
Abstract
Controlled ubiquitin-mediated protein degradation is essential for various cellular processes. GLI family regulates the transcriptional events of the sonic hedgehog pathway genes that are implicated in almost one fourth of human tumors. GLI3 phosphorylation by Ser/Thr kinases is a primary factor for their transcriptional activity that incurs the formation of both GLI3 repressor and activator forms. GLI3 processing is triggered in an ubiquitin-dependent manner via SCFβTrCP1 complex; however, structural characterization, mode of action based on sequence of phosphorylation signatures and induced conformational readjustments remain elusive. Here, through structural analysis and molecular dynamics simulation assays, we explored comparative binding pattern of GLI3 phosphopeptides against βTrCP1. A comprehensive and thorough analysis demarcated GLI3 presence in the binding cleft shared by inter-bladed binding grooves of β-propeller. Our results revealed the involvement of all seven WD40 repeats of βTrCP1 in GLI3 interaction. Conversely, GLI3 phosphorylation pattern at primary protein kinase A (PKA) sites and secondary casein kinase 1 (CK1) or glycogen synthase kinase 3 (GSK3) sites was carefully evaluated. Our results indicated that GLI3 processing depends on the 19 phosphorylation sites (849, 852, 855, 856, 860, 861, 864, 865, 868, 872, 873, 876, 877, 880, 899, 903, 906, 907 and 910 positions) by a cascade of PKA, GSK3β and CSKI kinases. The presence of a sequential phosphorylation in the binding induction of GLI3 and βTrCP1 may be a hallmark to authenticate GLI3 processing. We speculate that mechanistic information of the individual residual contributions through structure-guided approaches may be pivotal for the rational design of specific and more potent inhibitors against activated GLI3 with a special emphasis on the anticancer activity.
Similar content being viewed by others


Introduction
Protein modification by ubiquitination plays a central role in multiple cellular processes including signal transduction, cell cycle progression, and metabolic pathways1. The addition of ubiquitin to numerous fundamental components is involved in the regulation of Hedgehog (Hh) signaling pathway2. Hh signaling encompasses a wide range of cellular and molecular mechanisms, such as protein trafficking, protein-protein interactions and post-translational modifications including phosphorylation, lipidation and proteolytic cleavage3. The family of secreted Hh signaling molecules plays essential roles in the morphogenesis, homeostasis, cell growth and patterning of numerous embryonic structures of animals ranging from flies to humans4,5. It also controls morphogenesis and homeostasis of various tissues comprising vertebrate and invertebrate epidermal appendages5,6. In Drosophila, Hh signaling is stimulated through a transcription factor Cubitus interruptus (Ci), known to have a central zinc-finger DNA-binding domain4,7. In vertebrates, the function of Ci has been expanded to three GLI proteins: GLI1, GLI2 and GLI38. GLI proteins activate the transcription of several target genes that are involved in numerous aspects of tumorigenesis9,10. GLI3 primarily coordinates Hh signaling by functioning as activator or repressor depending upon the presence or absence of Hh11. GLI homologues exhibit distinctive but overlapping roles. GLI1 works only as a transcriptional activator, whereas GLI2 and GLI3 can act as activators as well as repressors of Hh target genes4,12.
Any imbalance of Hh pathway activity including the key transcription effector GLI has been implicated in many human disorders including cancer9,11. The constitutive activation of the Hh signaling pathway is a potential mediator of colon, glioma, medulloblastoma, basal cell carcinoma, lung cancer, esophageal cancer, gastric cancer, pancreatic cancer, breast cancer and tumors10,12,13,14,15,16. GLI activity is controlled by diverse regulatory processes; among them most prominent is the ubiquitin-mediated proteolysis. Ubiquitin modification of the GLI transcription factors is a vital mechanism to suppress Hh pathway activity. In the absence of Hh, limited degradation of GLI3 converts it into carboxyl-terminal truncated form that functions as a transcriptional repressor of Hh pathway17.
GL3 processing entails its extensive phosphorylation by three Ser/Thr kinases i.e. protein kinase A (PKA), glycogen synthase kinase 3 (GSK3), and casein kinase I (CKI) and component of SCF (SKP1, Cullin, F-box containing complex) E3 ligase i.e. βTrCP1, CUL1 (Cullin1) and RBX1 (RING-box protein 1)7,8,18. In the absence of Hh signaling, GLI3 processing is provoked by PKA-dependent phosphorylation, which is prerequisite for the later phosphorylation events by GSK3 and CK1 leading to direct binding and ubiquitination by SCFβTrCP1 16,17. In the presence of Hh signaling, GLI3 is released from PKA/GSK3-mediated phosphorylation to activate Hh target genes1,17,18 as shown in Fig. 1. Recently, it has been reported that GLI3 processing is mediated by sequential phosphorylation of βTrCP1-binding sites through PKA, GSK3β and CK1 enzymes16,17. GLI3 central domain comprises four PKA sites; the first PKA site is flanked by a CK1 site, while second, third and fourth sites are flanked by both GSK3 and CK1 sites8. GSK3β and CK1 trigger multiphosphorylation of GLI3, where GSK3β phosphorylates Ser 4 residue N-terminal to a phosphoserine, while CK1 phosphorylates Ser 3 residue C-terminal to a phosphoserine17. βTrCP1 explicitly binds to its substrates via the phosphodegron motif i.e. DSpGX2–4Sp, where Sp denotes phosphorylated serine and X denotes any residue19. βTrCP1 binding recruits the ubiquitination machinery to its substrates ensuing either degradation or processing. GLI3 binding necessitates 4 βTrCP1-binding sites (SSASTIS, SSAYLS, SSGISPCFS and DSYDPIS; referred as motifs 1–4) that are related to DSGX2–4S motif lying in the substrates of βTrCP117. GLI3 processing depends on the phosphorylation of 4 PKA sites (Ser849, Ser865, Ser877 and Ser907), further phosphorylated by CK1 (Ser852, Ser868, Ser880 and Ser910) and GSK3β (Ser861, Ser873 and Ser903). CK1-phosphorylation at Ser855 (motif-1) primes further phosphorylations by GSK3β at secondary sites including Ser856 and Ser860 (motif-2), Ser872 (motif-3) and Ser899 (motif-4) residues in addition to phosphorylation at Ser864, Ser876 and Ser906 residues17.

