
Abstract
Fourteen juvenile scalloped hammerhead sharks (Sphyrna lewini; SHS) were captured between November and December 2014 in the Rewa Delta in Fiji, and assessed for intestinal microflora characterisation using 16S rRNA amplicon sequencing by Illumina Miseq. The microbial population revealed a fluctuating dominance between the Enterobacteriaceae and Vibrionaceae families, namely Citrobacter and Photobacterium spp. Other related marine operational taxonomic units were closely related to Afipia felis, Chloroflexus aggregans, Psychrobacter oceani, Pontibacter actiniarum and Shigella sonnei. Two sharks had distinctive profiles that were dominated by known pathogens, namely Aeromonas salmonicida and Klebsiella pneumonia. The presence of a Methanosaeta species, and of Shigella and Psychrobacter, would suggest sewage contamination because of a spill that occurred on the 6th of December 2014. This study successfully establishes a baseline for future research.
Similar content being viewed by others
Introduction
The scalloped hammerhead shark (SHS), Sphyrna lewini, is a circumglobally distributed large apex predator species common to tropical and warm temperate coastal and semi-pelagic marine environments. Due to overfishing and habitat destruction, the species is considered to be among the most globally threatened sharks and was declared endangered by the International Union for Conservation of Nature (IUCN) Red List in 20071. It is also currently listed on Appendix II of the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES)2. Several SHS populations have been heavily exploited worldwide by both inshore and offshore fisheries3.
In Fiji waters, the species has been reported to aggregate in the Austral summer in seven estuarine areas on the main islands of Viti and Vanua Levu4. Marie, et al.5, recently confirmed that the Rewa Delta is a critical habitat for SHS by documenting its year-round presence. However, they found significant seasonality of occurrence and abundance in the area, with a parturition period during the wet austral spring and summer seasons between October and March with a population peak between December and February. Subsequently, Vierus, et al.6 showed that neonates and juveniles of SHS also aggregate in the Ba estuary during the same period. The evidence presented in these studies unequivocally suggest the need for developing and strengthening shark conservation and management measures within Fiji, as several estuaries may well be nursery areas for SHS. As is common for estuaries, the Rewa Delta (Fig. 1) is characterised by large fluctuating salinities, a fresh water layer, especially during the Austral summer, high turbidity, and an abundance of small crustaceans, fish, eels, and shellfish7. These conditions are characteristic of shark nurseries, where high levels of primary production increase population productivity and enhance young sharks’ chances of survival5,8,9.

Geographical location of sampling sites in the Rewa Delta, and location of sewage discharge during the spillage that occurred in December 2014, Viti Levu, Fiji (The map has been prepared by Mr Sione Kaituu using online resources from QGIS, version 3.6.0 https://www.qgis.org/en/site/). Grey dots reflect specific places where samples were collected, and the diameter is proportional to the number of sharks taken at that particular site.
Detailed knowledge of SHS ecology, behaviour and habitat requirements, particularly during the first stage of their life, is still limited10. In addition, there are indications that contamination by human and animal waste from villages lining the riverbanks may have shifted the balance of the ecosystem of the Suva Lagoon by increasing nutrient and bacterial load in water and organisms11,12,13. The main contributors of nutrients in Laucala Bay associated with sewage are the effluent of the Kinoya sewage treatment plant released at the sea outfall located in Laucala Bay (Fig. 1), and the human and animal waste from villages on the banks of the Rewa River12. Fish and most shellfish are directly affected by degraded environments through their static feeding behaviour, while juvenile scallop hammerhead sharks experience low abundance of food or consume contaminated prey14.
Fish diseases caused by enteric bacteria have been reported in eutrophic waters associated with faecal pollution15, thus sharks feeding on prey living in sewage-polluted waters would reflect the bacterial load present in those waters. Studying the effect of the microbiome in conjunction to other factors is important in evaluating the environmental quality of critical habitats of endangered species. There are many ways to monitor pollution in the marine ecosystem, one of which is the use of indicator microorganisms. For example, faecal coliform such as Escherichia coli are indicators of contamination of water with faecal matter from humans or warm-blooded animals. This further implies that other pathogenic bacteria belonging to species of Salmonella, Shigella, Pseudomonas, and Streptococcus can also be present14,16. The intestine constitutes an ideal niche for microorganisms due to its readily available source of carbon, minerals, and solutes that are conducive to growth. Because, there is evidence that the microbial colonisation of the intestine of vertebrates starts after hatching or birth, it is influenced by the environmental factors that surround the habitat where a newly hatched or born individual begins to feed17. Consequently, investigation of bioindicators in aquatic animals usually involves the characterisation of the intestinal microbial profile at different life stages17,18.
For this bioindicator method to be successful, a baseline needs to be established to differentiate between normal colonisers and potential pathogens. Emerging molecular methods for analysing microbial communities allow high-resolution assessments of complex communities. Such protocols usually include culture-independent microbial profiling based on 16S ribosomal RNA (16S rRNA), which is not limited by cultivability, and can often detect even the least abundant members of the microbial community19. To the best of our knowledge, no data on the intestinal microbial profile of scallop hammerhead sharks are available in the literature, while studies on the gut microbiome of other shark species are limited. Exceptions are for striped burrfish spinner sharks (Carcharhinus brevipinna), atlantic sharp nose sharks (Rhizoprionodon terraenovae), and sandbar sharks (Carcharhinus plumbeus)20,21,22,23.
