
Abstract
The ability to engineer plant genomes has been primarily driven by the soil bacterium Agrobacterium tumefaciens but recently the potential of alternative rhizobia such as Rhizobium etli and Ensifer adhaerens OV14, the latter of which supports Ensifer Mediated Transformation (EMT) has been reported. Surprisingly, a knowledge deficit exists in regards to understanding the whole genome processes underway in plant transforming bacteria, irrespective of the species. To begin to address the issue, we undertook a temporal RNAseq-based profiling study of E. adhaerens OV14 in the presence/absence of Arabidopsis thaliana tissues. Following co-cultivation with root tissues, 2333 differentially expressed genes (DEGs) were noted. Meta-analysis of the RNAseq data sets identified a clear shift from plasmid-derived gene expression to chromosomal-based transcription within the early stages of bacterium-plant co-cultivation. During this time, the number of differentially expressed prokaryotic genes increased steadily out to 7 days co-cultivation, a time at which optimum rates of transformation were observed. Gene ontology evaluations indicated a role for both chromosomal and plasmid-based gene families linked specifically with quorum sensing, flagellin production and biofilm formation in the process of EMT. Transcriptional evaluation of vir genes, housed on the pCAMBIA 5105 plasmid in E. adhaerens OV14 confirmed the ability of E. adhaerens OV14 to perceive and activate its transcriptome in response to the presence of 200 µM of acetosyringone. Significantly, this is the first study to characterise the whole transcriptomic response of a plant engineering bacterium in the presence of plant tissues and provides a novel insight into prokaryotic genetic processes that support T-DNA transfer.
Similar content being viewed by others


Introduction
The soil-borne plant pathogen Agrobacterium tumefaciens underpins Agrobacterium-mediated transformation (AMT) as the primary platform for the generation of engineered crop varieties, which cover ~189.8 million hectares globally1 (ISAAA, 2018). Achieved via the transfer of the T-DNA section of A. tumefaciens’ Ti plasmid into a targeted plant cell, the suitability of non-Agrobacterium strains to facilitate this process of horizontal gene transfer was first described by2 in 1997. Based on investigations around the potential of non-Agrobacterium strains to overcome some of the technical limitations (e.g. genotype dependency) of AMT, previous studies have examined Rhizobium sp. NGR234, Sinorhizobium meliloti and Mesorhizobium loti, collectively termed Transbacter™ 3. Evidence of T-DNA transfer into plant cells was shown for all three-bacterial species, but transformation efficiencies were considered too low for mainstream applications4. The non-pathogenic bacterium Ensifer adhaerens OV14 has been used in Ensifer-Mediated Transformation (EMT) to transfer DNA into rice5, canola6, cassava7 and potato8. Recently, Rhizobium etli9 and Ochrobactrum haywardense10 have also been added to the list of non-Agrobacterium spp capable of transforming plant cells. A complementary exploration of the transcriptional activation of vir genes contained on the R. etli p42a has provided further insight into similar and distinctive features compared to their A. tumefaciens counterpart11.
At a structural level, whole genome comparisons have identified key differences between A. tumefaciens and some of the non-Agrobacterium spp. For example, a comparative analysis of the 7.7 Mb genome of E. adhaerens OV14 against A. tumefaciens C58 (5.67 Mb) and the symbiont Sinorhizobium meliloti 1021 (6.7 Mb), revealed that E. adhaerens OV14 is equipped with homologs to several chromosomal-based genes, which are known to be essential for AMT. Of significance, genes that are deemed non-essential but exert a positive influence on the ability to transform a plant genome, while absent from the S. meliloti strain 1021 genome were found present in the genome of E. adhaerens OV1412. The same study noted that phylogenetically E. adhaerens OV14 and A. tumefaciens C58 reside in separate superclades of the Rhizobiacae family, which has been recently confirmed13. A syntenic analysis of the three studied bacteria revealed that E. adhaerens OV14 is void of functional vir gene orthologs across its four replicons. Yet, when E. adhaerens OV14 carries a Ti equivalent-plasmid (e.g. pCAMBIA 5105) containing a complement of vir genes it acquires the ability to accommodate, transfer and successfully integrate stable copies of T-DNA. The processes supporting this remain unknown, as indeed does the rate of vir gene transcription relative to whole genome activity and the role (if any) of individual E. adhaerens OV14 replicons in supporting EMT.
While multiple studies have investigated the processes that directly support the transfer and stable integration of T-DNA into host chromatin14,15,16,17,18,19,20,21,22,23, a complete understanding of the eukaryotic and indeed the complementary prokaryotic networks that support intranuclear targeting has yet to be fully attained22. To date, gene expression studies conducted to better understand the functional transformation machinery within Agrobacterium and non-Agrobacterium species have been restricted to a limited number of genes11,17,24,25,26,27, or oriented towards the plant’s response in the presence of the bacterium28,29,30,31,32. For example, the importance of the plant phenolic acetosyringone in eliciting the induction of virulence genes within A. tumefaciens is well understood24,25, while chromosomal genes such as att, exoC, chvA, chvD, chvB, and chvE are known to support processes central to transformation such as bacteria-plant cell attachment, exopolysaccharide production, secretion and sugar transportation18. Yet, in spite of the relevance of genetic engineering to global crop production, little else has been achieved in regards to characterizing whole transcriptional activity in plant transforming bacteria during the transformation process itself.
In response, here we report the first transcriptome analysis of a bacterium with the capacity for plant transformation. By surveying the response of the core E. adhaerens OV14 transcriptome as well as the unitary plasmid pCAMBIA 5105 (henceforth pC5105) following plant root inoculation, the temporal expression patterns of 25 vir genes (residing on pC5105) in parallel to the activity of the 7074 transcriptionally active sequences within the E. adhaerens OV14 genome were examined. The impact of this research is relevant to both the non-Agrobacterium and the Agrobacterium research communities interested in the molecular mechanisms supporting bacterial-mediated transformation of plant cells and provides a baseline dataset from which further functionality studies can now be completed.
Results
Rate and degree of EMT infection in A. thaliana roots
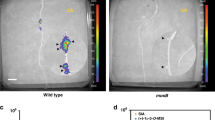
Optimum transient T-DNA transfer (87.2%, Fig. 1) was observed five days after inoculation with E. adhaerens OV14 containing pC5105 (EOV14_5105). Recorded as the percentage of blue foci visible on treated A. thaliana roots, the first signs of transient transformation were noted by D1 (3.9%), which increased significantly (P < 0.05) through to D3 (33.7%) and D5 (87.2%), decreasing slightly to 84.2% at 7 days post-treatment (Fig. 1). In regards to the degree of staining on treated roots (class I, II or III) class I was the highest recorded for each time point and peaked at D5. Class II staining was consistently lower in occurrence compared to class I but was also optimum at D5, while class III was the lowest recorded form of staining.

Progression of transient T-DNA transfer in A. thaliana roots co-cultivated with EOV14_5105 in a 7-day time course as measured via histochemical GUS staining. Incidence of GUS transient staining measured as percentage of blue foci ranked as: roots with a single spot (Class I), roots with <50% stained (Class II); roots with >50% stained (Class III) and total number of roots stained (T). D0, D1, D2, D3, D5, D7 indicate the days after co-cultivation. Error bars indicate standard error from three biological replicates.
Transcriptomic activity in EOV14_5105 following co-cultivation with A. thaliana roots
Thirty-six sequencing libraries/samples were created with raw reads ranging from 10 to 12 million per sample and a total of 406.9 million reads were recorded across the experiment. An average of 97.1% of reads were successfully mapped against the reference genome of EOV14_5105 plus the pC5105 plasmid (Supplementary Table S1). A total of 7074 and 29 genes were identified for EOV14_5105 and the plasmid pC5105 respectively, with 3756 genes on chromosome 1, 1774 on chromosome 2, 1441 associated with pOV14b and 103 with pOV14c. A total of 2333 DEGs were identified in the present study. After extracting the DEGs at each specific timepoint, 9 were identified at D0, 84 by D1, 167 by D2, 404 by D3, 707 by D5 and 962 by D7 (Fig. 2), with a greater overall proportion of down-regulated compared to up-regulated genes. The highest fold increase (9.3-fold) in the number of DEGs (84 at D1 v. 9 at D2) was observed in the transition from D0 to D1 (Fig. 2), with the increase in the subsequent time points ranging from 1.5- to 2.5-fold.

Schematic representation detailing the relative number of differentially expressed genes (DEGs) identified within EOV14_5105 following exposure to A. thaliana root tissues (BR) at 0, 1, 2, 3, 5, and 7 days (D0, D1, D2, D3, D5, D7) after inoculation. Details of the identification of these genes are depicted in Supplementary Fig. S3.
In order to observe how the DEGs were distributed across replicons in E. adhaerens OV14 and the plasmid pCAMBIA5105, the genes observed in Fig. 2 were sorted according to their corresponding replicon. The results indicated that the DEGs were represented across all four replicons, with the highest proportion of genes located on chromosome 1 and 2 followed by pOV14b (pb) and pOV14c (Fig. 3a). Approximately one fifth of DEGs recorded were associated with pCAMBIA5105 during the time course (Fig. 3a). Relative to the total number of genes on each respective replicon, the proportion of DEGs increased steadily through to D5–D7 (Fig. 3b). Meanwhile, during the first day of co-cultivation (D1) the proportion of DEGs in plasmid pOV14b (pb) was superior compared to the proportion of chromosomal-derived DEGs. This phenomenon was also observed in D5 and D7 for the proportion of down-regulated genes.

