
Abstract
Hybridization is a common phenomenon, yet its evolutionary outcomes remain debated. Here, we ask whether hybridization can speed adaptive evolution using resynthesized hybrids between two species of Texas sunflowers (Helianthus annuus and H. debilis) that form a natural hybrid in the wild (H. annuus ssp. texanus). We established separate control and hybrid populations and allowed them to evolve naturally in a field evolutionary experiment. In a final common-garden, we measured fitness and a suite of key traits for these lineages. We show that hybrid fitness evolved in just seven generations, with fitness of the hybrid lines exceeding that of the controls by 14% and 51% by the end of the experiment, though only the latter represents a significant increase. More traits evolved significantly in hybrids relative to controls, and hybrid evolution was faster for most traits. Some traits in both hybrid and control lineages evolved in an adaptive manner consistent with the direction of phenotypic selection. These findings show a causal pathway from hybridization to rapid adaptation and suggest an explanation for the frequently noted association between hybridization and adaptive radiation, range expansion, and invasion.
Similar content being viewed by others

Introduction
Although historically regarded as a transitory or rare phenomenon1,2,3, natural hybridization is currently recognized as common in plants (occurring globally in 40% of families4 and involving up to 25% of species in some floras)5, important in animals (frequency of interbreeding species: 0.1–3%)6 with up to 25% in some groups5, and increasingly found in fungi (reviewed in7). Arguing from theory, researchers have hypothesized for decades that hybridization can act as an evolutionary stimulus8,9,10,11,12,13. Evolution is constrained by the availability of standing genetic variation14,15. Novel genetic material can arise via two main mechanisms: either by new alleles from de novo mutations16,17 or via introgression of alleles from other populations or species (reviewed in18,19,20). In fact, hybridization has been shown to provide sources of new genetic material (reviewed in8,21, see examples in22,23) and there is mounting evidence in numerous systems that hybridization is associated with adaptation, speciation, and radiation8,9,10,24. For instance, in a meta-analysis, naturally-occurring hybrids had increased invasion potential (measured via proxies such as fecundity and size) relative to their progenitor nonhybrid species25. However, most of this evidence is correlational in nature, thus the causal nature of these relationships needs to be investigated empirically with field experiments.
Evolutionary experiments allow for the observation and characterization of evolutionary change in real time using baseline conditions that are known with a high degree of certainty. They can address many questions, including those related to adaptation, evolutionary tradeoffs, population genetic parameters, and other lineage-specific evolutionary hypotheses26,27,28. Conducting these experiments in a field setting allows for the assessment of changes under realistic conditions that include multidimensional selective pressures and relevant genotype-by-environment interactions29. Such studies (those that take place in the field, involve some type of manipulation, and last for multiple generations in situ) on the effects of hybridization have so far been carried out exclusively in crop-wild systems. These studies have documented that natural selection favors wild alleles and phenotypes in some Helianthus crop-wild hybrids30, and in Raphanus, crop-wild hybrids outperform nonhybrids in terms of survival and fecundity31,32,33,34. However, evolutionary outcomes of hybridization in crop systems may differ from those in wild systems, as environments are homogenized in the former35 and population genetic parameters and loci under selection differ between agricultural and wild settings36.
Here, we present a unique field experimental evolution study investigating whether natural hybridization can speed adaptation in the wild. We focus on the rate of adaptation (as well as the phenotypic endpoints) as this component of hybrid evolution has been little studied. As a model system, we used three taxa of annual Texas sunflowers, including the annual sunflower Helianthus annuus ssp. annuus, the hybrid-derived subspecies H. annuus ssp. texanus, and the cucumber-leaved sunflower H. debilis ssp. cucumerifolius. All taxa are annuals that are wild and native to North America: H. a. annuus is geographically widespread across nearly the entire continent, while H. a. texanus and H. debilis are both centered in Texas. The subspecies H. a. texanus is locally-adapted to the environmental conditions in Texas (see fitness comparisons in37) and has long been considered the product of natural introgression of Texas-adapted H. debilis alleles into the widespread species H. a. annuus38,39.
We used an eight-year field experiment to examine adaptive evolution in initial, multiple intermediate, and final generations of control (nonhybrid) and resynthesized hybrid (H. debilis × H. a. annuus backcrossed to H. a. annuus) populations in a common-garden setting. Examination of intermediate generations allows for fine-scale temporal resolution of evolutionary rates. We measure evolutionary changes in fitness and morphology, focusing on a suite of 27 ecophysiological, phenological, architectural, resistance/palatability, and herbivore damage traits to obtain a comprehensive picture of phenotypic evolution. We further use genomic data to detect allelic changes due to local gene flow. We ask: (1) does hybrid fitness evolve compared to controls? (2) do key traits evolve more rapidly in hybrids relative to controls?, and (3) can trait evolution be predicted by initial phenotypic distance from the locally-adapted phenotype?