As part of ongoing projects to understand and protect critical shark habitats in Fiji, this study generates baseline data about the intestinal microbial profile of a representative sample of SHS from the Rewa Delta using 16S rRNA Illumina MiSeq amplicon sequencing (MiSeq). The database thus generated should contribute to the reference library of intestinal colonisers of juvenile SHS and possibly support further studies on trends in microbiological communities and the identification of bioindicator microorganisms as impacted by pollution of the waterways.
Results
Bacterial diversity profile
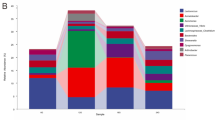
Since ANOSIM indicated that the variability among the technical repeats was negligible (R = 0.602, P = 0.001), an average was used to compute the bacterial diversity of each shark, which will be referred to by their catch number. The major microbial communities were identified to the family level and analysed for percentage relative abundance, with greater focus given to those that make at least 10% of the community in a minimum of one shark (Fig. 2). Enterobacteraceae was detected in high abundance in most sharks, while Vibrionaceae was seen to be more dominant in most sharks collected after the 9th of December 2014, namely SHS: 251 (44.2%), 265 (43.6%), 268 (17.5%) and 269 (76.0%). Samples from another shark had a distinctive profile: SHS 229 demonstrated an average of 68.8% relative abundance of Aeromonadaceae. The Shannon Index (1.07 ± 0.51) and Simpson index (0.39 ± 0.02) for SHS 229 was also lower in comparison with the other samples (Table 1), with a Chao 1 estimates of 4.67 ± 0.67, indicating less species richness and evenness. Similar observations were made for SHS 268 (Shannon Index:1.12 ± 0.61, Simpson Index: 0.37 ± 0.04 and Chao 1 estimate: 5.50 ± 0.50) and SHS 249 (Shannon Index:1.73 ± 0.73, Simpson Index: 0.50 ± 0.11 and Chao 1 estimate: 10.33 ± 2.18). A more diverse and evenly distributed microbial profile was attributed to SHS 251 (Shannon Index: 3.29 ± 0.10, Simpson Index: 0.85 ± 0.04 and Chao 1 estimate: 23.25 ± 5.41). In addition, members of the Moraxellaceae, Bradyrhizobiaceae, Pseudomonadaceae, Rhodobacteraceae, Staphylococcaceae, and Streptococcaceae were also detected among these samples.

Bacterial community profiles, including percentage relative abundances of the all identified taxonomic bacterial groups specified to the family level, found in selected juvenile hammerhead sharks captured in 2014 on the (a) 20–21 November, (b) 1 December, (c) 8–9 December. (d) 12 December and (e) 22 December, in the Rewa delta. The 2 OTUs with the highest relative abundance (Enterobacteriaceae and Vibrionaceae) and the OTU highly distinguished in SHS 229 (Aeromonadaceae) are highlighted in red in the figure legend.
Further comparison between the microbial profiles of the juvenile SHS sharks is illustrated in a Heatmap (Fig. 3). The Enterobacteriaceae microbes were further classified with major species of the genera Citrobacter, Shigella and Klebsiella. A bacterium closely related to the Citrobacter koseri (Accession No: HQ992945.1) was the most prevalent in most samples, ranging from a highest percentage relative abundance of 41.0% in SHS 194 to as low as 0.11% in SHS 268. While the presence of Shigella sp. (Accession No: KF225561.1) was observed in all samples at varying percentages, the other member of the Enterobacteriaceae family, associated with Klebsiella pneumoniae (Accession No: CP003200.1), was detected at highest relative abundance (19.9%) only in SHS 251. The data further suggested that the Vibrionaceae family is comprised mostly of Photobacterium spp. Other species associated with the top 15 operational taxonomic units (OTUs) identified in this experiment were Afipia spp. (Bradyrhizobiaceae), Cetobacterium spp. (Fusobacteriaceae), Chloroflexus spp. (Chlorothrixaceae), Psychrobacter spp. (Moraxellaceae) and Propionibacterium spp. (Propionibacteriaceae).

Heatmap of intestinal microbial communities in sharks with top 15 OTUs (relative abundance of at least 10% in a minimum of one sample). Samples are identified by their SHS capture number.