Relative distribution of DEGs in BR located across four replicons in E. adhaerens and the plasmid pCAMBIA5105. The DEGs from Fig. 2 sorted across the respective replicons from E. adhaerens and the plasmid pCAMBIA5105 (a). Detail of the number of DEGs found per replicon at each co-cultivation day (b). Proportions were calculated by dividing the number of DEGs for a specific replicon by the total number of genes for that replicon. Notations for replicons in E. adhaerens OV14: Chromosome 1 (Chr1); chromosome 2, (Chr2); plasmid pOV14b (pb); plasmid pOV14c (pc). Plasmid pCAMBIA5105 (p5105). Co-cultivation days of EOV14_5105 following exposure to A. thaliana roots from day 0 (D0) day 1, (D1) day 2, (D2) day 3, (D3) day 5 (D5) and day 7 (D7).
Visualizing how the DEGs from Fig. 2 were either independently expressed at specific timepoints or present across several timepoints, a total of 10, 8, 19, 116 and 146 up-regulated genes and 47, 19, 46, 62 and 316 down-regulated genes (located on the edges of the diagram) were found to be expressed at specific timepoints D1, D2, D3, D5 and D7 respectively (Fig. 4). A core set of five up-regulated and nine down-regulated genes were common to all timepoints with eight out of 14 of potential relevance in virulence and host defence evasion (Table 1). Day 0 (D0) is not shown in the Venn diagrams; only one out of six up-regulated genes (OV14_RS18105) was shared between D0 and D5 and no down-regulated genes were shared between D0 and any other time points. The full list of genes, accession numbers, location and function associated to the Venn diagrams are available in Supplementary Table S2.

Number of specific and common DEGs in bacteria treated with roots (BR) at different timepoints. Venn diagrams were constructed using differentially expressed (a) up- and (b) down-regulated genes, to illustrate the number of genes expressed per time point and the genes common to more than one timepoint. Labels in the Venn diagrams D1(BR), D2(BR), D3(BR), D5(BR) and D7(BR) indicate the number of co-cultivation days or days after inoculation of the bacteria treated with roots. Venn diagrams were constructed with the genes associated with a q-value < 0.05 for at least one-time point as previously illustrated in Supplementary Fig. S3. Only one up-regulated (OV14_RS18105) and one down-regulated gene (OV14_RS29425) was shared between timepoints D0-D5 and D0-D7 respectively, data not shown in the diagrams. The full list of genes, accession numbers, location and function associated to these Venn diagrams are available in Supplementary Table S2.
Gene ontology (GO) enrichment analysis of DEGs
Out of the 2333 DEGs initially identified, 1345 with complete CDS were used for the Gene Ontology (GO) analysis with 435, 699 and 752 sequences grouped in three main categories of cellular components (GO:0005575), biological processes (GO:0008150) and molecular functions (GO:0003674) using Blast2GO. In the cellular component, 72% and 67.3% of the sequences were associated with membrane components (GO:0016020 and GO:0044425), whereas 51.7% and 50.8% were associated with cell/cytoplasm components (GO:0005623 and GO:0044464). In the biological processes, the sub-categories of metabolic processes (GO:0008152), cellular processes (GO:0009987), single organism processes (GO:0044699) and localization (GO:0051179) were enriched 73.82%, 52.5%, 49.3% and 25.8% respectively. Finally, for metabolic function the enriched sub-categories were catalytic activity (GO:0003824), binding (GO:0005488), transporter activity (GO:0005215) and transcription factor activity (GO:0001071) with 69.2%, 48.1%, 12.3% and 9.9% respectively (Fig. 5). Further analysis using BlastKoala and KEGG indicated that out of the 1345 protein sequences uploaded, 1211 (>90%) showed evident orthology with sequences belonging to the α-proteobacteria class and 44.6% were fully annotated in the KEGG database, with the overrepresented functional categories corresponding to Environmental Information Processing (EIP), Cellular Processes (CP) and Genetic Information Processing (GIP).

Distribution of the gene ontology (GO) categories assigned to the EOV14_5105 transcriptome using Blast2GO. 1345 complete CDS were used for the Gene Ontology (GO) analysis with 435, 699 and 752 sequences grouped in three main categories of cellular components (GO:0005575), biological processes (GO:0008150) and molecular functions (GO:0003674) using Blast2GO.
Transcriptional activity of genes located on the pC5105 unitary plasmid
In the absence of A. thaliana roots but presence of 200 µM of acetosyringone, a higher level of transcription was recorded across the suite of vir genes on the pC5105 plasmid (Supplementary Table S3), relative to their expression following co-cultivation with roots (Supplementary Table S4). While 200 µM acetosyringone was present in both B and BR treatments, a general (q > 0.05) decrease in the FPKM values in the BR treatment compared to B was consistent across all the vir genes. Significant differences in expression (q < 0.05) when comparing B and BR within timepoints were only found for virE1(D3), virE2(D5), virK(D7), virB6(D7), virD1(D7).
Comparing expression values across timepoints independent of treatment we found that 21 out of 25 vir genes (virA, virJ, virB1, virB2, virB4, virB5, virB7, virB8, virB9, virB10, virB11, virG, virC1, virC2, virD1, virD2, virD4, virD5, virE1, virE2, virE3) were significantly induced in the transition from D0 to D1 (Fig. 6), with minimum FPKM values ranging from 326.85 and 372.47 (in D0) to maximum values of 75693 and 45249 (in D1) for B and BR respectively. (Supplementary Tables S3 and S4). Additionally, significant differences in expression were observed for virA and virG (D1–D2, treatment B), virB6 and virE2 (D5–D7, treatment B) and virA and virC1 (D1–D2, treatment BR). (Supplementary Tables S3 and S4).

FPKM expression values of 25 vir genes from plasmid pCAMBIA5105. Gene expression of twenty-five vir genes measured across 6 timepoints in (a) EOV14_5105 only (Bacteria only, B). Time points: D0B, D1B, D2B, D3B, D5B and D7B indicate the days after co-cultivation in bacteria without roots: day1 (D1B), day2 (D2B), day3 (D3B), day5 (D5B) and day7 (D7B) and (b) EOV14_5105 co-cultivated with A. thaliana roots (Bacteria plus roots, BR). Time points: D0BR, D1BR, D2BR, D3BR, D5BR and D7BR indicate the days after co-cultivation in bacteria with A. thaliana roots: day1 (D1BR), day2 (D2BR), day3 (D3BR), day5 (D5BR) and day7 (D7BR). Asterisks indicate where significant changes in expression were observed across timepoints (indicated by connecting lines) for each respective vir gene.
Of the genes induced, those belonging to virA, J, B, G, C and D operons showed peak expression at D1 irrespective of treatment, whereas virK was significantly down-regulated at the same timepoint (Fig. 6). For virB6 and virB7 expression peaks were noted at D2 and D3 respectively in the absence of roots while in the presence of root tissue peak expression shifted to D1 in both cases. For genes within the virE operon, maximal FPKM values in virE1, virE2 and virE3 occurred at D3 and D2 in B (Fig. 6a) and at D7, D5 and D5 respectively in BR (Fig. 6b). Interestingly, and contrary to what was commonly observed in most of the vir genes from this study, in virE1 from BR (Fig. 6b), peak expression was noted at D7, compared to a peak of expression at D3 in the absence of roots (Fig. 6a).
Validation of RNAseq data with relative gene expression analysis
Correlation analysis of the log2fold changes between B and BR treatments from generated RNAseq and qRT-PCR data of nine selected genes (virB1, virK, OV14_RS30665, OV14_RS18490, OV14_RS22505, OV14_RS31975, OV14_RS30910, OV14_RS16760 and trbD) returned a positive significant relationship: r = 0.71, p-value < 2.56E-08 (Fig. 7 and Supplementary Fig. S1). The genes used for validation were selected because of their potential relevance in the process of transformation (plant-bacteria interaction), and their differential patterns of expression, which are suitable for comparing both techniques (RNAseq-vs-qPCR). Relative expression analysis (Supplementary Fig. S7) indicated strong up-regulation of virB1 from D0 to D1 and then consistent down-regulation up to D7 for both the B and BR treated samples (Supplementary Fig. S7a). In contrast, virK was down-regulated from D0 to D1 and then upregulated from D3 through to D7 (Supplementary Fig. S7b). Genes OV14_RS30665-FMN-binding protein (Supplementary Fig. S7c) and OV14_RS18490-DUF2325 (Supplementary Fig. S7d) showed a significant decrease in expression from D0 to D1, remaining relatively constant at a lower relative expression levels up to D7. Gene OV14_RS22505 which encodes an exopolysaccharide production repressor-type protein (exoX) increased its expression from D1 to D7 (Supplementary Fig. S7e). The membrane-related protein gene OV14_RS31975 located in pOV14b exhibited a constant expression throughout the time course (Supplementary Fig. S7f). The expression of OV14-RS30910 (cell-envelope protein) located on pOV14b was up-regulated from D0 to D1 (Supplementary Fig. S7g) as was OV14_RS16760 (chvD) which encodes a chromosomal virulence D ortholog in EOV14_5105 (Supplementary Fig. S7h). Separately, the trbD (conjugal-transfer protein) gene with 97% identity with A. tumefaciens trbD was steadily up-regulated from D0 to D7 (Supplementary Fig. S7i).

Validation of RNAseq data with corresponding quantitative real time qRT-PCR analyses. Correlation plot between RNAseq and qRT-PCR data for log2fold changes between BR and B conditions of nine genes in six consecutive time-points. Pearson’s correlation coefficient = 0.71, p-value < 2.56E-08.