Results
Hybrid fitness evolution in a field experiment
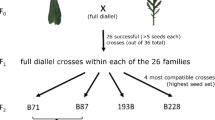
We synthesized a hybrid population by creating an F1 hybrid (H. a. annuus × H. debilis) and back-crossing it to H. a. annuus, resulting in BC1 individuals with approximately 75% H. a. annuus and 25% H. debilis genetic backgrounds to mimic the hypothesized genetic composition of the ancestors of the natural hybrid lineage38 (Fig. 1a). We established separate plots with 500 sunflower individuals in two locations approximately 14.5 km apart in central Texas in 2003, Lady Bird Johnson Wildflower Center (LBJ) and the Brackenridge Field Laboratory (BFL) (see Methods for details). At LBJ, we established both hybrid (BC1) and control (H. a. annuus) lines (separated by 260 m), but due to space limitations only a hybrid line was established at BFL. For all experimental lineages, the initial allelic composition was derived from sources hundreds of kilometers distant from the study site (see Methods and Supplementary Fig. 1), and indeed neither hybrid nor control populations were locally-adapted relative to local H. a. texanus (see generation 1 fitness in Fig. 2a). Thus, the design simulates a colonization event of a novel region, coupled (or not) with a hybridization event. Wild Helianthus neighbors were removed every spring to limit movement of alleles into or between the experimental plots (see Methods for details). This helped to reduce, but not eliminate, gene flow from local sources (see Supplementary Methods, Supplementary Fig. 2). Populations were allowed to reproduce naturally through generation 8; each generation we collected leaves from 96 individuals for genetic analyses and stored achenes (referred to as seeds hereafter) for common garden trials. In 2017, we established a final common garden at LBJ with multiple generations of controls and LBJ hybrids, the final generation of the BFL hybrid lineage, and multiple wild H. a. texanus accessions for comparison (Fig. 1b).

Field experimental evolution design. (a) Resynthesized hybrids were created by crossing H. a. annuus with H. debilis to form an F1 generation, then backcrossing this F1 generation to H. a. annuus to establish a hybrid BC1 seed stock. Sizes of illustrated inflorescences are approximately proportional to actual. (b) Initial populations of 500 individuals of Control (H. a. annuus, orange background) and Hybrid (BC1, dark blue background) were established in 2003 at separate plots at Lady Bird Johnson Wildflower Center (LBJ). A second Hybrid line was established at the Brackenridge Field Laboratory (BFL, light blue background). Lines were allowed to establish and reproduce naturally in situ for seven generations. Each generation, seeds were collected and stored from 96 randomly-chosen individuals per line. In 2017, a final common-garden was planted at LBJ with both control and hybrid seeds from generations one and five through eight for LBJ, generations one and eight for BFL, and accessions of locally-adapted H. a. texanus (not shown) for comparison.

Hybrid fitness evolves through time. Mean fitness values (seed production) +/− SEM for control (orange) and hybrid (blue) lines are shown. Hybrid fitness evolved at both LBJ (dark blue) and BFL (light blue) (95% credible intervals for modeled slope in Bayesian analysis are positive and do not overlap zero). Control fitness did not change (95% credible interval overlaps zero). The solid black line is the locally adapted wild hybrid (H. a. texanus) mean fitness value for comparison.
We tested our fundamental hypothesis, that hybridization can increase the rate of adaptation, by comparing the rate of fitness change across seven generations between hybrid and control populations. We estimated fitness as the total seed output of each individual (see Methods for details). We built Bayesian linear models in JAGS (Plummer 2003) to regress standardized fitness on generation and interpret significant positive slopes (β) as evolution of increased fitness. Control fitness did not change across generations (slope = 0.001, 95% credible interval = [−0.047, 0.047], Fig. 2). Hybrid fitness significantly increased through generational time (for LBJ hybrids: slope = 0.154, 95% credible interval = [0.096, 0.211], for BFL hybrids: slope = 0.115, 95% credible interval = [0.052, 0.180], Fig. 2). See Supplementary Table 1 for raw fitness values. Using a conservative Bayesian approach, we found that at LBJ control fitness exceeded hybrid fitness at generation 1 (mean standardized difference = −0.289, significant at 80% credible level [−0.535, −0.047], Fig. 2). Control and hybrid fitness values did not significantly differ in generations 5–7 at LBJ, but hybrid fitness exceeded control fitness in generation eight (by 56%; mean standardized difference = 0.320, significant at 95% credible level [0.007, 0.656], Fig. 2, Supplementary Table 2). Generation eight BFL hybrids had slightly higher fitness (14%) than LBJ controls, but this was not a significant difference (mean = 0.099, 95% credible interval = [−0.179, 0.429]. Thus, while both hybrid lineages were able to overcome low fitness levels in the early-generation (common in hybrids40,41 see meta-analysis in25) and evolve significantly, they significantly exceeded control fitness in one of the cases. The larger increase in performance of LBJ vs. BFL hybrids over controls may reflect local adaptation in the former to the common garden site, since the final common-garden was planted at home site for LBJ control and hybrid lines but was 14.5 km distant from the home site of the BFL hybrids.
More traits evolved significantly in hybrids than in controls
We tracked evolution of ecophysiological, phenological, architectural, resistance/palatability, and herbivore damage traits (see Table 1, Methods). To determine which traits evolved through time, we ran the same Bayesian linear regression models as we did for fitness separately for each of the 27 traits. In the control population, six traits out of 27 evolved with strong support (significant at the 95% credible level), while two additional traits evolved with moderate support (significant at the 80% credible level) (Fig. 3a). In the LBJ hybrid population, 16 traits out of 27 evolved with strong support and an additional three traits evolved with moderate support, while in the BFL hybrid population seven traits evolved with strong support and an additional 6 traits evolved with moderate support (Fig. 3a). The number of traits evolving in controls and LBJ hybrids differed significantly for traits that evolved with strong support (Χ2 = 4.55, df = 1, p = 0.033) and for those with both strong and moderate support (Χ2 = 4.48, df = 1, p = 0.034). The number of traits evolving in controls and BFL hybrids did not differ significantly for traits evolving with strong support (Χ2 = 0.07, df = 1, p = 0.782) or traits evolving with both strong and moderate support (Χ2 = 1.19, df = 1, p = 0.275). See Supplementary Table 1 for raw trait data summaries and Supplementary Table 3 for full results from the Bayesian regression analyses.