Compilation of the closely related species to the top five OTUs, in order of highest percentage relative abundance for each SHS (Table 2), indicated that either a Citrobacter sp. or a Photobacterium sp. was the top OTU. Also, commonly detected were Shigella spp. Afipia spp. and Chloroflexus spp. This analysis also revealed distinctive populations in three SHS, including SHS 229, where the top OTU was Aeromonas spp. (68.8%). As already mentioned, Klebsiella sp. (19.9%) was seen in SHS 251, which also had Cetobacterium sp. (16.6%) as the second most relative abundant species. The emergence of a Shewanella sp. (10.5%) was observed in SHS 268, the microbial profile of which was dominated by Photobacterium sp. (76.0%). Staphylococcus and Streptococcus species were also detected, but in lower relative abundance. For example, among the top 10 OTUs, a Staphylococcus sp. closely related to Staphylococcus warneri (Accession No: KY623039.1) was linked to SHS: 169 (1.0%), 171 (1.0%) and 194 (1.6%). Similarly, a Streptococcus sp. linked to Streptococcus tangierensis (Accession No: KF999656.1) was shown in profiles for SHS 190 (4.4%), 194 (2.4%), and 230 (2.5%).
Changes in microbial profiles
The emergence of a member of the Methanosaetaceae family was the only archaea detected in low relative abundance in one shark captured on the 12th of December 2014 (SHS 251 - 0.4%), and in 2 out of the 3 sharks captured on the 22nd of December 2014 (SHS 265 - 0.5% and SHS 269 - 0.14%). These data were included in the Principal Component Analysis (PCA) against external factors of time of capture, diet, shark individuality and umbilical site (Fig. 4). Samples collected from 20th of November 2014 to 9th of December 2014, mostly clustered together in the PCA, while a shift to the right was observed from the 12th of December 2014. Individuals SHS 249, SHS 251, SHS 262, SHS 268 and SHS 269, concurred with the dominance of Photobacterium sp. and, in some samples, the emergence of the archaeal methanogen. BLAST analysis related the Methanosaetaceae with a species of the genus Methanosaeta (Accession No: HM972512.2). In addition, a shift along PC2 was observed for SHS 229 sampled on the 8th of December 2014, suggesting it to be radically different from all other samples. ANCOVA (Table 3) confirmed that no significant shift could be attributed to the age of the individual, estimated through the degree of healing of the umbilical scars (see Methods) and the diet at the time of capture. However, a trend was observed within the shark population (R2 = 0.9, p = 0.001, Table 3) and with the date it was caught (R2 = 0.5, p = 0.001, Table 3) (Fig. 5).

Principal Component analysis of the OTUs for individual juvenile SHS, captured on different dates (differentiated by colour) during November and December 2014, with varying degree of healing of the umbilical site (differentiated by shape), showing a shift along PC1 with samples collected after the 9th of December namely SHS 249 (purple triangle), SHS 251 (purple squares, SHS 265 (pink circles close to the cluster), SHS 268 (pink triangles) and SHS 262 (pink circles furthest to the cluster). Also shown is a shift along PC2 for sample SHS 229 (mint green circles).

Component scores generated by Principal Component Analysis (PCA), with time of catch as the main factor, to determine the correlation between 11 major bacterial species closely related to the identified OTUs in the intestine of SHS, over a 32 days period.
The component scores further indicated a positive correlation with time for Photobacterium damselae (Accession No: MH423606.1), Shewanella baltica (Accession No: CP002383.1), Aeromonas salmonicida (Accession No: LT628040.1), and Cetobacterium somerae (Accession No: MG428863.1). The opposite was observed for C. koseri, Shigella sonnei, Afipia felis (Accession No: HF970590.1), Chloroflexus aggregans (Accession No: CP001337.1), Psychrobacter oceani (Accession No: MH989594.1), S. tangierensis, and Micrococcus caseolyticus (Accession No: MG996517.1) (Fig. 5).
Discussion
Our investigation on the intestinal microbiome of juvenile SHS from the Rewa Delta showed a diverse microbial community, with bacteria including members of the families Enterobacteraceae, Vibrionaceae, Propionibacteriaceae, Aeromonadaceae, Moraxellaceae, Bradyrhizobiaceae, Rhodobacteraceae, Staphylococcaceae, Streptococcaceae, Methanosaetaceae, Bradyrhizobiaceae, Fusobacteriaceae, Chlorothrixaceae, Moraxellaceae and Pseudomonadaceae. These are commonly known intestinal inhabitants of terrestrial and marine vertebrate species including humans. Major members of the groups identified in this study have also previously been found in Sharpnose, Spinner and Sand sharks, although their role as gut microbiota has yet to be confirmed23,24. While many have been associated with digestive physiology in other species, research in such areas for shark species is limited25, thus making accurate determination of their function difficult.
Two OTUs accounted for a major proportion of microbial communities in all tested sharks, which BLAST analysis related to C. koseri and P. damselae, belonging to the Enterobacteriaceae and Vibrionaceae family, respectively. Both taxa have previously been associated with elasmobranch species such as bull, tiger, sharpnose, spinner and sandbar sharks20,21,22,23. C. koseri is a gram-negative anaerobic bacterium that has yet to be associated with the gut microbiome of sharks, but has been linked with their oral microbial communities and infections in humans that resulted from shark bites (as have S. warneri) and low water quality in the marine environment20,26. While C. koseri has been shown to have the ability to digest glucose, the importance of this function in the gut of SHS is yet to be established27. In comparison, P. damselae is believed to be a normal member of some sharks’ gut microorganisms, such as the sharpnose, spinner, and sandbar sharks23. In this study, both are presented as part of the intestinal habitat of the juvenile SHS due to their presence in all sharks tested. The fluctuation of dominance between these two species, over time, may reflect an environmental shift, such as changes in temperature, or food and water quality. Both species are also known opportunistic pathogens, with P. damselae responsible for ulcers and haemorrhagic septicaemia in brown sharks, dolphins, and shrimps22. It is important to note that the species level identification in this study relies on BLAST matches to partial 16S rRNA sequences; therefore, it only serves as a guidance for further investigations in an attempt to validate the presence of these potential pathogens with appropriate marker genes.
