USA) to the PCR reaction (incubated at 37 °C for 2 h), and gel-elution of a 581 bp fragment. DIG-labeled probes were hybridized at 54 °C overnight and stringency washed at 68 °C.
Western blot analysis
Expression of eGFP in transgenic P. tricornutum lines was validated by probing a western blot of 20 µg crude protein extract resolved on a 10% SDS-PAGE gel with an anti-TAG(CGY)FP antibody (Evrogen, Moscow, Russia) at a dilution of 1:5000 and a secondary antibody, goat anti-rabbit IgG conjugated to horseradish peroxidase (Bio-Rad, Hercules, USA) at a dilution of 1:3000. Chemiluminescence development of the western blot bound antibody was done using an EZ-ECL Kit (Biological Industries, Beit Haemek, Israel) according to the manufacturer’s instructions and imaged with a MicroChemi detection system (DNR Bio-Imaging Systems Ltd., Jerusalem, Israel).
Fluorescence microscopy
In vivo characterization of lines expressing eGFP was verified by fluorescence microscopy with filter sets 38 HE and 16 for visualization of eGFP and chlorophyll auto-fluorescence, respectively, using a Zeiss Imager A2 microscope (Carl Zeiss MicroImaging Inc., Germany) with a Zeiss AxioCamMRc mounted digital camera.
References
Field, C. B. et al. Primary production of the biosphere: Integrating terrestrial and oceanic components. Science. 281, 237–240 (1998).
Falkowski, P. & Raven, J. Aquatic Photosynthesis. (Princeton University Press, 2007).
Wang, J.-K. & Seibert, M. Prospects for commercial production of diatoms. Biotechnol. Biofuels 10, 16 (2017).
Bozarth, A., Maier, U.-G. & Zauner, S. Diatoms in biotechnology: modern tools and applications. Appl. Microbiol. Biotechnol. 82, 195–201 (2009).
Levitan, O., Dinamarca, J., Hochman, G. & Falkowski, P. G. Diatoms: A fossil fuel of the future. Trends in Biotechnology 32, 117–124 (2014).
Li, D.-W. et al. Constitutive and chloroplast targeted expression of acetyl-CoA carboxylase in oleaginous microalgae elevates fatty acid biosynthesis. Mar. Biotechnol. 20, 566–572 (2018).
Xue, J. et al. Glucose-6-phosphate dehydrogenase as a target for highly efficient fatty acid biosynthesis in microalgae by enhancing NADPH supply. Metab. Eng. 41, 212–221 (2017).
Levitan, O., Dinamarca, J., Zelzion, E., Gorbunov, M. Y. & Falkowski, P. G. An RNA interference knock-down of nitrate reductase enhances lipid biosynthesis in the diatom Phaeodactylum tricornutum. Plant J. 84, 963–973 (2015).
Zou, L. G. et al. High-efficiency promoter-driven coordinated regulation of multiple metabolic nodes elevates lipid accumulation in the model microalga Phaeodactylum tricornutum. Microb. Cell Fact. 17, 54 (2018).
Niu, Y.-F. et al. Improvement of neutral lipid and polyunsaturated fatty acid biosynthesis by overexpressing a type 2 diacylglycerol acyltransferase in marine diatom Phaeodactylum tricornutum. Mar. Drugs 11, 4558–4569 (2013).
Hamilton, M. L., Haslam, R. P., Napier, J. A. & Sayanova, O. Metabolic engineering of Phaeodactylum tricornutum for the enhanced accumulation of omega-3 long chain polyunsaturated fatty acids. Metab. Eng. 22, 3–9 (2014).
Wang, X. et al. Enrichment of long-chain polyunsaturated fatty acids by coordinated expression of multiple metabolic odes in the oleaginous microalga Phaeodactylum tricornutum. J. Agric. Food Chem. 65, 7713–7720 (2017).
Hamilton, M. L. et al. Towards the industrial production of omega-3 long chain polyunsaturated fatty acids from a genetically modified diatom Phaeodactylum tricornutum. PLoS One 10 (2015).
D’Adamo, S. et al. Engineering the unicellular alga Phaeodactylum tricornutum for high-value plant triterpenoid production. Plant Biotechnol. J. 17, 75-87 (2018).