Hybridization leads to changes in the direction and magnitude of trait evolution. (a) Heatmaps for trait value changes through time for each of 27 different traits in controls and hybrids (LBJ and BFL). Color indicates whether the trait values increased (blue) or decreased (pink) through time. Squares with an “a” (“adaptive”) indicate that both selection gradients (measured in generation 1) and evolution through time were statistically significant and in the same direction, evidence that these traits evolved adaptively. Shading intensity increases with the absolute value of the posterior estimate. (b) Heatmaps measuring the degree to which traits in control and hybrid populations evolved at different rates, with either the absolute value of control (orange) or hybrid (blue) trait change estimates being steeper, and shading intensity increasing with a greater difference in steepness. Across both panels, squares outlined in black were significant at the 95% credible level, those outlined with black dashed lines were significant at the 80% credible level, and those with no outline were not significant.
Some traits evolved adaptively
Although traits evolved in both controls and hybrids, not all evolution was necessarily adaptive in nature, as it could have been driven by demographic factors such as genetic drift or by genetic correlations with other traits under selection; we note that many traits were correlated (Supplementary Fig. 3), but the strength of correlations did not differ between controls and hybrids. To assess which instances of trait evolution were adaptive, we compared the direction of trait changes with a phenotypic selection analysis (PSA) performed in 2003, the initial year of the experiment37,42. If both the evolution of the trait and the selection gradient (β, the partial regression coefficients from a multiple regression, see Methods) were significant and in the same direction, we interpreted this as evidence for adaptive evolution (see Supplementary Table 4 for PSA results). In controls, two out of six traits with significant selection gradients fit these criteria for adaptive evolution, while in LBJ and BFL hybrids, five out of ten and three out of six traits with significant selection gradients fit these criteria (Fig. 3a), though this difference in proportions between treatments is not significant (LBJ versus controls: Χ2 = 0.033, df = 1, p = 0.855, BFL versus controls: Χ2 = 0.033, df = 1, p = 0.855). Conversely, we examined how many of the traits that evolved significantly also evolved adaptively. In controls two out of five traits that evolved did so in an adaptive manner, while four out of 16 did so for LBJ hybrids and three out of six did so for BFL hybrids (Fig. 3a). These differences in proportion were not significant (LBJ versus controls Χ2 = 0.025, df = 1, p = 0.874, BFL versus controls Χ2 = 0.011, df = 1, p = 0.916). Note that not all traits measured in 2017 were included in the 2003 phenotypic selection analysis.
Rates of trait evolution are faster in hybrids relative to controls
We resampled estimates from the posterior distributions of the Bayesian slope estimates of trait change through time to create new posterior distributions comparing trait evolution in hybrids versus controls, subtracting the absolute value of control slope estimates from the absolute value of hybrid slope estimates, a conservative approach43. For eight traits the LBJ hybrids had steeper slope values than controls (with strong support) and four additional traits showed moderate support for a steeper slope (Fig. 3b). For two traits the BFL hybrids had steeper slope values than controls (with strong support) and four additional traits showed moderate support for a steeper slope Fig. 3b). There were zero traits for which the controls had strong support for steeper slopes, though two traits had moderate support (LBJ, Fig. 3b). Differences in the number of traits evolving faster in LBJ hybrids versus controls were significant when examining those with strong support (Χ2 = 8, df = 1, p = 0.005) and those with both strong and moderate support (Χ2 = 7.14, df = 1, p = 0.007). Differences in trait evolution for the BFL hybrids versus controls were not significant when examining those with strong support (Χ2 = 2.00, df = 1, p = 0.157) or those with both strong and moderate support (Χ2 = 2.00, df = 1, p = 0.157). As an additional metric, we measured the rate of trait evolution in haldanes (proportional change over generational time elapsed)44. Control traits evolved faster than LBJ hybrids in only three traits (mean absolute rate = 0.003 haldanes, range 0.000–0.012), while the LBJ hybrids evolved faster in the remaining 24 traits (mean absolute rate = 0.011 haldanes, range = 0.001–0.028), a significant difference (Χ2 = 16.3, df = 1, p < 0.001). Controls evolved faster than BFL hybrids in ten traits, while the BFL hybrids evolved faster in the remaining 17 traits (mean absolute rate = 0.007 haldanes, range 0.001–0.018), again a significant difference (Χ2 = 9.80, df = 1, p = 0.002) (Table 1).
Distance from the locally-adapted phenotype predicts trait evolution
Traits evolved in both hybrids and controls at different rates (above). We asked whether the initital phenotypic distance to the locally-adapted taxon (H. a. texanus) could predict the rate and direction of trait evolution. To do so, we computed the distance from the standardized average trait value of our experimental plants in generation 1 (hybrids and controls separately) to the average trait value of H. a. texanus planted in the garden. We used a simple linear regression to relate distance to the slopes from the Bayesian trait evolution analyses. Control and hybrid (both LBJ and BFL) evolutionary rates were both positively and significantly correlated with distance from the locally-adapted phenotype, meaning that traits with initial values far from those of the local phenotype tended to evolve faster than traits starting with trait values similar to the local phenotype, and in a direction toward the values of H. a. texanus (Fig. 4). However, the control estimate (coefficient = 0.053, p = 0.001) was not as strong as the LBJ hybrid estimate (coefficient = 0.120, p < 0.001), but similar to the BFL hybrid estimate (coefficient = 0.065, p < 0.001). This pattern indicates that the LBJ hybrid population was able to approach a locally-adapted phenotype more rapidly than controls, even when hybrids and controls started at an equal level of presumed maladaptation (i.e., for a given initial phenotypic distance between the experimental populations and H. a. texanus, the estimated rate of evolution is faster for hybrids than controls, Fig. 4a). See Supplementary Table 5 for values used.