Other OTUs identified and previously seen in marine taxa were A. felis, C. aggregans, P. adeliensis (Accession No: AJ539105.1), P. actiniarum (Accession No: CP021235.1) and S. sonnei. While not directly linked to sharks, A. felis bacteria have been associated with infection in free-living amoeba and are often isolated from hospital water28,29,30. C. aggregans are phototrophic bacteria native to marine environment and are often associated with ‘microbial mats’31,32. The genus Pontibacter belongs to the phylum Bacteroidetes, members of which are common colonisers of fish intestine and include the isolate P. actiniarum, first characterised from the Sea of Japan as a gram-negative, aerobic bacterium33. If not present since birth, published information would suggest that these bacteria originate from preys ingested and could thus colonised the gut of the juvenile SHS sharks.
Two bacterial species dominated the intestinal microbiota of two individual shark sampled. A closer analysis with BLAST related them to A. salmonisidas in SHS 229, and K. pneumoniae in SHS 248. Both microbes are described as opportunistic pathogens associated with nosocomial respiratory tract and urinary tract infections in humans and they were observed in lesions found in the gills and intestine of a dead black tip reef shark15,34,35,36. Being two isolated cases, the probability of these bacteria being indigenous to the intestinal microbial community of juvenile SHS is quite low. A possible explanation could be that these sharks were diseased, with the said pathogens eventually dominating the gut microbial biota.
A temporal shift in community was observed in the PCA plot, towards Methanosaeta sp. and P. damsalea. Date of capture was identified as a significant factor affecting PC1. The emergence of Methanosaeta sp. was a major contributor to PC1. There is no indication of the presence of this methanogen as a native archaeon in the gut microbiome of any shark species studied so far. However, the role of these archaea, namely Methanosaeta concilii, in waste degradation, is well documented37,38 and its presence has previously indicated sewage or effluent contamination of waterways7,11. It was later revealed that, midway through this study (6th of December 2014), a major sewage spill occurred in the Cunningham River, Suva, which discharged about 200 Ls−1 of untreated waste water into Laucala Bay. This discharge continued unabated for 18 days, until temporary control measures were implemented that led to a Government Environmental Emergency Declaration that prohibited swimming and fishing in the affected waters. Even though this discharge released untreated sewage to Laucala Bay and not to the Rewa Delta, which is about 6 km away and in the opposite direction of the trade winds and prevalent currents, the fact that we find a shift in the microbial community after the spill could be explained by two mechanisms. First, contaminated prey may have moved from Laucala Bay to the Rewa Delta via the Vunidawa River that connects Laucala Bay or second, newly born and juvenile individuals move in search of prey and fed in areas reached by the sewage12.
With this incident in mind, a preliminary screening of the results was carried out for other potential evidence of sewage pollution. The presence of indicator species such as P. adeliensis and S. sonnei was considered. Originally isolated from fast ice around the Antarctica region, strains of Psychrobacter spp. have been proven effective indicators of pollution in sites with industrial, agricultural and urban effluents39. It has also been isolated from marine taxa and other aquatic environments contaminated with hydrocarbons40,41. S. sonnei requires specific pH and temperature ranges to survive, and its ideal host is the human gastrointestinal tract. Shigellosis has always been associated with contaminated water, as well as contaminated seafood, and is spread easily in crowded and unhygienic conditions42,43,44. The presence of these species in all of the samples might imply that they may be indigenous to this shark species, or could also be an indicator of constant contamination of the waters of the Rewa Delta with existing pollutants associated with nearby agricultural and sewage effluents. Constant contamination from untreated sewage has been reported previously in Laucala Bay and Rewa River and is associated with two main factors. First, the Kinoya sewage treatment plant was built for a population of 77,000 persons but nowadays it supports a population of about 120,000. Second, more than 40% of the main Suva population still uses septic tanks without the ability to remove nutrients and pathogens12. Furthermore, some microbial growth is known to be favoured by sewage effluents that increase organic content and nutrient concentrations, as well as decreases in salinity16, which might explain the shift in microbe community in sharks after the sewage spill observed in juvenile SHS caught after the 9th of December 2014.