Schematic illustration of GLI3 translocation, processing, and degradation via SCF (SKP1, Cullin, F-box protein complex). In the absence of hedgehog (right panel), Ptch1 constitutively inhibits Smo, preventing its ciliary localization. In this state, GLI proteins are retained in a complex with Sufu, where PKA, CK1ε, and GSK3 phosphorylate them. Phosphorylated GLI3 binds the SCF complex that is partially ubiquitinated and processed by the proteasome into GLI3-R, which translocates to the nucleus and represses transcription of Hh target genes, including Akt, Gli1, Ptch1 etc., prior to degradation by an unknown E3 ligase complex. In the presence of Hh (Left panel), PKA phosphorylation is inhibited. The activated form of GLI3 not only prevents it from processing but also permits its subsequent transport to the nucleus to allow activation of transcription of Hh target genes. Following this, GLI3-A is ubiquitinated by the SPOP/CUL3 complex and degraded by the proteasome. P, phosphate group; Ptch1, Patched 1; SUFU, Suppressor of Fused; SMO, Smoothened; PKA, Protein kinase A, GSK3, Glycogen synthase kinase 3; CSKI, Casein kinase 1; CUL1, Cullin 1; CUL3, Cullin 3; RBX1, RING-box protein 1; SKP1, S-Phase Kinase-Associated Protein 1; SPOP, Speckle Type BTB/POZ Protein; GLI3-R, GLI3 repressor; GLI3-A, GLI3 activator.
Despite the contemporary range of βTrCP1 substrates, molecular significance of βTrCP1 binding to the multiple motifs in GLI3 is not clearly understood. Here in current study, we utilized in silico approaches to elucidate the structural basis of GLI3 phosphorylation in the regulation of βTrCP-mediated protein degradation. Unlike other substrates of E3 ligases, GLI3 requires PKA, GSK3 and CKI accessory proteins for phosphorylation and ubiquitination by βTrCP1. Our detailed structural analyses highlighted significant conformational switches and noteworthy residual contributions upon phosphorylation and binding. Thus by exploring the association of βTrCP1 and GLI3, our study may provide invaluable insight in understanding the Hh relationship with cancer pathogenesis.
Methodology
Data set
Primary protein sequence of GLI3 (UniProt ID: P10071) was retrieved through UniProtKB/Swiss-Prot database (http://www.uniprot.org/). Three dimensional (3D) coordinates of human βTrCP1 (PDB ID: 1P22; 2.95 Å resolution) were retrieved through Protein Data Bank20. 3D-structure of GLI3 central domain (846–910AA) was predicted by PEP-FOLD21, which is a de novo approach, used for the prediction of small peptide structures. MolProbity tool22 was utilized to validate 3D model followed by model refinement using WinCoot23. Structure editing (phosphorylation) was carried out using UCSF Chimera 1.11.224.
Phosphopeptides were generated by adding phosphate groups to serine residues lying at the degradation motif through UCSF Chimera 1.11.2. In GLI3PKA phosphopeptide, phosphorylation was performed at Ser849, Ser865, Ser877 and Ser907, GLI3GSK3β phosphopeptide exhibited phosphorylation at Ser861, Ser873 and Ser903 residues, while in GLI3CKIϵ phosphopeptide, Ser852, Ser868, Ser880 and Ser910 residues were phosphorylated. In addition to these residues, GLI3-β1 peptide exhibited phosphorylation at Ser855 residue, GLI3-β2 peptide was generated by the addition of phosphate groups at Ser856, Ser860 and Ser864 residues, GLI3-β3 peptide contained phosphate groups at Ser872 and Ser876 residues, GLI3-β4 peptide exhibited phosphate groups at Ser899 and Ser906 residues, while GLI3-β1–4 was generated by adding phosphate groups at Ser855, Ser856, Ser860, Ser864, Ser872, Ser876, Ser899 and Ser906 residues. Structures were optimized by energy minimization through UCSF Chimera 1.11.2 and GROMACS25 using Amber force field. Energy minimization was performed using the steepest descent algorithm for 50000 steps (with a lower dt value of 0.001). Ideally, the maximum force Fmax (gradient of the potential) should be less than 1000 kJ mol−1 nm−2. In our case, Fmax <1000 was achieved at 543 and 687 steps with potential energy values of −1.07452e + 06 and −1.284026e + 06 for βTrCP1 in complex with GLI3-β1–4 and GLI3-β4 peptides, respectively. All systems were converged at same range.