Distance from the locally-adapted phenotype predicts trait evolution. For both control (orange) and hybrid (blue) lineages, the rates of evolution for individual traits (using the slope values from the Bayesian evolution analyses) are significantly correlated with the distance from the initial mean trait value (BC1 and initial H. a. annuus generation for hybrids and controls, respectively) to the mean trait value of H. a. texanus. Each point represents an individual trait. Note that the same control lineage originally grown at LBJ (orange) is shown in both figure panels. (a) LBJ hybrid lineage vs. control. (b) BFL hybrid lineage vs. control.
Hybrid populations were susceptible to local gene flow
We found outside alleles in most, but not all hybrid samples (note that outside alleles could not be surveyed in the control population, see Methods). Although the proportion of outside alleles varied between individuals, a fraction of hybrid samples (14 LBJ and 17 BFL) had virtually no outside alleles (<0.5%), suggesting these samples had no outside ancestry and supporting the validity of the test statistic. For the LBJ experimental hybrid population, the percentage of admixed individuals increased with generation, from 0% in generation 1 (the BC1 generation), to 78% in generation 3 and 100% by generation 6 (Supplementary Fig. 2a). The BFL hybrid population had a similar trajectory, from 83% in generation 3 to 100% in generation 7 (Supplementary Fig. 2). The average percentage of novel alleles increased from generation 3 to generation 5, but subsequently stabilized at ~5%, suggesting fewer migrants in later generations (Supplementary Fig. 2b,c).
Discussion
Hybridization can result in adaptive introgression of fitness-enhancing alleles from one species into another45. While de novo mutations are biased toward deleterious effects and occur at relatively low rates over time, introgressing alleles can be introduced simultaneously in large numbers. Further, introgressing alleles have been tested by natural selection in the donor species, albeit in a different genetic background, and thus may be more likely to be beneficial. This mechanism is one potential explanation for the observed rapid increase in fitness and evolution of key traits in hybrids observed in our experiment. Adaptive introgression has been proposed in many examples (reviewed in18,19,46), including this system. Heiser originally proposed that H. a. texanus is the product of natural hybridization between H. debilis and H. a. annuus, with incorporation of genetic material from H. debilis allowing for the southward range expansion of H. annuus into Texas.
Previous work in this system found evidence for adaptive introgression of traits related to abiotic tolerance42 and herbivore resistance37 and identified putative QTL underlying these traits47. Our experiment finds rapid evolution in both traits and fitness in resynthesized hybrids, potentially due to the introgression of traits and alleles from H. debilis. Traits in all broad categories (ecophysiology, phenology, architecture, resistance/palatability, and herbivore damage) tended to evolve faster in hybrids than in controls (Fig. 3). The potential adaptive nature of these traits has been addressed in more detail in previous work (see37,42), but briefly we note that the damage done by seed predators (seeds killed by midges, MidgeDam; seeds damaged by Isophrictis holes, HoleDam; and seeds killed by seed weevils, GSW) is a strong selective pressure, apparently resulting here in decreased rates of damage in later generations. Other changes in ecophysiology or architecture may be associated with adaptation to the local abiotic environment, although we did not test for specific causation here. For instance, in both hybrid lines, specific leaf area (SLA) evolved higher values, perhaps in response to the warmer conditions in the study area relative to those experienced by the more northern H. a. annuus parent. Additionally, phenotypic distance from the locally-adapted phenotype (H. a. texanus) is correlated with the speed of evolution, indicating that this experiment may be “replaying” the natural history of hybridization, introgression, and adaptation in this system.
A competing (but not mutually-exclusive) explanation for the observed rapid evolution in hybrids is that, relative to the control genome, the hybrid genome may have permitted more rapid introgression of local alleles from wild H. a. texanus in the local environment. While we reduced local gene flow by removing wild Helianthus individuals near our experimental plots, we did not eliminate it, and novel alleles began to accumulate in the hybrid populations, stabilizing at around 5% of loci by generation 6 at both LBJ and BFL (Supplementary Fig. 2). The genomes of the original BC1 hybrid plants, and plants of subsequent generations, may have been more porous than those of controls due to a low diversity of self-incompatibility (SI) alleles, as has been found in Senecio squalidus in the British Isles48. In this scenario, a large fraction of the incoming pollen from local sunflowers would have had unique SI alleles relative to experimental hybrids, and thus would have had fertilization advantage. An alternative mechanism for increased porosity posits that hybrids may have had relatively high genetic load (relative to controls) via outbreeding depression (as evidenced by low initial fitness of the hybrids, Fig. 2), which in turn would lead to higher than neutral introgression of local alleles49,50. Under this mechanism, offspring sired by local pollen would have had higher fitness, resulting in a reduction in genetic load and an increased frequency of local alleles in subsequent generations. Rather than an experimental artifact, this porosity is likely a general a feature of natural hybridization events, where initial populations of interspecific hybrids are small in size and are likely to have one or both of the above-described genomic features (low SI allele diversity, high genetic load). We posit that historical formation of the wild hybrid H. a. texanus may have been shaped by this process as well, with high porosity and allele-sharing between multiple, independently formed early-generation hybrid populations contributing to rapid spread of advantageous alleles.
We found higher rates of trait evolution in hybrids relative to controls, both in terms of the slope of change over time (Fig. 3b) and in evolutionary rates measured in haldanes (Table 1). The latter rates allow comparisons with previous studies in plants over similar generational time periods. Bone and Farres51 compiled microevolutionary rates in plants across 23 studies, 15 of which include estimates in haldanes. Rates ranged from 0–0.808, with a mean of 0.158 haldanes. Most rates fell at the lower end of the distribution (<0.05 haldanes) with a median of 0.047 haldanes and only four studies with rates higher than 0.5 haldanes51. The rates we estimated in our study (means of 0.0003, 0.011, and 0.007 haldanes for controls, LBJ hybrids, and BFL hybrids, respectively, range 0 to 0.028), are well within this wide range.