Genetic pathways in E. adhaerens OV14 responsive to the presence of plant tissue
ABC-transporters and bacterial secretion systems
A total of 1345 protein accessions were uploaded in BlastKOALA of which 44.9% had an annotated function in the KEGG database. The KEGG database provided us with a KEGG identifier linked to each protein ID uploaded, with three main categories found: (i) genetic information processing (ii) cellular processes and (iii) processing of genetic information (Supplementary Table S5). Under the environmental information processing category the sub-category membrane transporters is composed of two sub-groups: ABC-transporters and Bacterial Secretion System. Within the ABC-transporters group there were 54 KEGG Orthology (KO) identifiers, associated with a total of 72 protein ID sequences. This represented the highest number of identifiers found in a single sub-group. Most of the genes from this group were related to oxidative phosphorylation; however, four KO identifiers including eight protein ID sequences were related to quorum sensing (Supplementary Table S5). The genes (livK, livH, livM and livF) identified that relate to quorum sensing act together in the same biosynthetic pathway with two accessions of livK-1 and livK-2 (OV14_RS24100 and OV14_RS08630) reporting contrasting expression patterns. In general terms, livK-1 was induced and livK-2 was repressed across timepoints (Fig. 8a,b). LivF, livH and livM were found to be down-regulated in respect to D0 under both B and BR conditions (Supplementary Fig. S8). Examining the effect of root tissue, significant (P < 0.05) changes occurred towards the end of the time course (D5 and D7) for livK-1 and livK-2 (OV14_RS24100, OV14_RS08630), livH (OV14_RS18800) and livM (OV14_RS18795) (Fig. 8a,b and Supplementary Fig. S8). Only livK-2 (OV14_RS08630) was up-regulated in BR respect to B, with the other genes from this group showing a reduced expression in the presence of roots.

Temporal FPKM expression values for genes belonging to the categories environmental information processing, cellular processes and processing of genetic information as determined using the KEGG database. D0 to D7 indicates abbreviations for co-cultivation days as described in materials and methods. Asterisks indicate evidence of significant differential expression using Tophat2 v2.1.1 (p < 0.05) between EOV14_5105 un-treated (B) and treated with roots (BR) for three biological replicates. Red and green bars indicate the gene expression level for EOV14_5105 un-treated (B) and treated bacteria (BR) respectively in regards to (a) livK (OV14_RS24100); quorum sensing, (b) livK (OV14_RS08630); quorum sensing, (c) tatA; cell motility and bacterial chemotaxis (d) fliG, (e) motB, (f) MCP, (g) cheA and flagellar assembly (h) flgE.
For the bacterial secretion system sub-category, a single identifier with a single representative gene (tatA) was identified. Highly expressed at D0, tatA activity abruptly decreased by D1. This continued through to D7 irrespective of the treatment (B, BR). tatA was the only gene associated with a bacterial secretion system that was found to be differentially expressed in the presence of root tissues (Fig. 8c).
Bacterial chemotaxis and flagellar assembly
The genes fliG, motB, mcp, cheA, fliM, fliNY/fliN, and cheY associated with bacterial chemotaxis were found to be differentially expressed under the BR treatment. All these genes exhibited a significant induction up to D1, after which genes were strongly repressed. Specifically for D1, the abundance of transcripts was higher in BR compared to B (Fig. 8d–g [fliG - cheA] and Supplementary Fig. S9 [fliM - cheY]). In the presence of roots the genes fliG, fliM and mcp exhibited significant down regulation at D2, motB, cheA and fliNY at D2 and D3 and cheY at D3 and D7 (Fig. 8d–g and Supplementary Fig. S9). Additionally, 14 DEGs involved in the structural formation of flagella were also identified (Supplementary Table S5). The genes flgE, flgC, flgF, flgG, flgK, flgL, fliC, fliF, fliL. flgC, flgI, fliF, fliG and fliM similarly to those previously mentioned for bacterial chemotaxis, recorded highly induced FPKM values at D1, which were down-regulated in the subsequent co-cultivation days in bacteria treated and untreated with roots (Fig. 8h [FlgE] and Supplementary Fig. S10 [FlgC – fliM]). Groups of genes showed significant down-regulation in bacteria treated with roots (BR) at different timepoints during co-cultivation, the genes flgC, flgF, fliC, fliF showed significant down-regulation at D2, fliL at D3, flgE, flgG, flgL at D2 and D3 and finally flgK at D2, D3 and D7. The expression profiles of the genes flgC, flgF, flgG, flgK, flgL, fliC, fliF, flil. and KEGG diagram of the genes involved in flagellar assembly can be found in Supplementary Fig. S10. Genes involved in intertwined processes such as bacterial chemotaxis and flagellar assembly showed overall a similar expression pattern in the presence and absence of plant tissue throughout the co-cultivation period.
Genes considered (by sequence homology) in12 to be potentially involved in the transformation process include chvA, chvB, exoC, gabD, attL, blcB, blcC, celA, celB, celC, celE, celG, pcs, pmtA, choX choV, choW, kdgF ligE, chvD, chvE, gguA, gguB, gguC, chvH, serB, miaA, acvB katA, ParA/VirC1, trbB, trbC, trbD, trbE, trbJ, trbK, trbL, trbF, trbG, trbH, trbI, aopB, vbp2-3, vbp1. Out of the previously mentioned genes, aopB and trbD exhibited significant down-regulation in presence of plant tissue (BR) in later stages of transformation with EOV14_5105 (Supplementary Table S6). aopB in A. tumefaciens is regulated by the chvD/chvI two component system and is indirectly involved in virulence, whereas trbD is related to plasmid mobilization between bacterial cells.
Homologous symbiosis-related genes showing differential expression in E. adhaerens OV14
Based on the in silico comparison between S. meliloti and EOV14_5105 symbiosis-related protein sequences from previous reports33,34, we found genes with comparable expression trends after exposure to plant tissues (Table 2). In 2009 Gurich et al. observed that suppression of flagellum production and thus down-regulation of flagella-related genes in S. meliloti plays an important role for the effective nodule formation of M. truncatula roots. Here, the flagella-related genes OV14_RS07290, OV14_RS07305 and OV14_RS07300 found in EOV14_5105 (showing >45% identity with S. meliloti) were heavily repressed in the BR treatment (Table 2). In fact, our previously depicted results in Fig. 8d–h and Supplementary Figs S9 and S10, show how genes related to assembly and functionality of flagella were heavily repressed in the BR treatment.
Discussion
The genome of E. adhaerens OV14 has been previously characterized and compared with that of the alternative rhizobia A. tumefaciens C58 and S. meliloti 102112. Yet, prior to this study the relative activity of the E. adhaerens OV14 genome in the presence and absence of plant tissues was undetermined. Indeed, the results presented here are novel not only in their capacity to provide insight into the genetic processes underway in a non-Agrobacterium spp but they are also the first survey of whole genome transcription in a plant engineering bacterium following co-cultivation with plant tissues.
Plant tissue conditions used were as per detailed in the Arabidopsis root assay35, which was a modified version of the original A. tumefaciens-based assay36. This provided a rapid and simple method of co-cultivating EOV14_5105 with root tissues and critically allowed for the rapid isolation of EOV14_5105 cells at each timepoint. Based on this assay, an optimum rate of transient transformation with A. thaliana roots was achieved after 5 days of co-cultivation. This contrasts with A. tumefaciens, where the optimal co-cultivation period for most plant species is typically 2–3 days18. The extended co-cultivation period is a requirement of EMT that has been used in the transformation of the recalcitrant rice variety IR645 and in several different plant species (e.g. potato, barley, wheat and oilseed rape) transformed using EOV14_5105 in our laboratory.
Here, the number of DEGs increased across timepoints in the presence of roots. The highest fold-change in the number of DEGs occurred between D0 and D1 co-cultivation. It is reasonable to think that a large proportion of genes potentially related to the plant-bacteria interaction and specifically to plant transformation are triggered at the early stages of the plant-prokaryote interaction. In A. tumefaciens, virulence genes directly related to transformation are triggered in the presence of plant tissue or phenolic compounds within 4 to 12 hours after inoculation24,25,26. Also, chromosomal genes such as chvE/I, aopB, rbsC, flaA, flaB, mcpA, cheY2 among others, have been found to be differentially expressed under acidic conditions suitable for transformation within 7 hours37. While, in S. meliloti nodulation, conjugal transfer and invasion related genes can be induced in bacteria under symbiotic conditions as early as 18 hours post inoculation38,39,40. These findings reinforce our assumption that many of the DEGs triggered here within the first 24 hours could be related to an early plant-bacteria sensing/attachment interaction and/or might be precursor genes that act as initiation factors involved in transformation-related pathways downstream in the process.
In broad terms the DEGs found in the BR treatment were represented across all four replicons from EOV14_5105. This supports the conclusion of Rudder12 who proposed the presence of chromosomal homolog genes from A. tumefaciens within EOV14_5105. However, when dissecting the process within the different co-cultivation days, an overrepresentation of DEGs derived from pOV14b was observed at day 1 (D1), which concurs with A. tumefaciens and S. meliloti, where most of the genes related to virulence and symbiosis respectively are also located on plasmid structures18,41.