Molecular docking analysis
In order to explore the binding pattern of βTrCP1 and GLI3 peptides (GLI3-un, GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4), molecular docking was accomplished by HADDOCK26. Prior to docking analysis, 3D structures of βTrCP1 and GLI3 peptides were submitted to CPORT server to predict the interface residues27. CPORT provides a list of active and passive residues of the interaction interfaces that are further employed for the preparation of ambiguous interaction restraints (AIR) by HADDOCK28,29. Docking simulations were carried out with default parameters, among them HIS protonation state, random removal of restraints, number of structures to dock and refine, electrostatic scaling constant, restraint energy constants, scoring parameters, temperature and time steps of refinement processes were defined automatically by the interface of HADDOCK server30. HADDOCK produces 1000 docked structures through experimental data to drive docking. HADDOCK scores each model using Equation 1 and retains the top 200 solutions for consequent flexible refinement, where EAIR, Eelec, EvdW and Edesolv are the restraints violation , electrostatic, van der Waals and desolvation energies, respectively. BSA is the buried surface area and Edata indicates the energy of other restricted data. HADDOCK score is weighted as the sum of the following four terms: electrostatic energy (weight: 0.2), Van der Waals energy (weight: 1.0), desolvation energy (weight: 1.0) and restraint violation energy (weight: 0.1).
Furthermore, selected models are subjected to a semiflexible refinement followed by water refinement step in torsion angle space and explicit water shell, respectively. These parameters are scored using Equations 2 and 3, respectively. The solutions are clustered using a 7.5 Å limit created on their pairwise ligand interface. Root mean square deviation (RMSD) values and cluster ranks are rendered to the average score of the top four structures in each cluster.
The best docked complexes of top ranked clusters were selected and visualized by UCSF Chimera 1.11.2 to analyze their conformational readjustments. Residual interactions such as hydrogen bonds, hydrophobic, electrostatic interactions and bond lengths were evaluated by employing DIMplot embedded in LigPlus31. Furthermore, these complexes were subjected to molecular dynamic simulations for detailed analysis.
Molecular dynamics simulation analysis
In order to gain a deep insight into the mechanism of GLI3 phosphodegron recognition by apo-βTrCP1 and its bound forms with GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4, these complexes were subjected to molecular dynamics (MD) simulation assays to evaluate the stability, folding, conformational changes and dynamic behavior of interacting proteins. Groningen Machine for Chemicals Simulations (GROMACS) 5.1.4 package was used to perform MD simulation assay. All MD simulations were performed by GROMOS96 43a1 force field32 to acquire the equilibrated system. All systems were solvated using SPC water model33 in a periodic box, followed by the addition of Na+ and Cl− counter ions to neutralize the system. Before MD simulations, systems were subjected to energy minimization to remove initial steric clashes using 1000 steps of steepest descent algorithm via a tolerance of 10 KJ/mol/nm−1. Systems were equilibrated for 1000 ps at 300 K and 1 bar pressure in NVT34 and NPT35 ensembles, respectively and their equilibration profiles were evaluated (Fig. S1). The hydrogen bond length constraints were employed at a time step of 2 fs for numerical integration with Verlet leap-frog algorithm36. PME (Particle Mesh Ewald) algorithm37 was employed to evaluate the long-range (LR) electrostatic interactions. Finally, MD simulations were run for 40 ns time scale. Stability and time-dependent behavior of each system were investigated using system conformations extracted every 5 ns from the MD trajectories and analyzed using UCSF Chimera and GROMACS tools. GROMACS modules such as g_rms, g_rmsf, g_energy and g_hbond were utilized to analyze the stability and behavior of each system. The secondary structure database (DSSP) was utilized to analyze the time-dependent secondary structure fluctuations in the bound complexes38.
Binding free energy calculation
Poisson-Boltzmann or generalized Born and surface area continuum solvation (MM/PBSA) method39 was employed to calculate the binding free energy of the system. This method provides inclusive energy composition and improves docking energy via incorporating protein flexibility. The binding energy of ligand-protein complex was calculated using the following equation:
Gcomplex is the total free energy of the protein-ligand complex; Gprotein and Gligand are total energy of separated protein and ligand in solvent, respectively. The free energy for each individual Gcomplex, Gprotein and Gligand were estimated by:
x is the protein-ligand complex. EMM is the molecular mechanics energy and Gsolvation is free energy of solvation. The molecular mechanics potential energy was calculated in vacuum as following:
Ebonded is bonded interaction including bond, angle, dihedral and improper interactions and Enon-bonded is non-bonded interactions that consist of van der Waals (Evdw) and electrostatic (Eelec) interactions.The solvation free energy (Gsolvation) was estimated as the sum of electrostatic solvation free energy (Gpolar) and apolar solvation free energy (Gnon-polar):
Gpolar was computed using the Poisson-Boltzmann (PB) equation40 and Gnon-polar was calculated using a solvent accessible surface area (SASA) as follows:
γ is a coefficient related to surface tension of the solvent and b is fitting parameter.
Results
Structural evaluation of GLI3 peptides
The predicted structures of GLI3 peptides (GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4) were evaluated by Ramachandran plots (Fig. S2), where blue colour indicated favorable region (sterically allowed regions), while no outliers were observed. Approximately, 92–95% residues were resided in the blue region. Additionally, parameters like peptide bond planarity, non-bonded interactions, Cα-tetrahedral distortion, main chain H-bond energy values and overall G-factors for the predicted models were lying in the favourable ranges. GLI3 peptide structures optimized through GROMACS tool were further evaluated by RAMPAGE41.