Here we show that, despite initial low fitness values, Helianthus hybrids rapidly evolved higher fitness than non-hybrid controls. Further, key traits associated with ecophysiology (such as specific leaf area, leaf succulence, leaf length-width ratio), phenology (bud initiation time, seed maturation time), architecture (floral disk diameter, branching height), and herbivore resistance (seeds killed by midges or holes) evolved faster in hybrids relative to controls, perhaps enabling hybrids to acquire resources and produce viable offspring more effectively. Our results provide experimental support for the long-hypothesized connection between hybridization and more rapid adaptive evolution and provide a set of potential mechanisms for the long-noted association of hybridization with diverse evolutionary phenomena including adaptive radiations, invasions, and range expansions. However, an important caveat is that we have been able to examine only one control and two hybrid populations, given logistical constraints inherent in conducting a large field experiment over eight years. Replication in studying the evolutionary outcomes of hybridization will need to be built up across (rather than within) studies, as has been done for other evolutionary questions requiring intensive field-based investigation. For instance, a series of experiments transplanted populations of the Trinidadian guppy Poecilia reticulata between environments, there by manipulating exposure to predators52,53,54,55. Each study was conducted using either microcosms or field systems in different rivers or tributaries, building on each other and on previous observational work to form a more complete picture of life history and color pattern evolution in response to predators (reviewed in28). Replication of field experiments within our Helianthus system and other plant and animal systems should provide additional evidence on the degree to which hybridization can speed adaptive evolution, while elucidating details regarding the context-dependent nature of hybrid evolution.
Methods
Establishment of hybrid and control lineages
Our design simulated the arrival of a non-locally-adapted taxon (H. a. annuus) to the study area from the north, followed either by hybridization with the local H. debilis (BC1 hybrid lines) or not (H. a. annuus control line). These populations then evolve in, and exchange genes with, a matrix of other hybrid populations, similar to the expected scenario during the historical formation of H. a. texanus. Note that the parental species H. debilis is native to the study area, and thus would not be an informative non-hybrid control for the rate of local adaptation, since it is presumably already locally adapted. The BC1 generation was obtained by first mating H. debilis from Texas to wild H. a. annuus from Oklahoma to produce F1 progeny in the greenhouse (Supplementary Table 6, see details in37). To produce enough BC1 seed for replicate field populations, a single progeny from the F1 generation was selected and propagated vegetatively to produce 14 F1 clones. A single H. a. annuus pollen donor from north Texas (the recurrent parent) was then mated to the F1 clones. Controls were field-collected H. a. annuus from the recurrent parent population “RAR59” (see Table 1 in37), consisting of roughly 10 seeds from each of 50 maternal sibships.
Seeds for both hybrids and controls were germinated, planted in field soil in peat pots, and grown in the greenhouse for one month before transplanting. One pair of control (H. a. annuus) and hybrid (BC1) populations were established at Lady Bird Johnson Wildflower Center (LBJ, 30.184°N, −97.877°W), with plots separated 260 meters and a dense copse of trees to limit gene flow between plots. To assess the generality of hybrid responses in different parts of the H. a. texanus range, a second hybrid population was established approximately 14.5 km away at the Brackenridge Field Laboratory (BFL, 30.282°N, −97.780°W). Space limitations prevented the establishment of a control population at BFL. All three populations were initiated with 500 individuals in late March 2003. The source populations for the BC1 line are far away from LBJ (H. debilis F1 parent: ~300 km; H. a. annuus F1 parent: ~650 km; H. a. annuus recurrent parent: ~375 km; see Supplementary Fig. 1, Supplementary Table 6), as is the source population for the control line (~375 km), so both types of experimental populations should be less locally adapted to the establishment sites than is either H. a. texanus or the parental species H. debilis. Populations were allowed to evolve naturally without human interference, with two exceptions. First, as annual sunflowers are early-successional species and require annual soil disturbance to maintain population sizes, we disturbed each plot each winter using a rototiller. Second, to provide some isolation to the experimental populations, we reduced (but did not eliminate, see Supplementary Fig. 2) the rate of local gene flow, by pulling all wild sunflowers that emerged within a 250 m buffer surrounding each plot prior to flowering each year.
Each year, we collected seeds and leaves from 96 individuals per population for use in the final common-garden and for genetic analyses, respectively. Seeds were stored at 20 °C in paper coin envelopes in sealed plastic tubs with drierite (W. A. Hammond DRIERITE Co., Ohio, USA) to maintain low humidity. The experiment had to be terminated after eight generations because of a change in land use at the host site.
Final common-garden
Stored seeds were germinated at the University of New Mexico (UNM). These included seeds from hybrid and control lineages, as well as seeds from wild H. a. texanus (see Supplementary Table 6 for source population information). Germination protocols are described in detail in37 and included hand-scarification of seeds germination on filter paper, and transplantation into peat pellets (Jiffy J3675, Oslo, Norway), followed by roughly one month of growth in the UNM greenhouses. Over ten thousand seeds were scarified for this study, with an average germination rate (across all lines) of 36%. The final common garden was planted at LBJ between April 2 and April 4, 2017. One-month old seedlings were transported to Texas and planted 90 cm apart in rows 1 m apart. We aimed to plant 60 individuals per lineage for final generation and wild H. a. texanus lines and 30 per lineage intermediate generations, using approximately equal numbers of seeds per maternal line to avoid overrepresenting any individual half-sibling families. Due to variable germination rates, the actual number of individuals planted varied (Supplementary Table 7). In particular, low seed availability and low germination rates (the latter likely due to age-associated mortality in storage, presumably associated with seed pathogens acquired from the field) meant that we were unable to include generations two through four in the final common garden. Note that in contrast, generation one plants were formed in the greenhouse, and therefore this generation was not exposed to these putative seed pathogens. The garden included 1002 plants total, 615 used in this study. Heavy rainfall just prior to planting meant that plants were able to successfully establish without planned hand-watering.