In this study we found a core set of 14 DEGs consistently activated from D1 through to D7. Eight are related to virulence and/or symbiosis. The up-regulated gene OV14_RS24990 with BLAST identity of 74% and 67% with S. meliloti and A. tumefaciens identified as Tetratricopeptide repeat protein (TPR) present in prokaryotic and eukaryotic organisms, mediates protein–protein interactions and is related to the virulence potential of pathogenic bacteria such as Y. pestis and P. aeruginosa42. The gene OV14_RS30440 (71% identity with S. meliloti) contains an adenylate/guanylate cyclase domain. In Agrobacterium and Rhizobium this domain is associated with cellulose production which helps in cell aggregation and anchoring to plant surfaces thereby facilitating colonization43. Finally, OV14_RS16135 (72% identity with S. meliloti) is a nucleoside-diphosphate sugar epimerase. Sugar nucleotides are relevant for the biosynthesis of sugar-containing bacterial cell structures. In pathogenic bacteria, this structure can lead to the evasion of the bacteria from the host immune system44. In S. meliloti, nucleoside-diphosphate sugar epimerase can be involved in cell envelope biogenesis, carbohydrate transport and metabolism45. Two steadily down-regulated genes (OV14_RS23300 and OV14_RS05820) are related to rhizopine metabolism. Rhizopines are secreted plant compounds, which are synthesized by bacteroids inside the plant-nodule in Rhizobium-legume interactions46. The production of certain rhizopines can confer selective advantage to free-living cells located in the vicinity of the rhizosphere. In 2014 Krysciak47 and colleagues discovered that genes responsible for rhizopine catabolism in S. fredii NGR234 were repressed in the presence of N-acyl-homoserine-lactones. They hypothesized that the downregulation is due to a link between rhizopine metabolism and quorum sensing signals mediated by the activation of the conjugative gene traR47. In EOV14_5105, traR- OV14_RS28560 (82% identity with S. meliloti) (located in pOV14c) was up-regulated in the presence of roots at D1 (p = 0.0061; q = 0.234). The possibility of a link between the down-regulation of the rhizopine-related genes by traR gene and its involvement in transformation in EOV14_5105 requires further clarification. The genes OV14_RS18300 (91% identity with S. meliloti) coding for inositol 2-dehydrogenase and OV14_RS05810 (92% identity with S. meliloti) for 5-dehydro-2-deoxygluconokinase are involved in inositol catabolism. Catabolism of inositol is essential for the utilization of rhizopines as a carbon source in S. meliloti L5–3048 and inositol 2-dehydrogenase is also involved in the formation of healthy nodules in soybean colonized by S. fredii49. Lastly, the gene OV14_RS05820 a sugar ABC sugar transporter (93% identity with S. meliloti) containing ribose (RbsA) and galactose (MglA) import ATP-binding motifs was identified. Sugars available extracellularly in bacteria can be transported into the cytoplasm by ABC transporters50. In A. tumefaciens the ABC transporter chvE-MmsAB is crucial for sugar utilization and virulence51. Bacteria can optimize energy usage by taking in certain available sugars; for this, repression of specific sugar uptake pathways is required52. The mechanism by which the OV14_RS05820-sugar transporter is downregulated throughout all timepoints in the presence of roots and its involvement in transformation remains to be elucidated.
In A. tumefaciens the vir genes are tasked with the transfer of T-DNA complex from bacteria to host cells18,19,53,54. In this study, E. adhaerens OV14 was equipped with an A. tumefaciens derived suite of vir genes on the pC5105 plasmid. The induction effect of acetosyringone on the activation of the virulence genes on the Ti plasmid from A. tumefaciens has been long known25 and this phenomenon has recently been described in R. etli11 and O. haywardense (patent WO2017040343A1)55. With the current EOV14_5105 genotype, the supplementation of co-cultivation media with acetosyringone is an important step for improving infection efficiency8. Here, all virulence genes (except virK) exhibited low levels of basal expression at D0, with a subsequent induction at D1, which was significant and in agreement with reports from21,22,56,57. In line with data reported by24,25,58, virA showed modest levels of induction in the presence of acetosyringone. This differs with the results obtained by17, where virA was expressed at basal constitutive levels and it was not found to be induced by acetosyringone. This difference can be attributed to the plasmid background present in our bacteria. In octopine (pTiA6) and nopaline (pTiC58)-type plasmids, expression of virA is constitutive17 and weakly inducible58,59,60 respectively. In our case, pCAMBIA5105 is derived from the ‘supervirulent’ succinaminopine-type plasmid pTiBo542, which is acetosyringone dependent61. Overall, these results highlight the effect of acetosyringone on EOV14_5105 gene expression relative to the limited number of vir genes from A. tumefaciens and R. etli evaluated to date in response to this compound11,62,63,64.
While both B and BR treatments included acetosyringone, when co-cultivated with roots a decrease (non-significant) in vir gene transcription within EOV14_5105 was recorded in BR. Bolton and collaborators originally reported this phenomenon with tobacco protoplasts24. The underlying reason for this downregulation in EOV14_5105 is most likely due to a feedback system of repression arising from a MAMP triggered elicitation of the plants immune system65,66. For example, it is known that salicylic acid67,68 and ethylene-associated69 defense responses in plants can shut down the expression of the vir regulon suppressing Agrobacterium-mediated transformation. EOV14_5105 possesses the salicylate-hydroxylase gene (OV14_RS11845) which could potentially degrade plant derived salicylic acid. However, OV14_RS11845 was not differentially expressed in bacteria treated with roots, suggesting that additional genes might be involved in the general down-regulation of the vir operon in the presence of roots. In considering known Agrobacterium MAMPs such as peptidoglycan70 and the EF-Tu elongation factor71, EOV14_5105 does indeed possess homologs of several peptidoglycan-binding type proteins and EF-Tu, but neither registered significant levels of expression in the BR treatment under the conditions of this study. Meanwhile the absence of a functional Type III secretion system in EOV14_5105 would suggest the absence of a delivery system for the secretion of specialized effector proteins directly into plant cells, which is a recognized mechanism for stimulating a plant’s defense response72. Of course, the stress induced by the physical cutting of the roots would have led to the release of defense compounds such as reactive oxygen species (ROS) among others that can decrease the activity of several metabolic pathways in bacteria73,74, which may have led to a general decrease in vir gene expression. Supporting this theory is the observation that the gene OV14_RS09075, a superoxide dismutase-sodA associated with the detoxification/elimination of ROS75,76 showed a significant induction in EOV14_5105 exposed to roots at D3, D5 and D7.
In A. tumefaciens, the VirB1 protein has been found to be extracellularly secreted, associated with the T-complex and linked to local lysis of the peptidoglycan wall77. This protein exhibited high levels of induction under our experimental conditions. The lipoprotein VirB7 stabilizes other VirB proteins and is associated with VirB2 in the T-pilus78,79. It is reasonable to assume therefore that it’s expression once initiated would be relatively constant during periods of bacterium-plant co-cultivation. In our results, whereas the expression of virB7 significantly decreased in untreated bacteria, it remained stable in bacteria treated with roots. virE1 acts as a chaperone for virB2 facilitating its stabilization and solubilization80,81 while virE2 and virE3 are imported into the host cell together with virE2 also assisting in T-complex stability82,83,84. In light of the critical roles played by virE2 and virE364,82,85 the delayed peak-expression of these genes, could explain the additional two days co-cultivation typically required in transformation experiments with EOV14_5105 compared to A. tumefaciens to obtain comparable infection efficiencies. Separately, a genetic complementation study has revealed that virB operon genes from the plasmid pTiA6 can mutually co-stabilize each other and that virB1/virB2/virB3/virB4 and virB7/virB8/virB9/virB10, are probably translationally coupled86. Interestingly, our expression results showed a clear differentiation in the expression pattern of both group of genes after exposure to acetosyringone, suggesting that there might also be a feedback system acting between these group of genes within pCAMBIA5105.
Vir genes aside, the whole genome response triggered in EOV14_5105 following contact with roots provides an opportunity to survey complementary genes that support T-DNA transfer. For example, the majority of genes from the cellular component category were membrane-associated and the KEGG database analysis retrieved 55 genes related to metabolic pathways that could be relevant to the transformation process. Genes from the liv operon are involved in the transport of branched-chain amino acids in E. coli87 and R. leguminusarium88. Branched-chain amino acids are essential for symbiosis in bacterioid structures in R. leguminusarium88. LivH/LivM and LivF/LivG are hydrophobic and hydrophilic components of the membrane structure respectively. LivK is a substrate binding protein, LivF is an ATP binding protein and LivH is part of the inner membrane complex encoding for a large hydrophobic protein. According to12 key genes involved in nodulation and nitrogen fixation are absent in EOV14_5104. This leads to a series of questions: are liv genes fulfilling a function related to N2 fixation in EOV14_5105? Is the intake of branched-chain amino acids essential for transformation in EOV14_5105? Or is the differential expression of the liv genes related to plant transformation?
An intact TAT secretion system is vital for A. tumefaciens infection of plant cells89 with the tat operon involved in the transfer of folded proteins from the cytoplasm into the cell periplasm90, cell division, chemotaxis and flagellar biogenesis89. EOV14_5105 tatA was significantly down-regulated at D1 in the presence of roots and approximately 34-fold in response to acetosyringone through the time course. Interestingly, this gene reached FPKM values >40,000 which is equivalent to expression levels observed with the virulence genes. The significant decrease in tatA activity after exposure to roots and acetosyringone suggests that there is a repression of accessory protein export systems possibly to re-direct energy towards transformation-specific protein export machinery from the vir genes. Crosstalk between Tat and Sec secretions systems has been shown in Streptomyces91, also sequential regulation of secretion systems has been evidenced using Boolean modelling in S. typhymorium between elements of T3SS and T6SS92.
The processes of biofilm formation, flagellar assembly and quorum sensing are closely intertwined and related to virulence93,94. In A. tumefaciens biofilm formation deficiency is correlated with a reduced virulence phenotype83 and a decreased ability to colonize roots in S. meliloti95. In a heterologous system using E. coli to evaluate A. tumefaciens genes, a mutation in crp dramatically reduced the expression of the lacZ gene under the control of the promoter from the virB operon96. Crp (cyclic AMP receptor protein) controls biofilm production in pathogenic bacteria97,98,99,100,101 and not surprisingly in this study crp was significantly increased in the presence of root tissues from D2 through to D7, suggesting a possible contribution of biofilm formation in enhancing surface attachment of EOV14_5105 with host tissues. The presence of functional flagella on EOV14_5105 has been confirmed (Rathore et al. unpublished) and 14 DEGs involved in flagella structural assembly and motor responses were identified here. All these genes were initially induced by acetosyringone and subsequently repressed after 24 h in the presence and absence of roots. Reduced motility of bacteria in response to high concentrations of plant inducers helps to maintain a close contact between bacteria and plant tissue possibly contributing in the process of transformation102.