Conclusions
We have used culture-independent molecular techniques for the characterisation of the intestinal microbial community of fourteen juvenile SHS from a nursery site of the Rewa estuary in the Fiji Island of Viti Levu. This initial study provides baseline information previously lacking for this species in Fiji and in the South Pacific region. While most of the bacteria characterised have previously been identified in other shark or marine species, the bacterial community also included many known opportunistic pathogens. Determining whether these bacterial pathogens are part of the indigenous intestinal microbiome of SHS warrants further investigation. The unfortunate sewage spill that occurred during the sampling period could account for the presence of some known indicator microorganisms, namely Methanosaeta spp., Shigella spp. and Psychrobacter spp. It indicates that this technique can successfully identify bioindicator microorganisms associated with polluted environments.
Methods
Study site
The Rewa Delta (RD) (178.55°E, −18.15°S, Fig. 1) is the largest fluvial system in the Fiji Islands; it is found in the largest island of the country, Viti Levu, and it originates from the Rewa River, Fiji’s longest river. The RD is characterised by strong currents and high wave actions because of the collision between river runoff and incoming waves/tides via the reef channel. This interaction gives the RD estuarine habitat conditions, such as large fluctuating salinities, a freshwater layer, high turbidity, and tidal waves7,45, which collectively make 45% of the RD inaccessible for sampling.
Sampling
Sampling sites for the current study were located on the RD, and encompassed one third of total Rewa Delta (Fig. 1). Local licensed fishermen caught the sharks studied in this experiment accidentally as bycatch during their regular fishing trips, thus no animals were intentionally sacrificed for the purpose of the current study and no IACUC or equivalent was needed. Three sharks were collected within a day of each other on three different periods, as illustrated in Table 4, except for November 2014, when only 2 sharks were available. The sharks were preserved on ice during transport for a maximum of 3 hours before immediate deep freezing at −80 °C upon return to the laboratory. Prior to DNA extraction, the intestine of each shark, including the proximal, spiral and distal region, excluding the stomach, were isolated from the specimen and any food content carefully removed and visually inspected and recorded for another study. The intestine was further cut into pieces, mixed, and separated to make 3 technical repeats for DNA extraction.
DNA extraction
DNA extraction was performed according to the Council for Scientific and Industrial Research (CSIR) protocol for the lysis of Corynebacterium species, with modifications46,47,48. In brief, approximately 2 g of the mixed intestinal sample was added to 500 μl of lysis buffer (20 mM Tris-HCL at pH 8.5, 2 mM EDTA at pH 8.0), with an additional 20 mg·ml−1 lysozyme (Thermofischer Scientific). The mixture was then incubated for at least an hour at 37 °C in a waterbath before 50 μg ml−1 of Proteinase K (Thermofischer Scientific) was added to the mixture and incubated for 30 mins. Sodium dodecyl sulphate (SDS) was added (10 ml; 20% v/v) and followed by further incubation at 65 °C for 90 mins. The supernatant was collected after centrifugation at 6 000 × g for 10 mins at room temperature, mixed with an equal volume of chloroform isoamyl alcohol (24:1 v/v), and incubated for 1 min at room temperature. The mixture was centrifuged again and the resulting supernatant was precipitated with 60% of its volume of isopropanol for 60 mins at room temperature. After centrifugation at 16 000 × g for 10 mins, the resulting pellet of crude nucleic acid was washed with 500 µl of 70% cold ethanol. The extracted genomic DNA was resuspended in Tris-EDTA buffer (10 mM Tris and 1 mM EDTA, pH 7.6) and stored at −20 °C. DNA extraction was carried out in triplicates for each individual shark sample. Extracted DNA was quantified by the Qubit® 3.0 Fluorometer.
Amplicon sequencing and data analysis
Amplification of the 16S rRNA gene region was verified using a universal primer set of 27 F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1392R (5′-ACGGGCGGTGTGTRC-3′)37 before submission of DNA to the Australian Centre for Ecogenomics (ACE, University of Queensland) for paired-end 16S rRNA amplicon sequencing by Illumina Miseq (Illumina Inc., USA). The amplification encompassed the V5 to V8 region of the 16S rRNA gene, using specific primers 803F (5′-TTAGANACCCNNGTAGTC-3′) and 1392Wr (5′-ACGGGCGGTGWGTRC-3′) containing Illumina adapter sequence as modified by ACE (University of Queensland, Australia).
Paired end sequencing data (ACE, University of Queensland), grouped to operational taxonomic units (OTUs) at 97% similarity, and aligned with the 16S rRNA identified sequences in the Greengenes database via the Quantitative Insights Into Microbial Ecology (QIIME) (Version 1.8.0) software package was received (ACE, University of Queensland). The resulting data were further processed in QIIME to calculate the alpha diversity including both the Shannon and Simpson indices as well as Chao 1 estimates, which is based on abundance. OTUs were further classified at the species level by BLAST analysis at 100% similarity (http://www.ncbi.nlm.nih.gov/). It is important to note that such identifications are subject to change over time should new sequences, with closer relationships, be uploaded in the database. A raw OTUs table was imported into R (v3.2.3) (R Core Team, 2015) and rarefied by function “rarefy_even_depth” of package ‘phyloseq’ (McMurdie and Holmes, 2013). The external factors of date, diet at the time of capture, shark individuality could potentially create a shift in the intestinal microbiome. In addition, at different stages of growth, juvenile SHS, as other viviparous sharks, either have the umbilical scar open, semi-open or healed. They could be more susceptible to influence from outside microorganisms if this scar is open or semi-open. Principle Component Analyses (PCAs) were performed on hellinger-adjusted OTUs tables and the above-mentioned factors as variables, with package ‘ampvis’49. Bar charts were generated in Microsoft Excel to illustrate the changes in the community profiles between the samples.