Trait measurements
Traits were measured as in37,42; see Table 1 for traits, abbreviations, and units. Briefly:
Ecophysiological traits
Specific leaf area (SLA, cm2 ∙ g−1) is the ratio of leaf area to mass and a measure of overall leaf construction costs (higher values associated with less cost per light-absorbing area). Leaf succulence (Succ) is calculated as (leaf wet mass − dry mass)/wet mass, while leaf dry matter content (LDMC) is calculated as leaf dry mass/wet mass. Leaf length to width ratio (LWR) is an estimate of the narrowness of the leaf and is calculated as leaf length/leaf width. Leaf water use efficiency (WUE) is the rate of carbon gained via photosynthesis per unit of water lost via transpiration, measured using δ13C. Leaf longevity (LeafLong) is the length of time that a leaf remains on the plant, measured in days. Two fully expanded leaves per plant were selected in the period before first flowering, one for WUE and one for the remaining leaf traits. An expanding leaf of approximately 4 cm was selected and tagged with a jewelry tag to track leaf longevity. Chlorophyll content (Chloro) was estimated using a SPAD meter (Spectrum Technologies, Aurora, IL), averaged across five measurements. Wet mass was measured on a microbalance, leaves were scanned using a flatbed scanner, and leaves were dried in a drying oven until constant mass was reached, and dry mass was then measured. One ca. 3 mg sample from dried leaf disk tissue (taken using a #7 cork borer) per plant was weighed in tin and analyzed for Carbon, Nitrogen, 13C, and 15N content at the University of New Mexico Center for Stable Isotopes. To reduce costs, only a subset of individuals was submitted for isotope analysis and leaf Carbon:Nitrogen ratio (CNratio) (Supplementary Table 7).
Phenology
Phenological status of all plants in the final common garden was assessed every third day beginning on May 1 until the majority of plants had senesced in early November. From these assessments, we calculated bud initiation time (DaysToBud) as the number of days between transplanting and the first appearance of the immature apical flowering head, seed maturation time (SMT) as the number of days between the end of stigma receptivity and achene maturity for the apical head, and plant longevity (Longevity) as the number of days between transplanting and mortality. For a small number of plants remaining alive after the last census date in November, we added one or two weeks to longevity based on the plant’s appearance (10 and 3 individuals, respectively). Eliminating these outliers from the analyses did not affect results.
Architectural traits
Disk diameter (DiskDiam, mm) is the diameter of the central disk of the apical flowering head measured during stigma receptivity. Height of the lowest branch (HtLow, cm) is the height of the lowest branching point on the primary stem. Bushiness (Bushy, a measure of higher-order branching) was estimated as the mean branch position of all flowering heads on the plant56, where heads originating on the main stem have a branch position of 1, heads from a primary branch have a branch position of 2, and heads from a secondary branch have a position of 3. Relative branch diameter (RelBrDiam) is a measure of investment in branches relative to the main stem and is estimated as the average branch diameter across all primary branches with diameters at the base > = 3 mm, divided by the stem diameter. Plant volume (Volume, cm3) is an estimate of overall plant size and is calculated using the equation for the volume of a cylinder, V = π × r2 × l, where r is half the diameter of the primary stem and l is the height of the plant.
Seed damage and fitness
For seed count and seed predation measurements, individual seed heads were enclosed with bags made of plastic mesh (DelStar Technologies, Delaware) after pollination but before seed release and collected throughout the season, with a goal of >4 heads per plant. At plant senescence, all remaining heads (both bagged and unbagged) were collected and counted. Damage to receptacles (the structures subtending the inflorescence) by head-feeding Lepidoptera was measured by counting the number of larval holes in a sample of one to eight mature receptacles per plant and taking the average (RecepDam). Seed damage was visually assessed under a dissecting microscope (Leica, Wetzlar, Germany). Seeds were sorted into categories including viable, parasitoid attacked (ParaDam), midge damaged (MidgeDam, the sunflower seed midge Neolasioptera helianthis; Diptera: Cecidomyiidae combined with parasitoid damage), hole damaged (HoleDam, Isophrictis sp.; Lepidoptera: Gelechiidae), and gray seed weevil damaged (GSW, Smicronyx sordidus; Coleoptera: Curculionidae). In total, 310,378 seeds were scored for this study (mean = 536 seeds per plant). Damage scores were calculated as fractions (number of seeds in each category/total seeds scored per plant). Viable seed production was chosen as the measure of fitness in these annual plants. Seed production was estimated by multiplying the total number of heads produced (bagged + unbagged) by the average number of viable seeds per bagged head.
Resistance/palatability traits
Densities of glandular (GlandDens) and nonglandular (HairDens) trichomes were measured on a single dried leaf disk per plant. Trichomes were counted using a Leica MZ 125 compound light microscope (Leica, Wetzlar, Germany) under 5x magnification with a 1 cm × 1 cm reticle and converted to densities (measuring a 0.2 cm × 0.2 cm area, 0.04 cm2). The ratio of leaf carbon to nitrogen (CNratio) was estimated using values from the Center for Stable Isotopes (see above).
Herbivore damage traits
Insect damage to leaves was scored twice for each plant, once in mid-June and once in late-July. Briefly, we scored percent cover of damage on three of the oldest leaves per plant caused by different types of herbivores and calculated a damage index D for each measured as percentage of leaf area, see37 for details. Composite damage indices for leaf-vascular-tissue feeders (SuckDam: Hemiptera, Homoptera) and for leaf chewers (ChewDam: Orthoptera, Lepidoptera, Diptera) were constructed by summing D scores for each of the component taxa. Stem and petiole damage were assessed in mid-June as a continuous trait, counting the number of lesions on all stems and petioles caused by Rhodobaenus weevils (Coleoptera: Curculionidae) (WeevilDam), and number of holes per plant caused by stem-boring larvae (StemBorer, Coleoptera, Lepidoptera).