Slattery, S. S. et al. An expanded plasmid-based genetic toolbox enables Cas9 genome editing and stable maintenance of synthetic pathways in Phaeodactylum tricornutum. ACS Synth. Biol. 7, 328–338 (2018).
Hempel, F. & Maier, U. G. An engineered diatom acting like a plasma cell secreting human IgG antibodies with high efficiency. Microb. Cell Fact. 11, 126 (2012).
Vanier, G. et al. Biochemical characterization of human anti-hepatitis b monoclonal antibody produced in the microalgae Phaeodactylum tricornutum. PLoS One 10, 1–19 (2015).
Hempel, F., Lau, J., Klingl, A. & Maier, U. G. Algae as protein factories: expression of a human antibody and the respective antigen in the diatom Phaeodactylum tricornutum. PLoS One 6, e28424 (2011).
Zaslavskaia, L. A. et al. Trophic conversion of an obligate photoautotrophic organism through metabolic engineering. Science 292, 2073–5 (2001).
Hamilton, M. L., Powers, S., Napier, J. A. & Sayanova, O. Heterotrophic production of omega-3 long-chain polyunsaturated fatty acids by trophically converted marine diatom Phaeodactylum tricornutum. Mar. Drugs 14 (2016).
Zaslavskaia, L. A., Lippmeier, J. C., Kroth, P. G., Grossman, A. R. & Apt, K. E. Transformation of the diatom Phaeodactylum tricornutum with a variety of selectable marker and reporter genes. J. Phycol. 386, 379–386 (2000).
Apt, K. E., Grossman, A. R. & Kroth-Pancic, P. G. Stable nuclear transformation of the diatom Phaeodactylum tricornutum. Mol. Gen. Genet. 252, 572–579 (1996).
Diner, R. E., Bielinski, V. A., Dupont, C. L., Allen, A. E. & Weyman, P. D. Refinement of the Diatom Episome Maintenance Sequence and Improvement of Conjugation-Based DNA Delivery Methods. Front. Bioeng. Biotechnol. 4, 65 (2016).
Karas, B. J. et al. Designer diatom episomes delivered by bacterial conjugation. Nat. Commun. 6, 6925 (2015).
Daboussi, F. et al. Genome engineering empowers the diatom Phaeodactylum tricornutum for biotechnology. Nat. Commun. 5 (2014).
Nymark, M. et al. CRISPR/Cas9 Gene Editing in the Marine Diatom Phaeodactylum tricornutum. Bio-Protocol 7 (2017).
Serif, M., Lepetit, B., Weißert, K., Kroth, P. G. & Rio Bartulos, C. A fast and reliable strategy to generate TALEN-mediated gene knockouts in the diatom Phaeodactylum tricornutum. Algal Res. 23, 186–195 (2017).
Stukenberg, D., Zauner, S., Dell’Aquila, G. & Maier, U. G. Optimizing CRISPR/Cas9 for the Diatom Phaeodactylum tricornutum. Front. Plant Sci. 9, 740 (2018).
Serif, M. et al. One-step generation of multiple gene knock-outs in the diatom Phaeodactylum tricornutum by DNA-free genome editing. Nat. Commun. 9, 3924 (2018).
De Riso, V. et al. Gene silencing in the marine diatom Phaeodactylum tricornutum. Nucleic Acids Res. 37 (2009).
Apt, K. E., Kroth-Pancic, P. G. & Grossman, A. R. Stable nuclear transformation of the diatom Phaeodactylum tricornutum. Mol. Gen. Genet. 252, 572–579 (1996).
Buck, J. M., Río Bártulos, C., Gruber, A. & Kroth, P. G. Blasticidin-S deaminase, a new selection marker for genetic transformation of the diatom Phaeodactylum tricornutum. PeerJ 6, e5884 (2018).
Brausemann, A. et al. Structure of phytoene desaturase provides insights into herbicide binding and reaction mechanisms involved in carotene desaturation. Structure 25, 1222–1232.e3 (2017).
Chamovitz, D., Sandmann, G. & Hirschberg, J. Molecular and biochemical characterization of herbicide-resistant mutants of cyanobacteria reveals that phytoene desaturation is a rate-limiting step in carotenoid biosynthesis. J. Biol. Chem. 268, 17348–17353 (1993).