Phosphopeptide binding and conformational transitions
In order to evaluate mechanism of substrate recognition by βTrCP1, GLI3 phosphopeptides were subjected to molecular docking analysis. Given a maximum number of 200 models for clustering, HADDOCK clustered 99 structures of βTrCP1-GLI3-un complex in 15 clusters, 86 structures of βTrCP1-GLI3PKA complex in 8 clusters, 66 structures of βTrCP1-GLI3GSK3β complex in 11 clusters, 72 structures of βTrCP1-GLI3CKIϵ complex in 11 clusters, 130 structures of βTrCP1-GLI3-β1 complex in 16 clusters, 115 structures of βTrCP1- GLI3-β2 complex in 15 clusters, 93 structures of βTrCP1- GLI3-β3 complex in 11 clusters, 117 structures of βTrCP1- GLI3-β4 complex in 15 clusters and 114 structures of βTrCP1-β1–4 complex in 14 clusters, representing 49.5%, 43.0%, 33.0%, 36.0%, 65.0%, 57.5%, 46.5%, 58.5% and 57.0% of water-refined models, respectively. The statistics of top 10 clusters (ranked on the basis of lowest overall energy and Z-score values) were shown by HADDOCK, out of which scores of the optimal clusters for each βTrCP1-peptide complexes are illustrated in Table 1. The more negative HADDOCK and Z-scores indicate a reliable interaction. Z-score is the quantitative measure of cluster standard from the average score.
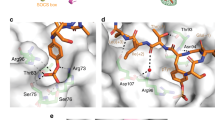
All βTrCP1-peptide complexes were carefully characterized to access their binding patterns. In case of βTrCP1-GLI3PKA complex, phosphopeptide exhibited binding with the 1st, 2nd and 7th WD40 repeats of βTrCP1 having a score of −17.6 (Fig. 2A). In contrast, GLI3GSK3β and GLI3CKIϵ peptides did not exhibit binding with βTrCP1 (Fig. 2B,C). In βTrCP1 and GLI3-β1–4 complex, phosphopeptide binding was observed at the upper interface of β-propeller (Fig. 2D). Thus GLI3 phosphorylation by all three enzymes (PKA, GSK3β and CKIε) resulted in accurate binding with βTrCP1 substrate binding site.

Binding orientation of β-propeller due to phosphopeptide binding. 7 WD40 repeats of βTrCP1, comprising 25 beta sheets are organized to form a circular structure (β-propeller). Optimal docked complexes of βTrCP1 bound (A) GLI3PKA (B) GLI3GSK3β, (C) GLI3CSKIϵ (D) GLI3-β1–4. βTrCP1 is shown in white colored ribbon, while GLI3 phosphopeptide is shown in red colored ribbon.
Next, we examined GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4-bound βTrCP1 complexes to explore the intricate details of phosphopeptide binding. Evidently, docking clusters of phosphopeptides at the substrate binding pocket of βTrCP1 revealed predominant binding affinities for WD40 repeats (Fig. 3). The individual residues involved in interactions were evaluated through DIMPLOT and UCSF Chimera 1.11.2. These residual contributions specified that almost all 7 WD40 repeats imparted equal propensity to bind with GLI3 phosphopeptides. Though, it is vague at the moment whether binding of peptide results in any notable modification in the βTrCP1functioning. The binding residues as listed in Table 2. In comparison to other complexes, βTrCP1-GLI3-β1–4 complex exhibited more number of hydrogen bonds. Phosphorylated residues (Sep873, Sep876, Sep877 and Sep880) of GLI3 contributed in interaction with all 7 WD40 repeats of βTrCP1. As reported by Wu et al., 200342, βTrCP1-specific residues (Tyr271, Arg285, Ser309, Leu311, Ser325, Leu351, Asn394, Arg431, Gly432, Ala434, Ser448, Leu472, Arg474, Tyr488 and Arg521) involved in phosphorylated β-catenin peptide binding were consistent in GLI3-βTrCP1 complex, where GLI3-β1–4 peptide binding was evident at the upper face of β-propeller (Fig. 3). These results indicate that βTrCP1 shares common region upon interaction with phosphor-substrates.

Binding mode and molecular interaction analysis of motif peptides. Optimal docked complexes of βTrCP1-bound (A) GLI3-β1 (B) GLI3-β2, (C) GLI3-β3 (D) GLI3-β4 and (E) GLI3-β1–4 peptides. βTrCP1 and GLI3 are shown in white and khaki colored ribbons with interacting residues in green and goldenrod colored ball and sticks, respectively.
Molecular dynamics simulation analysis
In order to permit elucidation of conformational transitions, dynamic behavior and stability of contacts, complexes of βTrCP1 and phosphorylated peptides (GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4) were further characterized by 40 ns molecular dynamics (MD) simulations. The stability of secondary structure elements and conformational changes of simulated complexes were assessed by plotting RMSD (Root Mean Square Deviation), RMSF (Root Mean Square Fluctuation), hydrogen bonding and binding energy plots. RMSD for each complex was measured throughout 40 ns time scale using apo-form as a reference. Overall RMSD analysis revealed stable behavior for all systems in a range of 2.0–4.2 Å (Fig. 4A). Dynamically, βTrCP1 bound GLI3-β1–4 complex displayed slight increase in deviations during the initial 10 ns time period, compared to other complexes (Fig. 4A). However, later on, backbone RMSD profile for GLI3-β1–4 was quite stable (3.5–4 Å). The pronounced changes in RMSD trend indicated variability in the structural rearrangements upon GLI3 phosphorylation. Correspondingly, Rg profiles of individual systems were consistent with their resultant RMSD profiles (Fig. 4B). A higher Rg value implies lower compactness of a system43,44,45,46,47. Consequently, βTrCP1-GLI3-β1–4 exhibited minor compactness than apo-form. Thus higher Rg values of complexes than that of apo-βTrCP1 suggested firmness in the synergic conformational adaptation owing to βTrCP1 interaction.