Statistical analysis: general
All analyses were carried out in R v3.3.157. Prior to analysis, of the 615 original plants, we filtered out early transplant deaths (n = 9), plants apparently damaged during seed scarification that never developed a root system (n = 10), and plants that lived less than 75 days because the majority of traits could not be measured on these plants (n = 35; note that including these plants in analyses did not affect fitness results). Additionally, when analyzing architectural traits, we excluded plants with weevil damage to the primary bud, as these resulted in abnormal growth architectures. See Supplementary Table 7 for final counts of each line analyzed. Trait values across control and hybrid lines were standardized to a mean of zero and standard deviation of one.
Trait evolution
We used Bayesian linear regression models to estimate the evolutionary change in phenotypic values of traits through time. We ran separate models for each trait (response variable, Eq. (1)), with generation as covariate, using separate slopes and intercepts for control and hybrid lines, where \({y}_{i}\) is the standardized value of each trait for individual \(i\), \({x}_{i}\) is the covariate generation:
β0j is the intercept term for treatment j (control or hybrid) and βj is the regression coefficient for treatment j. For the slope and intercept terms, we used normal priors with a mean of zero and variance of one, while the error term was assigned a gamma prior with shape and scale both equal to one. We implemented the models in JAGS v 4.3.058 and report results from five MCMC (Markov Chain Monte Carlo) chains run for 100,000 iterations (with a burnin of the first 25,000 discarded), thinned every 25th iteration, with 10,000 iterations saved for the posterior sample. We used traceplots to check for convergence, and verified that Rhat values were under 1.01 for all parameters59. Regression coefficients were deemed significant if the 95% credible intervals did not overlap zero. We used all 10,000 posterior samples to create posterior distributions for the difference between treatments by subtracting the values for the hybrid coefficient samples from the control coefficient samples and taking the absolute values of these differences. From these new distributions, we estimated means and 95% credible intervals, where intervals that did not overlap zero indicate that coefficients differ between treatments.
To explicitly compare fitness values for controls against hybrids for each generation (LBJ) or the final generation (BFL), we ran Bayesian models using the stan_glmer() function in the R library rstanarm60 using standardized fitness values as a response with a random terms for lineage. We increased adapt_delta to 0.999 to avoid divergent transitions and used N(0,100) priors. We ran each model using four chains for 2000 iterations each including a 1000 iteration burnin and checked Rhat values (<1.01) to ensure that the chains converged. We compared the posterior distributions by subtracting the samples of control fitness from the samples of the hybrid fitness, producing a mean and full distribution of the differences.
We calculated the allochronic evolutionary rates in haldanes, the proportional change over generational time elapsed (H, Eq. (2)), for both controls and hybrids using the method of 44:
where \({\bar{y}}_{8}\) is the mean natural log-transformed value of the trait in generation 8 and \({\bar{y}}_{1}\) is the mean natural log-transformed value of the trait in generation 1, sp is the pooled standard deviation of the samples, and g is number of generations passed (g = 7).
Phenotypic selection analysis
To determine whether evolution was adaptive for individual traits, we determined if the direction of trait evolution aligned with trait – fitness associations during 2003, the first field generation. We estimated selection differentials (s′) and selection gradients (β) from 2003 trait and fitness data separately for controls and hybrids at each home site (BFL and LBJ)37,42. Predictor variables were transformed to approach normality then standardized to a mean of 0 and standard deviation of 1 within each treatment. Linear selection differentials were estimated from the covariance of relative fitness within treatment (hybrid or control). Linear selection gradients were estimated from the partial regression coefficients from a multiple regression using the lm() function in R (see Supplementary Table 4 for traits used in the regression models). Note not all traits measured in 2017 were measured in 2003, e.g. Chloro, LWR, and ParaDam were not measured in 2003. Assumptions of normality were violated when using relative fitness values, so we generated 95% confidence intervals using bias-corrected bootstrap sampling of 10,000 replicates using the boot61 package in R. Phenotypic selection results for hybrids were published previously37,42, while results for controls are previously unpublished. In the current analyses, we included additional traits not included in previous analyses, so values of β (which depend on the phenotypic correlations of the focal trait with the other traits in the model, and with selection on those other traits) will necessarily differ somewhat from previously reported results. Full results are presented in Supplementary Table 4.
Predicting the speed of evolution
For each trait, we calculated the distance from the mean value of the initial (2003) generation hybrids and controls to that of H. a. texanus, which we assume to have a locally-adapted phenotype. Across all traits, we estimated the correlation between this distance and the slope from the Bayesian analysis (as a measure of the speed of evolution) using the lm() function in R.
Detection of local gene flow
We used genotyping-be-sequencing to determine if our experimental hybrid populations experienced local gene flow from outside the experiment (e.g., from naturally occurring H. a. texanus individuals) over the course of the study. We sequenced 90 samples from the original BC1 generation, plus 229 LBJ and 264 BFL samples from subsequent generations 3 to 8; material from generation 2 was not available. See Supplementary Methods for details on DNA extraction, library preparation, sequencing, and bioinformatics. Since our hybrid population is entirely derived from a single backcross, all variants should be found in the BC1 population at approximately 25%, 50% or 75% frequency. We did not attempt to examine the control population in this way, as its much higher starting level of allelic diversity would have made identification of outside alleles difficult or impossible. In our hybrid populations, variants that only appear in subsequent generations are likely to be the result of outside gene flow. To quantify this gene flow, we filtered for sites sequenced in at least 20 BC1 samples and catalogued all alleles present. We then looked in all subsequent generation samples and calculated the percentage of called variants that were not in our catalogue from the BC1s. See Supplementary Methods for full explanation of genetic work.