Statistical and correlation analysis
Analysis of variance in mixed categorical/continuous mode (ANCOVA) was done to test the significance of environmental and host biological parameters on microbial community using the function “Anova” of package ‘car’ in R. Specifically, PC values of overall communities were used as output, date of capture was treated as a coded continuous factor, and shark individuality, diet, umbilical scar as categorical factors and numerical variables, including weight as a continuous factor. The model was tested to identify the most parsimonious, by elimination of non-significant factors to the minimum parameter. A significance threshold of 0.05 was applied for rejection of the null hypothesis. Analysis of similarity (ANOSIM) was also conducted on microbial community profile between and within triplicate analysis of individual sharks with package ‘vegan’ in R (v3.2.3) (R Core Team, 2015).
References
Baum, J. et al. Sphyrna lewini (Northwest and Western Central Atlantic subpopulation). The IUCN Red List of Threatened Species 2007: e.T165293A6000960, https://doi.org/10.2305/IUCN.UK.2007.RLTS.T165293A6000960.en (2007).
CITES. cites.org. Available at, https://cites.org/eng/app/appendices.php Accessed 04 February 16 (2015).
Compagno, L. J. V. FAO species catalogue. Vol.4. Sharks of the world. An annotated and illustrated catalogue of shark species known to date. Part 2. Carcharhiniformes., Vol. 4 251–655 (FAO Fish Synop., 1984).
Rasalato, E., Maginnity, V. & Brunnschweiler, J. M. Using local ecological knowledge to identify shark river habitats in Fiji (South Pacific). Environmental Conservation 37, 90–97, https://doi.org/10.1017/s0376892910000317 (2010).
Marie, A. D., Miller, C., Cawich, C., Piovano, S. & Rico, C. Fisheries-independent surveys identify critical habitats for young scalloped hammerhead sharks (Sphyrna lewini) in the Rewa Delta, Fiji. Scientific Reports 7, 17273, https://doi.org/10.1038/s41598-017-17152-0 (2017).
Vierus, T. et al. Discovery of a multispecies shark aggregation and parturition area in the Ba Estuary, Fiji Islands. Ecology and Evolution (2018).
Singh, A. & Aung, T. Salinity, Temperature and Turbidity Structure in the Suva Lagoon, Fiji. American Journal of Environmental Sciences 4, 266–275 (2008).
Heupel, M. R., Carlson, J. K. & Simpfendorfer, C. A. Shark nursery areas: concepts, definition, characterization and assumptions. Marine Ecology Progress Series 337, 287–297 (2007).
Brown, K. T., Seeto, J., Lal, M. M. & Miller, C. E. Discovery of an important aggregation area for endangered scalloped hammerhead sharks, Sphyrna lewini, in the Rewa River estuary, Fiji Islands. Pacific Conservation Biology 22, 242–248, https://doi.org/10.1071/PC14930 (2016).
Hoffmayer, E. R., Franks, J. S., Driggers, W. B. III. & Howey, P. W. Diel vertical movements of a scalloped hammerhead, Sphyrna lewini, in the northern Gulf of Mexico. Bulletin of Marine Science 89, 551–557, https://doi.org/10.5343/bms.2012.1048 (2013).
Veitayaki, J. Vol. 1 229 (Institute of Pacific Studies, University of the South Pacific, 1994).
Singh, S., Aalbersberg, W. G. L. & Morrison, R. J. Nutrient Pollution in Laucala Bay, Fiji Islands. Water, Air, and Soil Pollution 204, 363–372, https://doi.org/10.1007/s11270-009-0050-8 (2009).
Morrison, R. J., Narayan, S. P. & Gangaiya, P. Trace Element Studies in Laucala Bay, Suva, Fiji. Marine Pollution Bulletin 42, 397–404, https://doi.org/10.1016/S0025-326X(00)00169-7 (2001).
Miranda, C. D. & Zemelman, R. Antibiotic Resistant Bacteria in Fish from the Concepción Bay, Chile. Marine Pollution Bulletin 42, 1096–1102, https://doi.org/10.1016/S0025-326X(01)00093-5 (2001).
Austin, B. The Bacterial Microflora of Fish, Revised. The Scientific World Journal 6, 931–945, https://doi.org/10.1100/tsw.2006.181 (2006).
Al-Bahry, S. N. et al. Coastal sewage discharge and its impact on fish with reference to antibiotic resistant enteric bacteria and enteric pathogens as bio-indicators of pollution. Chemosphere 77, 1534–1539, https://doi.org/10.1016/j.chemosphere.2009.09.052 (2009).
Smith, K., McCoy, K. D. & Macpherson, A. J. In Seminars in Immunology. 59–69 (Elsevier).