Time-dependent analysis of 40 ns MD simulations of apo- versus GLI3 peptide-bound βTrCP1. (A) RMSD plotted as a time function computed through least square fitting of backbone Cα-atoms. (B) Rg plots of individual simulated complexes along the course of 40 ns of MD simulation. (C) RMSF per residue plot for each trajectory file. (D) Comparison of the most fluctuating residues is indicated by bar chart. Apo and bound forms of βTrCP1 with GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4 are represented in blue, green, gold, orange, cyan and purple colors, respectively.
Subsequent RMSF analysis indicated residual fluctuations at the substrate binding cleft of WD40 repeats (Fig. 4C). βTrCP1 upon binding to GLI3-β1 exhibited significantly higher rate (3–5 Å) of fluctuations as compared to apo and other βTrCP1-bound phosphopeptide forms. In case of GLI3-β3 complex, major fluctuations (up to 3 Å) were detected in βTrCP1 residues, while residues involved in GLI3 binding were comparatively stable (Fig. 4D). In βTrCP1-GLI3-β2 complex, more fluctuations (1.6 Å) were observed in Gly308 and Gly388 residues, while βTrCP1 residues involved in binding remained stable during the course of simulation run. Correspondingly, in βTrCP1-GLI3-β4 complex, major fluctuations were detected in Gly388, His389, Ala392-Asn394, Gly408-Arg410, Lys430-Gly432 and Ser448 (1.7 Å) residues located in the immediate vicinity of binding region (Fig. 4D). Interestingly, all fluctuations were observed in the loop regions. In βTrCP1-GLI3-β1–4 complex, significant fluctuation (2 Å) was observed in Lys268 residue, while βTrCP1 binding residues namely, Arg285, Ser325, Leu343, His346, Cys347, Lys365, Arg367, Arg390, Arg410, Arg431, Gly432, Ala434, Ser448, Arg474 and Arg521 remained stable during the course of simulation run (Fig. 4D).
MD simulation trajectory files of βTrCP1-bound phosphopeptide complexes were subjected to energy calculation via LJ-SR (Lennard-Jones Short-Range) binding descriptor. LJ-SR are normal non-bonded interactions within the short-range cutoff. Overall, LJ-SR energy values were quite stable ranging between −10000 to −11500 kcal/mol (Fig. 5A). Analogously, coulomb short range energy values (Coul-SR) are used to assess the system’s equilibration along the simulation run. Coul-SR energy values (−81016 to −87087 kcal/mol) indicated the stability of systems. Furthermore, simulated trajectories of βTrCP1-bound GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4 were examined for hydrogen bond shifts. Inclusively, hydrogen bond interaction pattern remained stable during the entire simulation time (Fig. 5B). The presence of more intermolecular hydrogen bonds in GLI3-β1–4 as compared to other simulated systems indicated enhanced binding of βTrCP1 with GLI3-β1–4 phosphopeptide. Overall, H-bonding pattern inferred stable interactions in agreement with the RMSD distribution (Fig. 4A).

Binding energy and hydrogen bond versus time plots for 40 ns MD simulation. (A) LJ-SR binding energy profile. (B) Intermolecular hydrogen bonding pattern of βTrCP1-GLI3 complexes. GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4 are represented in green, gold, orange, cyan and purple colors, respectively.
Conformation change analysis
To monitor the structural changes in apo versus βTrCP1-bound systems, PDB files were extracted every 5 ns (5, 10, 15, 20, 25, 30, 35 and 40 ns) time interval from MD trajectories. During MD simulations, momentous conformational changes were observed at the proximity of central cavity, influencing the peptide binding. The conformational transitions occurring in the β-propellers of βTrCP1 were deeply examined at 30 ns to understand the changes in secondary structural elements (Table 3). Evidently, in GLI3-β1–4-bound βTrCP1, conversion of Thr381-Leu386 β-strand into loop was visible in comparison to other complexes (Table 3). Another change persuaded upon GLI3-β1–4 binding was the extension of 4 β-strands (Arg301-Leu303, Leu313-Tyr315, Ile492-Trp495 and Ile532-Ser534 regions) of βTrCP1 that induced more stability in binding propensity. Moreover, lengths of β12 (Val393-Asp399) and β14 (Phe422-Leu426) strands were reduced; however, these shrinkages did not alter the active site conformation. Another notable secondary structural amendment was witnessed in the loop region of βTrCP1, where Thr540-Trp544 region adopted a β-conformation upon binding to GLI3-β1, GLI3- β2 and GLI3-β1–4 phosphopeptides.
Through comparative analysis of βTrCP1-bound phosphopeptides, contributions of βTrCP1-specific Ser267, Lys268, Ala309 and His352 residues were observed in GLI3-β1–4 phosphopeptide binding (Fig. 6). To further characterize the βTrCP1 and GLI3 phosphopeptide interactions, we mapped βTrCP1-specific probable regions that could be prerequisite for GLI3 phosphopeptide binding. Evidently, two residues (Cys347 and Arg367) lying in 3rd WD40 repeat of βTrCP1 actively contributed in the phosphopeptide binding (Fig. 6). Additionally, Glu265, Arg285, Ser309, Ser325, Arg367, Arg390, Arg410, Lys430, Arg431, Ser448, Tyr488, Arg474 and Arg521 residues were directly involved in hydrogen bonding with phosphoserines of GLI3-β1–4.