Chemotaxis is a crucial step for A. tumefaciens’ colonization of the plant surface93,102. A. tumefaciens and S. meliloti chromosomal genes relevant for chemotaxis include MCP ((Methyl-accepting chemotaxis, which structurally resembles virA), cheA, cheY (resembles virG), fliG, fliM, fliNY and motB which act together in a pathway where the chemotactic stimuli are detected by the transmembrane MCP sensor protein93,103,104,105,106,107. Located on chromosome 1, all of the aforementioned genes had a strong response to acetosyringone. After D1 however, all these genes were heavily repressed, which coincides again with the observation that a high concentration of inducers (e.g. acetosyringone + plant-based inducers) can suppress chemotaxis and motility responses in A. tumefaciens102.
The rseP gene (Regulator of Sigma E, Protease) encodes a protease involved in the regulation of cell surface signaling systems (CSS) in P. putida108 and P. aeruginosa109. Little is known about the function of rseP in A. tumefaciens; however, it has been documented that similar proteins called Lon-proteases are required for normal growth, cellular morphology and full virulence110. Additionally, in S. meliloti Lon is involved in the regulation of exopolysaccharide synthesis and is required for effective nodulation in alfalfa111. In our results, rseP was triggered in the presence of roots, suggesting that these proteases are involved in the regulation of cell surface signaling systems in EOV14_5105. As an additional note on bacterial surface/secreted proteins, it has been found that exopolysaccharides (EPS) can suppress host defense responses towards S. meliloti112. Interestingly, in EOV14_5105 the gene OV14_RS22500 (exopolysaccharide biosynthesis protein-ebp) is significantly induced in the presence of roots at D5 and D7 (when T-DNA transfer is more pronounced). The suppression of host defense response by triggering the synthesis of exopolysaccharides in EOV14_5105 could therefore be enhancing host susceptibility by facilitating a closer interaction between plant and bacteria.
To summarize, the work detailed provides a novel insight into the genetic processes that underpin EMT across the examined timecourse. This study has characterized the temporal response of the EOV14_5105 genome to acetosyringone as well as to the presence of plant tissues. As the first published study describing the transcriptional response of a plant transforming bacterium in the presence/absence of plant tissues exposed to acetosyringone and with the recent identification of additional non-Agrobacterium species such as R. etli and O. haywardense, the presented datasets provide a benchmark for future studies focused on improving plant transformation processes in order to facilitate downstream applications.
Materials and Methods
E. adhaerens OV14_pC5105 growth conditions
Strain E. adhaerens OV14_pC5105 (henceforth EOV14_5105) was generated as described previously8 and cultivated overnight in 20 ml of YEP liquid media at 28 °C and 220 rpm. Thereafter, bacteria were harvested at O.D 1.0, centrifuged at 4000 rpm for 10 minutes and re-suspended in the same volume of filter sterilized Inoculation Media (IM; 4.4 g/l Murashige and Skoog salts plus Gamborg B5 vitamins, 200 mg/l hydrolysate casein, 10 g/l sucrose, 0.5 g/l MES, pH5.5). This bacterial suspension was incubated at 21 °C, 220 rpm for up to 60 minutes and used to inoculate Arabidopsis thaliana root tissues.
Plant material preparation and co-cultivation with EOV14_5105
The preparation and growth of A. thaliana roots for co-cultivation with EOV14_5105 was completed in-vitro as previously described35. A root-based assay was selected because of its logistical advantages and also because E. adhaerens is a soil bacterium and under natural conditions has been shown to colonize root systems8. For co-cultivation, roots of approximately 100 mm in length from up to ten plants per treatment were aseptically cut into ~5 mm fragments using a small amount of water in a petri dish. Cut roots were grouped into bundles that were transferred to solid co-cultivation media (4.32 g/l of MS salts, 1X vitamin solution (0.5 mg/l nicotinic acid, 0.5 mg/l pyridoxine, 0.5 mg/l thiamine-HCl), 100 mg/l myo-inositol (stock 100X), 0.5 g/l MES, 10 g/l sucrose, 7.5 g/l Agar technical No. 3 (Oxoid LP0011), adjusted to pH 5.5 and supplemented with 200 µM of sterile-filtered acetosyringone post-autoclaving after media temperature reached 50 °C. Unless otherwise indicated media were autoclaved at 121 °C for 20 min and the supplier company for the reagents was Ducehfa Biochemie®. Excess of water was removed, and roots were immediately inoculated with 1 ml of EOV14_5105 resuspended in Inoculation Media. Roots were then manually spread to an approximate 2 cm radius to maximize contact with the media. Plates were co-cultivated at 21 °C in the dark for the designated period: day 0 (D0), day 1 (D1), day 2 (D2), day 3 (D3), day 5 (D5) and day 7 (D7) days post-inoculation. For day 0 (D0), bacteria were briefly pipetted onto roots, immediately removed and used for RNA extraction. After each co-cultivation time, collected suspensions were stabilized with 3 ml of RNAprotect Bacteria Reagent ®(Qiagen) for 5 min, vortexed for 5 min and the bacterial suspension collected using a micropipette. The suspension was then centrifuged at 12,000 rpm for 5 min after which the pellet was immediately placed in liquid nitrogen and transferred to −80 °C. In order to account for the potential variation in gene expression due to the time of co-cultivation, untreated bacteria (control) were handled as above for each timepoint but in the absence of A. thaliana roots. Three biological replicates (derived from three independent EOV14_5105 starter cultures) were processed for both treated and un-treated bacteria at six timepoints covering D0, D1, D2, D3, D5 and D7 (Supplementary Fig. S6). EOV14_5105 bacteria treated with A. thaliana roots was denominated as BR (bacteria + roots) and EOV14_5105 bacterium un-treated with roots was denominated as B (bacteria only). Supplementary Fig. S6.
Separately, a portion of treated roots from each respective timepoint was collected and histochemically stained to monitor EOV14_5105 infection efficiency, based on transient GUS gene expression35. Infection efficiency was recorded by counting the presence/absence of blue stained foci along each root length (T), but also semi-quantitative measurements were made by counting the number of roots with a single blue foci (class I), the number of roots with <50% surface area stained blue (class II) and finally the number of roots with >50% and <100% surface area stained blue (class III). Significant differences between staining levels were evaluated using ANOVA complemented with Tukey-Kramer post-hoc test (Microsoft Excel®). Supplementary Fig. S2.
RNA isolation and quality verification
For RNA extraction, collected bacterial pellets were thawed on ice and then re-suspended in 300 µl of resuspension/lysis buffer (TE pH: 8.0, lysozyme 1 mg/ml and Proteinase K®, Qiagen). The following RNA extraction steps were performed in 1.5 ml vials using Trizol Reagent® as per manufacturer’s instructions. Pelleted RNA was washed with 70% ethanol, centrifuged, air dried (briefly, <1 min), re-suspended in ~30 µl of sterile Sigma® water and incubated at 65 °C for 10 min to evaporate residual ethanol. RNA was then precipitated and resulting pellets were once again washed with 70% ethanol, centrifuged, dried briefly, resuspended and incubated at 65 °C for 10 min. RNA quality was verified using a 2100 Bioanalyzer (Agilent Technologies; Santa Clara, CA, USA), with recorded RIN values for total RNA >7.4.
rRNA depletion, cDNA library preparation and next-generation sequencing
rRNA depletion, cDNA library preparation and RNA sequencing was performed externally by the Beijing Genomics Institute (BGI, China). Briefly, rRNA depletion and fragmentation was carried out using Ribo-Zero Magnetic Kit (Bacteria, EPICENTRE). RNA was fragmented into 130–170 nt and purified with RNAClean XP Beads (AGENCOURT). cDNA synthesis was generated using First Strand Master Mix and Super Script II reverse transcription (Invitrogen). Adapters ligation was achieved by combining the Adenylate 3’Ends DNA, RNA Index Adapter, Ligation Mix and then purifying with DNA with Ampure XP Beads (AGENCOURT). Several rounds of PCR amplification with PCR Primer Cocktail and PCR Master Mix were performed to enrich the cDNA fragments. Resulting PCR products were purified using Ampure XP Beads (AGENCOURT), with the final library validated via an Agilent 2100 bioanalyzer instrument (Agilent DNA 1000 Reagents) and using real-time quantitative PCR (qRT-PCR) (TaqMan Probe). Qualified libraries were amplified on cBot to generate the cluster on the flowcell (TruSeq PE Cluster Kit V3–cBot–HS, Illumina). The amplified flowcell was sequenced under paired ends on a HiSeq. 4000 System (TruSeq SBS KIT-HS, Illumina), with an expected read length of 100 bp. Depth coverage was calculated at 10–12 million reads per sample, with 36 samples in total (6 timepoints × 2 treatments (B, BR) × 3 independent replications).
Mapping and processing of sequencing data
Sequencing reads were trimmed using Trimmomatic v. 0.36 using paired end parameters. Mapping of reads to the E. adhaerens OV14 reference genome; acc. No. NZ_CP007236.1, NZ_CP007237.1, NZ_CP007238.1 and NZ_CP007239.1 and the plasmid pC5105 (EF042581) was carried out using Tophat2 v. 2.1.1 using parameters for paired end reads (tophat -p8 -G genes.gtf -o (name) indexgenome read_1.fq read_2.fq). Pairwise comparisons to identify DEGs were obtained using Cuffdiff v. 2.2.1 from the Cufflinks software package. Genes with a depicted [yes] in the excel Cuffdiff-output spreadsheet (False Discovery Rate-FDR or q_value < 0.05) were considered differentially expressed. Six pairwise comparisons were performed in Tophat2 in order to identify DEGs between both treatments (B, BR) at each time point (D0, D1, D2, D3, D5 and D7) (Supplementary Fig. S3). In addition, to visualize the induction dynamics exerted by acetosyringone across the timepoints, pairwise comparisons of pC5105 were carried out independently for both B and BR treatments across timepoints (Supplementary Fig. S4). As Tophat2 assigns the probability of observing each fragment in dataset uploaded, FPKM values found for the comparison between specific timepoints and across timepoints can vary because it corresponds to independent runs in the software113. After observing this phenomenon, we performed a Pearson correlation coefficient test using Microsoft Excel® which delivered a value of r = 0.99, indicating that both datasets clearly provide the same information (Supplementary Fig. S5).