Billroth, T. Untersuchungen über die Vegetationsformen von Coccobacteria septica und den Antheil, welchen sie an der Entstehung und Verbreitung der accidentellen Wundkrankheiten haben: Versuch einer wissenschaftlichen Kritik der verschiedenen Methoden antiseptischer Wundbehandlung. G. Reimer (1874).
Hiergeist, A., Gläsner, J., Reischl, U. & Gessner, A. Analyses of intestinal microbiota: culture versus sequencing. ILAR Journal 56, 228–240 (2015).
Interaminense, J. A. et al. Recovery and screening for antibiotic susceptibility of potential bacterial pathogens from the oral cavity of shark species involved in attacks on humans in Recife, Brazil. Journal of Medical Microbiology 59, 941–947, https://doi.org/10.1099/jmm.0.020453-0 (2010).
Abrahamian, F. M. & Goldstein, E. J. C. Microbiology of Animal Bite Wound Infections. Clinical Microbiology Reviews 24, 231–246, https://doi.org/10.1128/cmr.00041-10 (2011).
Rivas, A. J., Balado, M., Lemos, M. L. & Osorio, C. R. The Photobacterium damselae subsp. damselae Hemolysins Damselysin and HlyA Are Encoded within a New Virulence Plasmid. Infection and Immunity 79, 4617–4627, https://doi.org/10.1128/iai.05436-11 (2011).
Givens, C. E., Ransom, B., Bano, N. & Hollibaugh, J. T. Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Marine Ecology Progress Series 518, 209–223 (2015).
Trust, T. J., Bull, L. M., Currie, B. R. & Buckley, J. T. Obligate Anaerobic Bacteria in the Gastrointestinal Microflora of the Grass Carp (Ctenopharyngodon idella), Goldfish (Carassius auratus), and Rainbow Trout (Salmo gairdneri). Journal of the Fisheries Research Board of Canada 36, 1174–1179, https://doi.org/10.1139/f79-169 (1979).
Leigh, S., Papastamatiou, Y. & German, D. P. The nutritional physiology of sharks (2017).
Buck, J. D., Spotte, S. & Gadbaw, J. J. Bacteriology of the teeth from a great white shark: potential medical implications for shark bite victims. Journal of Clinical Microbiology 20, 849–851 (1984).
dos Santos, G. S. et al. Study of the Enterobacteriaceae Group CESP (Citrobacter, Enterobacter, Serratia, Providencia, Morganella and Hafnia): A Review (2015).
Brenner, D. J. et al. Proposal of Afipia gen. nov., with Afipia felis sp. nov. (formerly the cat scratch disease bacillus), Afipia clevelandensis sp. nov. (formerly the Cleveland Clinic Foundation strain), Afipia broomeae sp. nov., and three unnamed genospecies. Journal of Clinical Microbiology 29, 2450–2460 (1991).
Benamar, S., La Scola, B. & Croce, O. Genome Sequence of Afipia felis Strain 76713, Isolated in Hospital Water Using an Amoeba Co-Culture Procedure. Genome Announcements 2, e01367–01314, https://doi.org/10.1128/genomeA.01367-14 (2014).
Scola, B. L. & Raoult, D. Afipia felis in hospital water supply in association with free-living amoebae. The Lancet 353, 1330, https://doi.org/10.1016/s0140-6736(99)00906-x (1999).
Hanada, S., Hiraishi, A., Shimada, K. & Matsuura, K. Chloroflexus aggregans sp. nov., a filamentous phototrophic bacterium which forms dense cell aggregates by active gliding movement. International Journal of Systematic and Evolutionary Microbiology 45, 676–681 (1995).
Nübel, U., Bateson, M. M., Madigan, M. T., Kühl, M. & Ward, D. M. Diversity and Distribution in Hypersaline Microbial Mats of Bacteria Related to Chloroflexus spp. Applied and Environmental Microbiology 67, 4365–4371 (2001).
Nedashkovskaya, O. I. et al. Pontibacter actiniarum gen. nov., sp. nov., a novel member of the phylum ‘Bacteroidetes’, and proposal of Reichenbachiella gen. nov. as a replacement for the illegitimate prokaryotic generic name Reichenbachia Nedashkovskaya et al. 2003. International Journal of Systematic and Evolutionary Microbiology 55, 2583–2588, https://doi.org/10.1099/ijs.0.63819-0 (2005).
Briones, V. et al. Haemorrhagic Septicaemia by Aeromonas salmonicida subsp. salmonicida in a Black-tip Reef Shark (Carcharhinus melanopterus). Journal of Veterinary Medicine, Series B 45, 443–445, https://doi.org/10.1111/j.1439-0450.1998.tb00814.x (1998).
Gerding, D. N. et al. Nosocomial Multiply Resistant Klebsiella pneumoniae: Epidemiology of an Outbreak of Apparent Index Case Origin. Antimicrobial Agents and Chemotherapy 15, 608–615 (1979).