Structural details of βTrCP1 and GLI3 phosphopeptide binding. βTrCP1 is represented by light gray ribbon, while pale yellow ribbons represent phosphopeptide GLI3-β1–4 with interacting residues indicated by coral ball and stick mode. Illustration of four sequence motifs (β1 to β4) related to the βTrCP1 binding site are underlined that are phosphorylated by a putative cascade of PKA, GSK3β and CK1. PKA phosphorylated serines (phosphoserine) in the sequence motifs are colored in red. GSK3β phosphorylates serines (green) four residues N-terminal to a phosphoserine, while CK1 phosphorylates serines (blue) three residues C-terminal to a phosphoserine; both can chronologically multiphosphorylate GLI3 after priming. Middle panel shows the conservation pattern of βTrCP1 binding residues upon phosphopeptide binding. X-axis indicates the binding residues of βTrCP1 and Y-axis indicates the GLI3 phosphopeptides (GLI3-β1, GLI3-β2, GLI3-β3, GLI3-β4 and GLI3-β1–4). Dot represents the contribution of respective residue in binding to phosphopeptide.
Furthermore, PDB files were characterized to measure the conformational switches in GLI3 phosphopeptides upon binding to βTrCP1. All phosphopeptides exhibited quite stable binding patterns at 25 ns. Particularly, upon binding to βTrCP1, both helical regions (Ile854-Ser864 and Thr900-Glu908) were shortened in GLI3-β1–4 to accommodate it in the cavity formed by β-propellers (Fig. 7E). A profound conformational change was observed in Thr900-Glu908 helical region (Fig. 7D), as upon binding to βTrCP1, this helix was completely missing. This trend was observed throughout MD simulation run as evident from the analysis of time-dependent secondary structure fluctuations via DSSP module (Fig. S3). Another notable secondary structural amendment was witnessed in the loop region of GLI3, where Sep875-Glu878 region of GLI3-β2 adopted a α-helical conformation upon binding to βTrCP1 (Figs S3B and 7B). In βTrCP1-bound GLI3-β1, GLI3-β3, GLI3-β4 and GLI3-β1–4 peptides, this region remained structurally preserved (Fig. S3). Subsequent analysis of RMSF indicated residual flexibility of phosphorylated residues upon GLI3 binding to βTrCP1. In case of GLI3-β1 and GLI3-β3 binding, major fluctuations up to 10 Å and 4.5 Å were perceived in all phosphorylated residues (Fig. 7F). Correspondingly, GLI3-β2 and GLI3-β4 peptides exhibited minor rate (up to 2.8 Å) of fluctuations as compared to other simulated systems. In case of GLI3-β1–4, significant fluctuations were detected in Sep899, Sep903, Sep906, Sep907 and Sep910 residues (4–11 Å) to assist in binding, while phosphorylated residues involved in binding (Sep852, Sep855, Sep872, Sep873, Sep876, Sep877 and Sep880) were quite stable (Fig. 7E). These results specified that Sep899-Sep910 of GLI3-β1–4 exhibited more fluctuations thus suggesting that Sep899-Sep910 region of GLI3 may be crucial for βTrCP1 binding.

Conformational switches of the GLI3 phosphopeptide structure upon binding to βTrCP1. Phosphopeptides of (A) GLI3-β1, (B) GLI3-β2, (C) GLI3-β3, (D) GLI3-β4 and (E) GLI3-β1–4 are represented in green, gold, orange, cyan and purple colors, respectively. Phosphorylated residues via PKA, GSK3β and CSKI are shown by red, light green and blue colors, respectively in ball and stick mode. Secondary structures are illustrated above the corresponding plots. Coils delineate α-helices, while line specifies loop. (F) Comparative RMSF versus time plot of significant phosphorylated residues.
Binding free energy analysis
βTrCP1 complexes with GLI3-β4 and GLI3-β1–4 were employed to estimate binding free energy values using MM/PBSA method. GLI3-β1–4 peptide possessed more negative binding free energy as compared to GLI3-β4, suggesting higher binding affinity for βTrCP1 (Table 4). The van der Waals (Evdw), electrostatic (Eelec) interactions and nonpolar salvation (ΔGsol-nonpolar) energies negatively contributed, while polar solvation energy (ΔGsol-polar) contributed positively to the total binding energy (ΔGbinding). Our results demonstrated a dominant role of electrostatic interaction in stabilizing the βTrCP1 and GLI3-β1–4 association. The binding free energy decomposition analysis revealed multiple residual contributions (Fig. 8), which delineated a similar interaction pattern with βTrCP1. These data were consistent with the findings of RMSF analysis (Fig. 4C). In case of GLI3-β1–4 and βTrCP1 complex, predominant energy contributions were due to Arg285, Lys365, Arg367, Arg390, Arg410, Arg431, Arg474 and Arg521 residues (Fig. 8B). Notably, energetic contribution of key gatekeeper residues (Arg474 and Arg524) was significant in the overall interaction paradigm, as describe previously19. Sep849, Sep852, Sep868, Sep872 and Sep877 residues of GLI3-β1–4 were critical for βTrCP1 binding; however, active role of these residues was not observed in the binding of GLI3-β4 and βTrCP1 (Fig. 8D).

The binding free decomposition on per residue basis calculated from 40 ns MD trajectories by MM/PBSA method. Binding free energy decomposition at residue basis for βTrCP1 upon binding to (A) GLI3-β4 (B) GLI3-β1–4 peptides. Binding free energy decomposition on a per-residue basis for (C) GLI3-β4 (D) GLI3-β1–4.