Data mining and gene enrichment analysis
Exploration and visualization of the Cuffdiff output files was conducted in R studio using the package CummeRbund v. 0.1.3 and Microsoft Excel 2016. The DEGs depicted by [yes] in the excel Cuffdiff-output spreadsheet (q_value < 0.05) were retrieved for posterior analysis. Cuffdiff provides a log2fold change value in pairwise comparisons for each independent gene. Up- and down-regulated genes were retrieved based on a fold change >0 and <0 respectively. According to114 using the statistical correction of the q value using the FDR (False discovery rate: ≤0.05) can provide enough power to discriminate DEGs without the requirement of an extra cutoff value in the log2Fold change. To visualize which genes were exclusively expressed at certain time points and which ones were present in several time points, we listed the up- and down-regulated genes found separately from the pairwise comparisons for each independent time point (D0, D1, D2, D3, D5 and D7) and visualized the resulting groups of DEGs in two Venn diagrams using the online tool: http://bioinformatics.psb.ugent.be/webtools/Venn/ (Supplementary Fig. S3). Day 0 (D0) is not shown in the Venn diagrams; only one out of six up-regulated genes was shared between D0 and D5 and no down-regulated genes were shared between D0 and any other time points. The list of DEGs obtained from the Venn diagrams was associated with descriptive Gene Ontology (GO) terms using Blast2GO (v.4.1.5), via protein sequences and default parameters under the taxonomy filter for α-proteobacteria. At the same time, in order to retrieve maps of metabolic pathways potentially relevant for the transformation process, the resulting list of genes was analyzed with BlastKoala v. 2.1115 and KEGG116,117,118 (Kyoto Encyclopedia of Genes and Genomes) using default parameters within the taxonomy group prokaryotes-bacteria. BlastKOALA and KEGG allow for comparisons to be made for the functionality of individual genes or, in combination with larger number of genes, in complex metabolic pathways.
To investigate the potential commonality in the symbiotic relationship between S. meliloti and plant and that of EOV14_5105 and plant tissues a basic local alignment using BLAST (NCBI) of S. meliloti protein sequences with known function was carried out on the E. adhaerens OV14 genome. The protein ID and percentage of identity were recorded in order to evaluate the FPKM values corresponding to the retrieved-ortholologous protein in E. adhaerens OV14. This information was then used to evaluate if there was induction/repression of symbiosis-related genes in E. adhaerens OV14.
Gene validation using qRT-PCR
To validate RNAseq datasets, the expression patterns of nine DEGs were independently analyzed using qRT-PCR. RNA samples of EOV14_5105 cultivated with root tissues were treated with RQ1 RNase-free DNase (M6101, Promega) as per manufacturer’s instructions, with the exception that the incubation time was increased to 60 min (at 37 °C). cDNA was synthesized using Random Hexamers™ and SuperScript™III Reverse Transcriptase system (LifeTechnologies). qRT-PCR reactions were prepared with in triplicate with three technical replications and samples were processed on a Lightcycler96® platform (Roche) using QuantiFast-Sybr®Green kit (Qiagen). Reaction setup was as follows: 2× QuantiFast SYBR GreenPCR Master Mix (1X), Primer forward (1 uM), Primer reverse (1 uM) and 100 ng of DNA per reaction, final volume of 25 µl was completed with RNase free water. Cycling parameters were as follows: PCR initial heat activation: 5 min at 95 °C; 2 step cycling (40 cycles): Denaturation for 10 sec at 95 °C and combined annealing/extension 30 secs at 60 °C, melting curve: 10 sec at 95 °C, 60 sec at 60 °C and 1 s at 95 °C. For the relative quantification of transcripts, the 2−ΔCt method119 of was used, with target gene expression normalized against the E. adhaerens OV14 secG gene using the following equation:
The association of the RNAseq data v. qRT-PCR outputs was determined with Pearson’s correlation in Microsoft excel® (P < 0.05) using the log2fold values obtained from both data series.
Data Availability
BAM file alignments for the 36 libraries were deposited in the Sequence Read Archives (SRA) of NCBI under accession number BioProject PRJNA479665 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA479665). An Excel file including all the CuffDiff results has been included (Supplementary Table 7).
References
ISAAA. ISAAA Briefs brief 39 Global status of Commercialized biotech/GM Crops: 2008. ISAAA Briefs 135, https://doi.org/10.1017/S0014479706343797 (2016).
Hooykaas, P., Klapwijk, P., Nuti, M., Schilperoort, R. & Rorsch, A. Transfer of the Agrobacterium tumefaciens TI Plasmid to Avirulent Agrobacteria and to Rhizobium ex planta. J. Gen. Microbiol. 98, 477–484 (1977).
Broothaerts, W. et al. Gene transfer to plants by diverse species of bacteria. Nature 433, 629–633 (2005).
Wendt, T., Doohan, F., Winckelmann, D. & Mullins, E. Gene transfer into Solanum tuberosum via Rhizobium spp. Transgenic Res. 20, 377–386 (2011).
Zuniga-Soto, E., Mullins, E. & Dedicova, B. Ensifer-mediated transformation: an efficient non-Agrobacterium protocol for the genetic modification of rice. Springerplus 4, 600 (2015).
Rathore, D. S., Doohan, F. & Mullins, E. Capability of the plant-associated bacterium, Ensifer adhaerens strain OV14, to genetically transform its original host Brassica napus. Plant Cell. Tissue Organ Cult. 127, 85–94 (2016).
Chavarriaga-Aguirre, P. et al. The potential of using biotechnology to improve cassava: a review. Vitr. Cell. Dev. Biol. - Plant 52, 461–478 (2016).
Wendt, T., Doohan, F. & Mullins, E. Production of Phytophthora infestans-resistant potato (Solanum tuberosum) utilising Ensifer adhaerens OV14. Transgenic Res. 21, 567–578 (2012).
Lacroix, B. & Citovsky, V. A Functional Bacterium-to-Plant DNA Transfer Machinery of Rhizobium etli. PLoS Pathog. 12, 1–15 (2016).
Anand, A. & Todd, J Advancing Agrobacterium-Based Crop Transformation and Genome Modification Technology for Agricultural Biotechnology. Curr. Top. Microbiol. Immunol. 19, https://doi.org/10.1007/82_2018_97 (2018).
Wang, L., Lacroix, B., Guo, J. & Citovsky, V. Transcriptional activation of virulence genes of Rhizobium etli. J. Bacteriol. JB.00841–16, https://doi.org/10.1128/JB.00841-16 (2017).
Rudder, S., Doohan, F., Creevey, C. J., Wendt, T. & Mullins, E. Genome sequence of Ensifer adhaerens OV14 provides insights into its ability as a novel vector for the genetic transformation of plant genomes. BMC Genomics 15, 268 (2014).
Ormeño-Orrillo, E. et al. Taxonomy of rhizobia and agrobacteria from the Rhizobiaceae family in light of genomics. Syst. Appl. Microbiol. 38, 287–291 (2015).
Douglas, C. J., Halperin, W. & Nester, E. W. Agrobacterium tumefaciens mutants affected in attachment to plant cells. J. Bacteriol. 152, 1265–1275 (1982).
Douglas, C. J., Staneloni, R. J., Rubin, R. A. & Nester, E. W. Identification and genetic analysis of an Agrobacterium tumefaciens chromosomal virulence region. J. Bacteriol. 161, 850–860 (1985).
Klee, H. J., White, F. F., Iyer, V. N., Gordon, M. P. & Eugene, W. Mutational analysis of the virulence region of an Mutational Analysis of the Virulence Region of an Agrobacterium tumefaciens Ti Plasmid. J. Bacteriol. 153, 878 (1983).
Stachel, S. E. & Nester, E. W. The genetic and transcriptional organization of the vir region of the A6 Ti plasmid of Agrobacterium tumefaciens. EMBO J. 5, 1445–54 (1986).
Gelvin, S. B. Agrobacterium-Mediated Plant Transformation: the Biology behind the “ Gene-Jockeying “ Tool. Microbiol. Mol. Biol. Rev. 67, 16–37 (2003).
Tzfira, T. & Citovsky, V. Agrobacterium-mediated genetic transformation of plants: biology and biotechnology. Current Opinion in Biotechnology 17, 147–154 (2006).
Citovsky, V. et al. Biological systems of the host cell involved in Agrobacterium infection. Cell. Microbiol. 9, 9–20 (2007).
Gelvin, S. B. Plant Proteins Involved in Agrobacterium-Mediated Genetic Transformation. Annu. Rev. Phytopathol 48, 45–68 (2010).
Bourras, S., Rouxel, T. & Meyer, M. Agrobacterium tumefaciens Gene Transfer: How a Plant Pathogen Hacks the Nuclei of Plant and Nonplant Organisms. Phytopathology 105, 1288–1301 (2015).
Gelvin, S. B. Integration of Agrobacterium T-DNA into the Plant Genome. Annu. Rev. Genet. 51, 195–217 (2017).
Bolton, G. W., Nester, E. W. & Gordon, M. P. Plant Phenolic Compounds Induce Expression of the Agrobacterium tumefaciens Loci Needed for Virulence. Sci. reports 232, 983–985 (1986).
Stachel, S. E., Nester, E. W. & Zambryski, P. C. A plant cell factor induces Agrobacterium tumefaciens vir gene expression. PNAS 83, 379–383 (1986).