Breitenbach, J., Zhu, C. & Sandmann, G. Bleaching herbicide norflurazon inhibits phytoene desaturase by competition with the cofactors. J. Agric. Food Chem. 49, 5270–5272 (2001).
Apt, K. E. et al. In vivo characterization of diatom multipartite plastid targeting signals. J. Cell Sci. 115, 4061–4069 (2002).
Gruber, A. et al. Protein targeting into complex diatom plastids: functional characterisation of a specific targeting motif. Plant Mol. Biol. 64, 519–530 (2007).
Kilian, O. & Kroth, P. G. Identification and characterization of a new conserved motif within the presequence of proteins targeted into complex diatom plastids. Plant J. 41, 175–183 (2005).
Seo, S., Jeon, H., Hwang, S., Jin, E. & Chang, K. S. Development of a new constitutive expression system for the transformation of the diatom Phaeodactylum tricornutum. Algal Res. 11, 50–54 (2015).
Alonso, D. L., del Castillo, S., Grima, C. I., Cohen, E. M. & First, Z. Insights into improvement of eicosapentaenoic acid content in Phaeodactylum tricornutum (Bacillariophyceae) by induced mutagenesis. J. Phycol. 32, 339–345 (1996).
Steinbrenner, J. & Sandmann, G. Transformation of the green alga Haematococcus pluvialis with a phytoene desaturase for accelerated astaxanthin biosynthesis. Appl. Environ. Microbiol. 72, 7477–7484 (2006).
Sharon-Gojman, R., Maimon, E., Leu, S., Zarka, A. & Boussiba, S. Advanced methods for genetic engineering of Haematococcus pluvialis (Chlorophyceae, Volvocales). Algal Res. 10, 8–15 (2015).
Suarez, J. V., Banks, S., Thomas, P. G. & Day, A. A new F131V mutation in Chlamydomonas phytoene desaturase locates a cluster of norflurazon resistance mutations near the FAD-binding site in 3D protein models. PLoS One 9 (6), e99894, https://doi.org/10.1371/journal.pone.0099894 (2014).
Liu, J., Gerken, H., Huang, J. & Chen, F. Engineering of an endogenous phytoene desaturase gene as a dominant selectable marker for Chlamydomonas reinhardtii transformation and enhanced biosynthesis of carotenoids. Process Biochem. 48, 788–795 (2013).
Liu, J. et al. One amino acid substitution in phytoene desaturase makes Chlorella zofingiensis resistant to norflurazon and enhances the biosynthesis of astaxanthin. Planta 232, 61–67 (2010).
Liu, J. et al. Genetic engineering of the green alga Chlorella zofingiensis: A modified norflurazon-resistant phytoene desaturase gene as a dominant selectable marker. Appl. Microbiol. Biotechnol. 98, 5069–5079 (2014).
Huang, J., Liu, J., Li, Y. & Chen, F. Isolation and characterization of the phytoene desaturase gene as a potential selective marker for genetic engineering of the astaxanthin-producing green alga Chlorella zofingiensis (Chlorophyta). J. Phycol. 44, 684–690 (2008).
Wagner, T., Windhövel, U. & Römer, S. Transformation of tobacco with a mutated cyanobacterial phytoene desaturase gene confers resistance to bleaching herbicides. Zeitschrift fur Naturforsch. - Sect. C J. Biosci. 57, 671–679 (2002).
Arias, R. S., Dayan, F. E., Michel, A., Howell, J. & Scheffler, B. E. Characterization of a higher plant herbicide-resistant phytoene desaturase and its use as a selectable marker. Plant Biotechnol. J. 4, 263–273 (2006).
Martínez-Férez, I. M. & Vioque, A. Nucleotide sequence of the phytoene desaturase gene from Synechocystis sp. PCC 6803 and characterization of a new mutation which confers resistance to the herbicide norflurazon. Plant Mol. Biol. 18, 981–983 (1992).
Martinez-Ferez, I. M., Vioque, A. & Sandmann, G. Mutagenesis of an amino acid responsible in phytoene desaturase from Synechocystis for binding of the bleaching herbicide norflurazon. Pestic. Biochem. Physiol. 48, 185–190 (1994).