Discussion
Ubiquitination plays a crucial role in Hh signaling activity via GLI proteins2 that act as zinc finger transcriptional effectors and regulate vertebrate development18. In the absence of Hh, phosphorylation-mediated ubiquitination keeps GLI3 in the repressor form. Despite the fact that mechanistic role of phosphorylation-mediated GLI3 degradation is well-established, the structural-functional paradigm is largely unknown. Here, we explored the potential role of multisite phosphorylation in the selective binding of βTrCP1 and GLI3 phosphopeptides via in silico approaches. Evidently, key substrate binding residues (Arg285, Arg307, Ser327, Ile344, Cys347, Lys365, Arg367, Asn394, Ser448, Arg474 and Arg521) of βTrCP1 were consistent with the earlier studies19,42. RMSD analysis demonstrated stability (2–4 Å) in all systems at 12 ns. Further analysis elucidated multiple conformational changes that invoked specificities in the β-propeller upon phosphopeptide binding. The overall topology of β-strands remained preserved in the βTrCP1 structure (Table 3). A predominant transformation of β-strand (Thr381-Leu386) into loop conformation facilitated the binding via flexibility. Other prominent positional readjustments observed in the β-strands were localized in WD40 repeat-1 (Arg301-Leu303 and Leu313-Tyr315) and repeat-6 (Ile492-Trp495 and Ile532-Ser534), leading to GLI3-β1–4 binding. In RMSF analysis, βTrCP1 binding region (Arg285-Arg521) attained more stability upon binding to GLI3-β-4 (Fig. 4D).
In agreement to the previous observations17, where crucial role of GLI3 motif-4 has been reported in βTrCP1 binding and GLI3 processing, our findings indicate that motif-4-specific Ser899 phosphorylation invokes other phosphorylated serines to impart active role in binding to βTrCP1 (Fig. 8). The interaction of βTrCP1 and GLI3 was significantly influenced by the positional readjustments of residues lying in two helices (Ile854-Sep865 and Thr900-Sep907) due to phosphorylation of paired neighboring residues that induced flexibility differences through helix-loop inter-conversion. Generally, introduction of phosphate group targets loop conformation by rearranging the hydrogen bonding network of side chains lying at the vicinity of loop region48. These transitions in the surrounding regions render helical shifting into loop that acts as a conformational switch for the binding cleft geometry49. The presence of diverse hydrogen bonding pattern and conformational switching due to phosphorylation is crucial for the recognition of GLI3 by βTrCP1. Any change in this pattern may impair their binding affinity due to imbalanced phosphorylation level. It is however unclear at the moment how energy barrier overcomes the phosphorylation or other post-translational modification-induced conformational space.
GLI3 contains multiple binding sites for βTrCP1, where approximately, two-third of GLI3 contacts involve phosphorylated PKA sites and secondary CK1/GSK3 sites. The potential involvements of GLI3-specific primary (Ser852, Ser873, Ser877, Ser880 and Ser903) and secondary phosphorylated (Ser855, Ser872, Ser876, Ser906) residues in βTrCP1 binding indicate that both primary and secondary phosphorylations are required for βTrCP1 binding. Study of interdependent phosphorylation status through structural knowledge may expand the repertoire of GLI3 processing. Indeed, any mutation at the PKA-specific sites may significantly reduce the binding of βTrCP1 to GLI37. Taken together, our results are in good agreement with the experimental outcomes7,17. This study may uncover the spectrum of structural linkages in association with the kinase-mediated phosphorylation paradigm to illustrate the molecular basis of GLI3 processing in Hh signaling. Further studies will be needed to elaborate the effect of putative phosphorylation site mutations at structural level.
Data Availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
Jiang, J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle. 5, 2457–2463 (2006).
Hsia, E. Y., Gui, Y. & Zheng, X. Regulation of Hedgehog signaling by ubiquitination. Front Biol. 10, 203–220 (2015).
Cochrane, C., Szczepny, A., Watkins, D. & Cain, J. Hedgehog signaling in the maintenance of cancer stem cells. Cancers. 7, 1554–1585 (2015).
Pan, Y., Bai, C. B., Joyner, A. L. & Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26, 3365–3377 (2006).
Ingham, P. W. & McMahon, A. P. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087 (2001).
Hatsell, S. J. & Cowin, P. Gli3-mediated repression of Hedgehog targets is required for normal mammary development. Development. 133, 3661–3670 (2006).
Wang, B. & Li, Y. Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 103, 33–38 (2006).
Wen, X. et al. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol. 30, 1910–1922 (2010).
Sabol, M., Trnski, D., Musani, V., Ozretić, P. & Levanat, S. Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets. Int. J. Mol. Sci. 19, 2562 (2018).
Katoh, Y. & Katoh, M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 9, 873–886 (2009).
Huntzicker, E. G. et al. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 20, 276–281 (2006).
Trnski, D. et al. GSK3β and Gli3 play a role in activation of Hedgehog-Gli pathway in human colon cancer—Targeting GSK3β downregulates the signaling pathway and reduces cell proliferation. Biochim. Biophys. Acta, Mol. Basis Dis. 1852, 2574–2584 (2015).
Jia, J. et al. Phosphorylation by double-time/CKIε and CKIα targets cubitus interruptus for Slimb/β-TRCP-mediated proteolytic processing. Dev. Cell. 9, 819–830 (2005).
Steg, A., Amm, H. M., Novak, Z., Frost, A. R. & Johnson, M. R. Gli3 mediates cell survival and sensitivity to cyclopamine in pancreatic cancer. Cancer Biol. Ther. 10, 893–902 (2010).