Engstrom, P., Zambryski, P., Montagu, M. V. & Stachel, S. Characterization of Agrobacterium tumefaciens Virulence Proteins Induced by the Plant Factor Acetosyringone. J. Mol. Biol. 197, 635–645 (1987).
Pazour, G. J., Ta, C. N. & Das, A. Mutants of Agrobacterium tumefaciens with elevated vir gene expression (vir gene regulation/mutagenesis/virA mutants). Genetics 88, 6941–6945 (1991).
Gelvin, S. B. Agrobacterium and plant genes involved in T-DNA transfer and integration. Annu. Rev. Plant Physiol. Plant Mol. Biol 51, 233–256 (2000).
Ditt, R. F. et al. The Arabidopsis thaliana transcriptome in response to Agrobacterium tumefaciens. Mol. Plant. Microbe. Interact. 19, 665–81 (2006).
Anand, A. et al. Identification and characterization of plant genes involved in Agrobacterium-mediated plant transformation by virus-induced gene silencing. Mol. Plant-Microbe Interact. 20, 41–52 (2006).
Gohlke, J. & Deeken, R. Plant responses to Agrobacterium tumefaciens and crown gall development. Front. Plant Sci. 5 (2014).
Shih, P. et al. Differential Roles of Glucosinolates and Camalexin at Different Stages of Agrobacterium‐Mediated Transformation. Mol. Plant Pathol. 1–15, https://doi.org/10.1111/mpp.12672 (2018).
Capela, D., Filipe, C., Bobik, C., Batut, J. & Bruand, C. Sinorhizobium meliloti Differentiation During Symbiosis with Alfalfa: A Transcriptomic Dissection. Mol. Plant-Microbe Interact. 19, 363–372 (2006).
Gurich, N. & Gonza, J. E. Role of Quorum Sensing in Sinorhizobium meliloti -Alfalfa. Symbiosis. 191, 4372–4382 (2009).
Rathore, D., Doohan, F. & Mullins, E. Ensifer-mediated Arabidopsis thaliana Root Transformation (E-ART): A Protocol to Analyse the Factors that Support Ensifer-mediated Transformation (EMT) of Plant Cells. Bio-Protocol 7, 1–11 (2017).
Gelvin, S. B. Agrobacterium transformation of Arabidopsis thaliana roots: a quantitative assay. Methods Mol. Biol. 343, 105–113 (2006).
Yuan, Z., Liu, P., Saenkham, P., Kerr, K. & Nester, E. Transcriptome profiling and functional analysis of Agrobacterium tumefaciens reveals a general conserved response to acidic conditions (pH 5.5) and a complex acid-mediated signaling involved in Agrobacterium-plant interactions. J. Bacteriol. 190, 494–507 (2008).
Peters, N. K., Frost, J. W. & Long, S. R. A plant flavone, luteolin, induces expression of Rhizobium meliloti nodulation genes. Science (80). 233, 977–980 (1986).
Ampe, F., Kiss, E., Sabourdy, F. & Batut, J. Transcriptome analysis of Sinorhizobium meliloti during symbiosis. Genome Biol. 4, R15 (2003).
Zhang, X. & Cheng, H. Identification of Sinorhizobium meliloti Early Symbiotic Genes by Use of a Positive Functional Screen †. 72, 2738–2748 (2006).
Jones, K. M., Kobayashi, H., Davies, B. W., Taga, M. E. & Walker, G. C. How rhizobial symbionts invade plants: the Sinorhizobium–Medicago model. Nat. Rev. Microbiol. 5, 619–633 (2007).
Cerveny, L. et al. Tetratricopeptide repeat motifs in the world of bacterial pathogens: Role in virulence mechanisms. Infect. Immun. 81, 629–635 (2013).
Matthysse, A. G., Thomas, D. L. & White, A. R. Mechanism of cellulose synthesis in Agrobacterium tumefaciens. J. Bacteriol. 177, 1076–1081 (1995).
Sousa, Ślvia A., Feliciano, J. R. & Leitao, J. H. Activated Sugar Precursors: Biosynthetic Pathways and Biological Roles of an Important Class of Intermediate Metabolites in Bacteria. INTECH open 257–274, 10.5772163 (2011).
Stiens, M., Schneiker, S., Pühler, A. & Schlüter, A. Sequence analysis of the 181-kb accessory plasmid pSmeSM11b, isolated from a dominant Sinorhizobium meliloti strain identified during a long-term field release experiment. FEMS Microbiol. Lett. 271, 297–309 (2007).
Murphy, P. J., Wexler, W., Grzemski, W., Rao, J. P. & Gordon, D. Rhizopines-Their role in symbiosis and competition. Soil Biol. Biochem. 27, 525–529 (1995).
Krysciak, D. et al. RNA sequencing analysis of the broad-host-range strain sinorhizobium fredii NGR234 Identifies a large set of genes linked to quorum sensing-dependent regulation in the background of a trai and ngri deletion mutant. Appl. Environ. Microbiol. 80, 5655–5671 (2014).
Galbraith, M. P. et al. A functional myo-inositol catabolism pathway is essential for rhizopine utilization by Sinorhizobium meliloti. Microbiology 144, 2915–2924 (1998).
Jiang, G., Krishnan, A. H., Kim, Y. W., Wacek, T. J. & Krishnan, H. B. A functional myo-inositol dehydrogenase gene is required for efficient nitrogen fixation and competitiveness of Sinorhizobium fredii USDA191 to nodulate soybean (Glycine max [L.] Merr.). J. Bacteriol. 183, 2595–2604 (2001).
Higgins, C. F. ABC Transporters: From Microorganisms to Man. Annu. Rev. Cell Biol. 8, 67–113 (1992).
Zhao, J. & Binns, A. N. GxySBA ABC transporter of Agrobacterium tumefaciens and its role in sugar utilization and vir gene expression. J. Bacteriol. 196, 3150–3159 (2014).
Chen, S., Oldham, M. L., Davidson, A. L. & Chen, J. Carbon catabolite repression of the maltose transporter revealed by X-ray crystallography. Nature 499, 364–368 (2013).
Hooykas, P. & Baijersbergen, A. The Virulence System of Agrobacterium. Annu. Rev. Phytopathol. 32, 157–179 (1994).
Fronzes, R., Christie, P. J. & Waksman, G. The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7, 703–714 (2009).
Anand, A. et al. Patent WO2017040343A1 Ochrobactrum-mediated transformation of plants. 45 (2018).
Stachel, S. E. & Zambryski, P. C. virA and virG control the plant-induced activation of the T-DNA transfer process of A. tumefaciens. Cell 46, 325–333 (1986).
Chen, C. Y., Wang, L. & Winans, S. C. Characterization of the supervirulent virG gene of the Agrobacterium tumefaciens plasmid pTiBo542. MGG Mol. Gen. Genet. 230, 302–309 (1991).
Rogowsky, P. M., Close, T. J., Chimera, J. A., Shaw, J. J. & Kado, C. I. Regulation of the vir genes of Agrobacterium tumefaciens plasmid pTiC58. J. Bacteriol. 169, 5101–5112 (1987).
Winans, S. C., Kerstetter, R. A. & Nester, E. W. Transcriptional Regulation of the virA and virG Genes of Agrobacterium tumefaciens. J. Bacteriol. 170, 4047–4054 (1988).
Brencic, A., Xia, Q. & Winans, S. C. VirA of Agrobacterium tumefaciens is an intradimer transphosphorylase and can actively block vir gene expression in the absence of phenolic signals. 52, 1349–1362 (2004).
Cambia Labs. pCambia Vectors. BioForge Transbacter Project 3187, 1–13 (2015).
Sheikholeslam, S. N. & Weeks, D. P. Acetosyringone promotes high efficiency transformation of Arabidopsis thaliana explants by Agrobacterium tumefaciens. Plant Mol. Biol. 8, 291–298 (1987).
Godwin, I., Todd, G., Ford-Lloyd, B. & Newbury, H. J. The effects of acetosyringone and pH on Agrobacterium-mediated transformation vary according to plant species. Plant Cell Rep. 9, 671–675 (1991).
Gelvin, S. B. Agrobacterium virulence gene induction. Methods Mol. Biol. 343, 77–84 (2006).
Jones, J. D. G. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Nishimura, M. T. & Dangl, J. L. Arabidopsis and the plant immune system. Plant J. 61, 1053–1066 (2010).
Anand, A. et al. Salicylic Acid and Systemic Acquired Resistance Play a Role in Attenuating Crown Gall Disease Caused by Agrobacterium tumefaciens. PLANT Physiol. 146, 703–715 (2007).
Yuan, Z.-C. et al. The plant signal salicylic acid shuts down expression of the vir regulon and activates quormone-quenching genes in Agrobacterium. Proc. Natl. Acad. Sci. 104, 11790–11795 (2007).
Nonaka, S. et al. Ethylene production in plants during transformation suppresses vir gene expression in Agrobacterium tumefaciens. New Phytol. 178, 647–656 (2008).
Erbs, G. et al. Peptidoglycan and Muropeptides from Pathogens Agrobacterium and Xanthomonas Elicit Plant Innate Immunity: Structure and Activity. Chem. Biol. 15, 438–448 (2008).
Zipfel, C. et al. Perception of the Bacterial PAMP EF-Tu by the Receptor EFR Restricts Agrobacterium-Mediated Transformation. Cell 125, 749–760 (2006).
Block, A., Li, G., Qing Fu, Z. & Alfano, J. R. Phytopathogen type III effector weaponry and their plant targets. Brief. Bioinform. 11, 582–591 (2014).
Katz, A. & Orellana, O. Cell free protein synthesis. Protein Synthesis and the Stress Response. Intech 133 (2012).
Tsuda, K. et al. An efficient Agrobacterium-mediated transient transformation of Arabidopsis. Plant J. 69, 713–719 (2012).