Diancourt, L., Passet, V., Verhoef, J., Grimont, P. A. D. & Brisse, S. Multilocus Sequence Typing of Klebsiella pneumoniae Nosocomial Isolates. Journal of Clinical Microbiology 43, 4178–4182, https://doi.org/10.1128/jcm.43.8.4178-4182.2005 (2005).
Juste-Poinapen, N. M. S., Turner, M. S., Rabaey, K., Virdis, B. & Batstone, D. J. Evaluating the potential impact of proton carriers on syntrophic propionate oxidation. Scientific Reports 5, 18364, https://doi.org/10.1038/srep18364, http://www.nature.com/articles/srep18364#supplementary-information (2015).
Patel, G. B. & Sprott, G. D. Methanosaeta concilii gen. nov., sp. nov. (Methanothrix concilii) and Methanosaeta thermoacetophila nom. rev., comb. nov. International Journal of Systematic and Evolutionary Microbiology 40, 79–82 (1990).
Azevedo, J. S. N., Correia, A. & Henriques, I. Molecular analysis of the diversity of genus Psychrobacter present within a temperate estuary. FEMS Microbiology Ecology 84, 451–460, https://doi.org/10.1111/1574-6941.12075 (2013).
Coelho, F. et al. In Studies on environmental chemistry-biological responses to contaminants. Interdisciplinary Studies on Environmental Chemistry-Biological Responses to Contaminants. (eds Hamamura, N. et al.) 77–87 (2010).
Prabagaran, S. R., Manorama, R., Delille, D. & Shivaji, S. Predominance of Roseobacter, Sulfitobacter, Glaciecola and Psychrobacter in seawater collected off Ushuaia, Argentina, Sub-Antarctica. FEMS Microbiology Ecology 59, 342–355 (2007).
Mammina, C., Aleo, A., Romani, C. & Nastasi, A. Shigella sonnei biotype g carrying class 2 integrons in southern Italy: a retrospective typing study by pulsed field gel electrophoresis. BMC Infectious Diseases 6, 1–5, https://doi.org/10.1186/1471-2334-6-117 (2006).
Seol, S. Y. et al. Molecular characterization of antimicrobial resistance in Shigella sonnei isolates in Korea. Journal of Medical Microbiology 55, 871–877, https://doi.org/10.1099/jmm.0.46441-0 (2006).
Onyango, D. M., Wandili, S., Kakai, R. & Waindi, E. Isolation of Salmonella and Shigella from fish harvested from the Winam Gulf of Lake Victoria, Kenya. J. Infect Dev Ctries 3, 99–104 (2009).
Mohammed, S. W. C. & Coppard, S. E. Ecology and distribution of soft-sediment benthic communities off Viti Levu (Fiji). Marine Ecology Progress Series 371, 91–107, https://doi.org/10.3354/meps07618 (2008).
Yeates, C., Gillings, M., Davison, A., Altavilla, N. & Veal, D. Methods for microbial DNA extraction from soil for PCR amplification. Biological procedures online 1, 40–47 (1998).
Zhou, J., Bruns, M. A. & Tiedje, J. M. DNA recovery from soils of diverse composition. Applied and Environmental Microbiology 62, 316–322 (1996).
Engelbrektson, A. et al. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. The Isme Journal 4, 642, https://doi.org/10.1038/ismej.2009.153, https://www.nature.com/articles/ismej2009153#supplementary-information (2010).
Albertsen, M., Karst, S. M., Ziegler, A. S., Kirkegaard, R. H. & Nielsen, P. H. Back to Basics – The Influence of DNA Extraction and Primer Choice on Phylogenetic Analysis of Activated Sludge Communities. PLoS ONE 10, e0132783, https://doi.org/10.1371/journal.pone.0132783 (2015).
Acknowledgements
This work was primarily funded by The University of the South Pacific, Strategic Research Themes Program for Research and Innovation, (Grant No. F1006-R1001-71502-624 to CR, JP and MF). We acknowledged Semi Nagatalevu and Inoke Nagatalevu, for collecting the samples. We thank Sione Kaituu for producing Fig. 1. We acknowledged Aisake Batibasaga (Director of the Fisheries Department of the Ministry of Fisheries and Forest, and Joji Kalounivalu (Roko Tui, Rewa Provincial Council, Rewa) for their continued advice and/or support during the project.
Author information
Authors and Affiliations
Contributions
N.J., C.R., J.P. and M.F. designed the project and wrote the grant proposals which funded the project. C.R. organised and sampled all specimens. N.J. and L.Y. performed the analyses. All co-authors contributed to the writing and review of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Juste-Poinapen, N.M.S., Yang, L., Ferreira, M. et al. Community profiling of the intestinal microbial community of juvenile Hammerhead Sharks (Sphyrna lewini) from the Rewa Delta, Fiji. Sci Rep 9, 7182 (2019). https://doi.org/10.1038/s41598-019-43522-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43522-x
This article is cited by
-
Trophic level and proteobacteria abundance drive antibiotic resistance levels in fish from coastal New England
Animal Microbiome (2023)
-
Elasmobranch microbiomes: emerging patterns and implications for host health and ecology
Animal Microbiome (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