Mazumdar, T., DeVecchio, J., Agyeman, A., Shi, T. & Houghton, J. A. The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget. 2, 638 (2011).
Pandolfi, S. & Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: implications for cancer therapy. Expert Rev. Mol. Med. 17, e5 (2015).
Tempé, D., Casas, M., Karaz, S., Blanchet-Tournier, M. F. & Concordet, J. P. Multisite protein kinase A and glycogen synthase kinase 3β phosphorylation leads to Gli3 ubiquitination by SCFβTrCP. Mol. Cell. Biol. 26, 4316–4326 (2006).
Niewiadomski, P. et al. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 6, 168–181 (2014).
Shafique, S., Younis, S., Niaz, H. & Rashid, S. Elucidation, functional clustering and structural characterization of βTrCP1 substrates through a molecular dynamics study. Mol. BioSyst. 12, 2233–2246 (2016).
Berman, H. M. et al. The Protein Data Bank and the challenge of structural genomics. Nat. Struct. Mol. Biol. 7, 957–959 (2000).
Shen, Y., Maupetit, J., Derreumaux, P. & Tufféry, P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 10, 4745–4758 (2014).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2009).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Meng, E. C., Pettersen, E. F., Couch, G. S., Huang, C. C. & Ferrin, T. E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics. 7, 339 (2006).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 1, 19–25 (2015).
De Vries, S. J., van Dijk, M. & Bonvin, A. M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 (2010).
De Vries, S. J. & Bonvin, A. M. CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PloS One. 6, e17695 (2011).
Dominguez, C., Boelens, R. & Bonvin, A. M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 (2003).
Van Dijk, A. D., Boelens, R. & Bonvin, A. M. Data‐driven docking for the study of biomolecular complexes. FEBS J. 272, 293–312 (2005).
Van Dijk, A. D. & Bonvin, A. M. Solvated docking: introducing water into the modelling of biomolecular complexes. Bioinformatics. 22, 2340–2347 (2006).
Wallace, A. C., Laskowski, R. A. & Thornton, J. M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 (1995).
Duan, Y. et al. A point‐charge force field for molecular mechanics simulations of proteins based on condensed‐phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 (2003).
Campo, M. G. Structural and dynamic properties of SPC/E water. Pap.Phys. 2, 020001 (2010).
Labík, S. & Smith, W. R. Scaled particle theory and the efficient calculation of the chemical potential of hard spheres in the NVT ensemble. Mol. Simul. 12, 23–31 (1994).
McDonald, I. R. NpT-ensemble Monte Carlo calculations for binary liquid mixtures. Mol. Phys. 23, 41–58 (1972).
Van Gunsteren, W. F. & Berendsen, H. J. C. A leap-frog algorithm for stochastic dynamics. Mol. Simul. 1, 173–185 (1988).
Abraham, M. J. & Gready, J. E. Optimization of parameters for molecular dynamics simulation using smooth particle‐mesh Ewald in GROMACS 4.5. J. Comput. Chem. 32, 2031–2040 (2011).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 22, 2577–2637 (1983).
Kumari, R., Kumar, R., Open Source Drug Discovery Consortium & Lynn, A. g_mmpbsa–A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 54, 1951–1962 (2014).
Honig, B. & Nicholls, A. Classical electrostatics in biology and chemistry. Science. 268, 1144–1149 (1995).
Lovell, S. C. et al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Struct. Funct. Bioinf. 50, 437–450 (2003).
Wu, G. et al. Structure of a β-TrCP1-SKP1-β-catenin complex: destruction motif binding and lysine specificity of the SCFβ-TrCP1 Ub ligase. Mol. Cell. 11, 1445–1456 (2003).
Ali, W., Shafique, S. & Rashid, S. Structural characterization of β-catenin and RX-5902 binding to phospho-p68 RNA helicase by molecular dynamics simulation. Prog Biophys Mol Biol. 140, 79–89 (2018).
Shafique, S., Ali, W., Kanwal, S. & Rashid, S. Structural basis for Cullins and RING component inhibition: Targeting E3 ubiquitin pathway conductors for cancer therapeutics. Int. J. Biol. Macromol. 106, 532–543 (2018).
Shafique, S. & Rashid, S. Antiviral Drug Acyclovir Exhibits Antitumor Activity via targeting βTrCP1: Molecular Docking and Dynamics Simulation Study. J. Mol. Graph. Model. 72, 96–105 (2017).
Shafique, S. & Rashid, S. Structural basis for renal cancer by the dynamics of pVHL-dependent JADE1 stabilization and β-catenin regulation. Prog Biophys Mol Biol, https://doi.org/10.1016/j.pbiomolbio.2018.12.005 (2018).
Fakhar, M. & Rashid, S. Targeted inhibition of Klotho binding to fibroblast growth factor 23 prevents hypophosphetemia. J. Mol. Graph. Model. 75, 9–19 (2017).
Groban, E. S., Narayanan, A. & Jacobson, M. P. Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS Comput. Biol. 2, e32 (2006).
Birck, C. et al. Conformational changes induced by phosphorylation of the FixJ receiver domain. Structure. 7, 1505–1515 (1999).
Author information
Authors and Affiliations
Contributions
Conceptualization, S.R.; Investigation, data analysis, writing–original draft, S.S.; Writing–Review & Editing, S.R.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shafique, S., Rashid, S. Structural basis of βTrCP1-associated GLI3 processing. Sci Rep 9, 6865 (2019). https://doi.org/10.1038/s41598-019-43392-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43392-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