Alquéres, S. et al. The bacterial superoxide dismutase and glutathione reductase are crucial for endophytic colonization of rice roots by Gluconacetobacter diazotrophicus PAL5. Mol. Plant. Microbe. Interact. 26, 937–45 (2013).
Fridovich, I. Superoxide dismutases. Annu Rev Biochem 44, 147–159 (1973).
Baron, C., Llosa, M., Zhou, S. & Zambryski, P. C. VirB1, a component of the T-complex transfer machinery of Agrobacterium tumefaciens, is processed to a C-terminal secreted product, virB1. J. Bacteriol. 179, 1203–1210 (1997).
Fernandez, D., Spudich, G. M., Zhou, X. R. & Christie, P. J. The Agrobacterium tumefaciens VirB7 lipoprotein is required for stabilization of VirB proteins during assembly of the T-complex transport apparatus. J. Bacteriol. 178, 3168–3176 (1996).
Sagulenko, V., Sagulenko, E., Jakubowski, S., Spudich, E. & Christie, P. J. Virb7 lipoprotein is exocellular and associates with the Agrobacterium tumefaciens T pilus. J. Bacteriol. 183, 3642–3651 (2001).
Zhao, Z., Sagulenko, E., Ding, Z. & Christie, P. J. Activities of virE1 and the VirE1 Secretion Chaperone in Export of the Multifunctional VirE2 Effector via an Agrobacterium Type IV Secretion Pathway. Society 183, 3855–3865 (2001).
Dym, O. et al. Crystal structure of the Agrobacterium virulence complex VirE1-VirE2 reveals a flexible protein that can accommodate different partners. Proc. Natl. Acad. Sci. USA 105, 11170–5 (2008).
Tzfira, T. & Citovsky, V. In Molecular Biology Inteligence unit 229, https://doi.org/10.1007/0-387-27747-1 (Plenium, 2005).
Tzfira, T. & Citovsky, V. In Agrobacterium: From Biology to Biotechnology 768 (2007).
García-Rodríguez, F. M., Schrammeijer, B. & Hooykaas, P. J. J. The Agrobacterium VirE3 effector protein: a potential plant transcriptional activator. Nucleic Acids Res. 34, 6496–6504 (2006).
Pitzschke, A. & Hirt, H. New insights into an old story: Agrobacterium-induced tumour formation in plants by plant transformation. EMBO J. 29, 1021–1032 (2010).
Berger, B. R. & Christie, P. J. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J. Bacteriol. 176, 3646–3660 (1994).
Adams, M. D., Wagner, L. M., Gibson, A. L. & Oxenderll, D. L. Nucleotide Sequence and Genetic Characterization Reveal Six Essential Genes for the LIV-I and LS Transport Systems of Escherichia coli. J. Biol. Chem. 265, 11436–11443 (1990).
Prell, J. et al. Legumes regulate Rhizobium bacteroid development and persistence by the supply of branched-chain amino acids. Proc. Natl. Acad. Sci. 106, 12477–12482 (2009).
Ding, Z. & Christie, P. J. Agrobacterium tumefaciens Twin-Arginine-Dependent Translocation Is Important for Virulence, Flagellation, and Chemotaxis but Not Type IV Secretion. J. Bacteriol. 185, 760–771 (2003).
Berks, B. C., Sargent, F. & Palmer, T. The Tat protein export pathway. Mol Microbiol. 35, 260–274 (2000).
Schaerlaekens, K. et al. Comparison of the Sec and Tat secretion pathways for heterologous protein production by Streptomyces lividans. J. Biotechnol. 112, 279–288 (2004).
Leung, K. Y., Siame, B. A., Snowball, H. & Mok, Y. K. Type VI secretion regulation: Crosstalk and intracellular communication. Curr. Opin. Microbiol. 14, 9–15 (2011).
Merritt, P. M., Danhorn, T. & Fuqua, C. Motility and chemotaxis in Agrobacterium tumefaciens surface attachment and biofilm formation. J. Bacteriol. 189, 8005–8014 (2007).
Heindl, J. E. et al. Mechanisms and regulation of surface interactions and biofilm formation in Agrobacterium. Front. Plant Sci. 5 (2014).
Amaya-Gómez, C. V., Hirsch, A. M. & Soto, M. J. Biofilm formation assessment in Sinorhizobium meliloti reveals interlinked control with surface motility. BMC Microbiol. 15, 58 (2015).
Lohrke, S. M., Yang, H. & Jin, S. Reconstitution of Acetosyringone-mediated Agrobacterium tumefaciens virulence gene expression in the heterologous host Escherichia coli. J. Bacteriol. 183, 3704–3711 (2001).
Fong, J. C. N. & Yildiz, F. H. Interplay between cyclic AMP-cyclic AMP receptor protein and cyclic di-GMP signaling in vibrio cholerae biofilm formation. J. Bacteriol. 190, 6646–6659 (2008).
Salazar, J. K. et al. Roles of a Novel Crp/Fnr Family Transcription Factor Lmo0753 in Soil Survival, Biofilm Production and Surface Attachment to Fresh Produce of Listeria monocytogenes. PLoS One 8 (2013).
Lu, X. H. et al. RsmA Regulates Biofilm Formation in Xanthomonas campestris through a Regulatory Network Involving Cyclic di-GMP and the Clp Transcription Factor. PLoS One 7 (2012).
Torres-Escobar, A., Juárez-Rodríguez, M. D., Lamont, R. J. & Demuth, D. R. Transcriptional regulation of aggregatibacter actinomycetemcomitans lsrACDBFG and lsrrk operons and their role in biofilm formation. J. Bacteriol. 195, 56–65 (2013).
Kalivoda, E. J., Stella, N. A., O’Dee, D. M., Nau, G. J. & Shanks, R. M. Q. The cyclic AMP-dependent catabolite repression system of Serratia marcescens mediates biofilm formation through regulation of type 1 fimbriae. Appl. Environ. Microbiol. 74, 3461–3470 (2008).
Wright, E. L., Deakin, W. J. & Shaw, C. H. A chemotaxis cluster from Agrobacterium tumefaciens. Gene 220, 83–89 (1998).
Shaw, C. H. Swimming Against the Tide: Chemotaxis in Agobacterium. BioEssays 13, 25–29 (1991).
Deakin, W. J., Sanderson, J. L. C. I., Goswami, T. & Shaw, C. H. The Agrobacterium tumefaciens motor gene, motA, is in a linked cluster with the flagellar switch protein genes, fliG, fliM and fliN. Gene 189, 139–141 (1997).
Wadhams, G. H. & Armitage, J. P. Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 5, 1024–1037 (2004).
Harighi, B. Role of cheY1 and cheY2 in the chemotaxis of A. tumefaciens toward acetosyringone. Curr. Microbiol. 56, 547–552 (2008).
Riepl, H. et al. Interaction of CheY2 and CheY2-P with the cognate CheA kinase in the chemosensory-signalling chain of Sinorhizobium meliloti. Mol. Microbiol. 69, 1373–1384 (2008).
Bastiaansen, K. C., Ibañez, A., Ramos, J. L., Bitter, W. & Llamas, M. A. The Prc and RseP proteases control bacterial cell-surface signalling activity. Environ. Microbiol. 16, 2433–2443 (2014).
Draper, R. C., Martin, L. W., Beare, P. A. & Lamont, I. L. Differential proteolysis of sigma regulators controls cell-surface signalling in Pseudomonas aeruginosa. Mol. Microbiol. 82, 1444–1453 (2011).
Su, S., Stephens, B. B., Alexandre, G. & Farrand, S. K. Lon protease of the alpha-proteobacterium Agrobacterium tumefaciens is required for normal growth, cellular morphology and full virulence. Microbiology 152, 1197–1207 (2006).
Summers, M. L., Botero, L. M., Busse, S. C. & McDermott, T. R. The Sinorhizobium meliloti Lon protease is involved in regulating exopolysaccharide synthesis and is required for nodulation of alfalfa. J. Bacteriol. 182, 2551–2558 (2000).
Zamioudis, C. & Pieterse, C. M. J. Modulation of Host Immunity by Beneficial Microbes. Mol. Plant-Microbe Interact. 25, 139–150 (2012).
Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L. & Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (2011).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc., https://doi.org/10.1038/nprot.2012.016 (2012).
Li, W. & Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28, 2000 (2000).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45, 353–361 (2017).
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K. & Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res 8, 590–595 (2019).
Schmittgen, T. & Livak, K. Analyzing real-time Analyzing real-time PCR data by the comparative C T method. 6, 1101–1108 (2008).
Acknowledgements
This publication is an output from research conducted with the financial support of Science Foundation Ireland under grant number SFI 11/RFP.1/GEN/3420 and the Teagasc Walsh Fellowship Scheme (Grant reference number 2011210).
Author information
Authors and Affiliations
Contributions
E.Z. was involved in the study as well as experimental design, performed experimental work, carried out data analysis and drafted the manuscript. D.F. and E.Z. carried out main RNAseq data analysis. F.D. and E.M. contributed to study design, experimental design, funding and drafting of the manuscript. All authors contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
E. Mullins and F. Doohan are authors of US Patent No. 9,365,858 and No. 9,822,375 which details the use of an isolated Ensifer adhaerens strain OV14 deposited under NCIMB Accession Number 41777 as a gene delivery system in the genetic transformation of plant material. Our manuscript has in no way been affected by this fact, nor has our participation in the work influenced in any manner the analysis of the generated datasets and/or the conclusions drawn.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zuniga-Soto, E., Fitzpatrick, D.A., Doohan, F.M. et al. Insights into the transcriptomic response of the plant engineering bacterium Ensifer adhaerens OV14 during transformation. Sci Rep 9, 10344 (2019). https://doi.org/10.1038/s41598-019-44648-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44648-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