Michel, A. et al. Somatic mutation-mediated evolution of herbicide resistance in the nonindigenous invasive plant hydrilla (Hydrilla verticillata). Mol. Ecol. 13, 3229–3237 (2004).
Chamovitz, D., Pecker, I. & Hirschberg, J. The molecular basis of resistance to the herbicide norflurazon. Plant Mol. Biol. 16, 967–974 (1991).
Linden, H., Sandmann, G., Chamovitz, D., Hirschberg, J. & Böger, P. Biochemical characterization of Synechococcus mutants selected against the bleaching herbicide norflurazon. Pestic. Biochem. Physiol. 36, 46–51 (1990).
Koschmieder, J. et al. Plant-type phytoene desaturase: Functional evaluation of structural implications. PLoS One 12, 1–26 (2017).
Falciatore, A., Casotti, R., Leblanc, C., Abrescia, C. & Bowler, C. Transformation of nonselectable reporter genes in marine diatoms. Mar. Biotechnol. 1, 239–251 (1999).
Dambek, M. et al. Biosynthesis of fucoxanthin and diadinoxanthin and function of initial pathway genes in Phaeodactylum tricornutum. J. Exp. Bot. 63, 5607–5612 (2012).
Frommolt, R. et al. Ancient recruitment by chromists of green algal genes encoding enzymes for carotenoid biosynthesis. Mol. Biol. Evol. 25, 2653–2667 (2008).
Moustafa, A. et al. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science 324, 1724–6 (2009).
Sarrion-Perdigones, A. et al. GoldenBraid: An iterative cloning system for standardized assembly of reusable genetic modules. PLoS One 6, e21622 (2011).
Andreou, A. I. & Nakayama, N. Mobius Assembly: A versatile Golden-Gate framework towards universal DNA assembly. PLoS One 13, e0189892 (2018).
Guihéneuf, F. et al. Cloning and molecular characterization of a novel acyl-CoA:diacylglycerol acyltransferase 1-like gene (PtDGAT1) from the diatom Phaeodactylum tricornutum. FEBS Journal 278, 3651–3666 (2011).
Seepersad, B., Ramnath, K., Dyal, S. & Mohammed, R. The use of Aniline Blue for the determination of dead phytoplankton, zooplankton and meroplankton in LC50 testings after 96 hours. A re-evaluation of the US Environmental Protection Agency methodology. J. Energy Resour. Technol. 126, 215 (2004).
States, D. J. & Gish, W. Combined use of sequence similarity and codon bias for coding region identification. J. Comput. Biol. 1, 39–50 (1994).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539–539 (2014).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Lefort, V., Longueville, J. E. & Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424 (2017).
Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997).
Hordijk, W. & Gascuel, O. Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 21, 4338–4347 (2005).
Waterhouse, A. et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1), W296–W303, (2018).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010).
Dellaporta, S. L., Wood, J. & Hicks, J. B. A plant DNA minipreparation: Version II. Plant Mol. Biol. Report. 1, 19–21 (1983).
Acknowledgements
The authors would like to thank Prof. Peter G. Kroth (Konstanz University, Germany) and Prof. Assaf Vardi (Weizmann Institute of Science, Israel), respectively, for sharing the pPhaT1 and pPhaT1::eGFP constructs. This research was partially funded by Israel Science Foundation grant number 1958/18.
Author information
Authors and Affiliations
Contributions
Y.T., S.L., A.Z. and I.K.G. designed and conceived the experiments. Y.T. performed the experiments and wrote the manuscript. Y.T. and R.Z. performed the in-silico modelling of protein structures. S.B. supervised the project. All authors analyzed the data, reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taparia, Y., Zarka, A., Leu, S. et al. A novel endogenous selection marker for the diatom Phaeodactylum tricornutum based on a unique mutation in phytoene desaturase 1. Sci Rep 9, 8217 (2019). https://doi.org/10.1038/s41598-019-44710-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44710-5
This article is cited by
-
The oleaginous astaxanthin-producing alga Chromochloris zofingiensis: potential from production to an emerging model for studying lipid metabolism and carotenogenesis
Biotechnology for Biofuels (2021)
-
Fucoxanthin biosynthesis has a positive correlation with the specific growth rate in the culture of microalga Phaeodactylum tricornutum
Journal of Applied Phycology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