References
Wagner, W. H. Biosystematics and evolutionary noise. Taxon 19, 146–151 (1970).
Schemske, D. W. Understanding the origin of species. Evolution 54, 1069–1073 (2000).
Mayr, E. A local flora and the biological species concept. American Journal of Botany 79, 222–238 (1992).
Whitney, K. D., Ahern, J. R., Campbell, L. G., Albert, L. P. & King, M. S. Patterns of hybridization in plants. Perspectives in Plant Ecology, Evolution and Systematics 12, 175–182 (2010).
Mallet, J. Hybridization as an invasion of the genome. Trends in Ecology & Evolution 20, 229–237 (2005).
Schwenk, K., Brede, N. & Streit, B. Introduction. Extent, processes and evolutionary impact of interspecific hybridization in animals. Philosophical Transactions of the Royal Society of London B: Biological Sciences 363, 2805–2811 (2008).
Albertin, W. & Marullo, P. Polyploidy in fungi: evolution after whole-genome duplication. Proceedings of the Royal Society of London B: Biological Sciences 279, 2497–2509 (2012).
Seehausen, O. Hybridization and adaptive radiation. Trends Ecol. Evol. 19, 198–207 (2004).
Stebbins, G. L. The role of hybridization in evolution. Proceedings of the American Philosophical Society 231–251 (1959).
Abbott, R. et al. Hybridization and speciation. Journal of Evolutionary Biology 26, 229–246 (2013).
Schierenbeck, K. A. & Ellstrand, N. C. Hybridization and the evolution of invasiveness in plants and other organisms. Biol Invasions 11, 1093 (2009).
Anderson, E. & Stebbins, G. L. Hybridization as an evolutionary stimulus. Evolution 8, 378–388 (1954).
Arnold, M. L. Natural Hybridization and Evolution. (Oxford University Press, 1997).
Barrett, R. D. H. & Schluter, D. Adaptation from standing genetic variation. Trends in Ecology &. Evolution 23, 38–44 (2008).
Schluter, D., Clifford, E. A., Nemethy, M. & McKinnon, J. S. Parallel evolution and inheritance of quantitative traits. The American Naturalist 163, 809–822 (2004).
Nei, M. Genetic polymorphism and the role of mutation in evolution. Evolution of genes and proteins 71, 165–190 (1983).
Nei, M. Mutation-driven evolution. (OUP Oxford, 2013).
Suarez-Gonzalez, A., Lexer, C. & Cronk, Q. C. B. Adaptive introgression: a plant perspective. Biology Letters 14, 20170688 (2018).
Hedrick, P. W. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology 22, 4606–4618 (2013).
Marques, D. A., Meier, J. I. & Seehausen, O. A combinatorial view on speciation and adaptive radiation. Trends in Ecology & Evolution (in press).
Anderson, E. Introgressive hybridization. Biological Reviews 28, 280–307 (1953).
Rieseberg, L. H. Evolution: replacing genes and traits through hybridization. Current Biology 19, R119–R122 (2009).
Lewontin, R. C. & Birch, L. C. Hybridization as a source of variation for adaptation to new environments. Evolution 20, 315–336 (1966).
Mallet, J. Hybrid speciation. Nature 446, 279–283 (2007).
Hovick, S. M. & Whitney, K. D. Hybridisation is associated with increased fecundity and size in invasive taxa: meta-analytic support for the hybridisation-invasion hypothesis. Ecology Letters 17, 1464–1477 (2014).
Kawecki, T. J. et al. Experimental evolution. Trends in Ecology & Evolution 27, 547–560 (2012).
Garland, T. & Rose, M. R. Experimental Evolution: Concepts, Methods, and Applications of Selection Experiments. (University of California Press, 2009).
Reznick, D. N. & Ghalambor, C. K. Selection in nature experimental manipulations of natural populations. Integrative and Comparative Biology 45, 456–462 (2005).
Mitchell, N. & Whitney, K. D. Can plants evolve to meet a changing climate? The potential of field experimental evolution studies. Am. J. Bot. 105, 1613–1616 (2018).
Corbi, J., Baack, E. J., Dechaine, J. M., Seiler, G. & Burke, J. M. Genome-wide analysis of allele frequency change in sunflower crop–wild hybrid populations evolving under natural conditions. Molecular Ecology 27, 233–247 (2018).
Hovick, S. M., Campbell, L. G., Snow, A. A. & Whitney, K. D. Hybridization alters early life-history traits and increases plant colonization success in a novel region. Am. Nat. 179, 192–203 (2012).
Campbell, L. G. & Snow, A. A. Competition alters life history and increases the relative fecundity of crop–wild radish hybrids (Raphanus spp.). New Phytologist 173, 648–660 (2007).
Campbell, L. G., Snow, A. A. & Ridley, C. E. Weed evolution after crop gene introgression: greater survival and fecundity of hybrids in a new environment. Ecology Letters 9, 1198–1209 (2006).
Snow, A. A. et al. Long-term persistence of crop alleles in weedy populations of wild radish (Raphanus raphanistrum). New Phytologist 186, 537–548 (2010).
Stukenbrock, E. H. & McDonald, B. A. The origins of plant pathogens in agro-ecosystems. Annu. Rev. Phytopathol. 46, 75–100 (2008).
Doebley, J. F., Gaut, B. S. & Smith, B. D. The molecular genetics of crop domestication. Cell 127, 1309–1321 (2006).
Whitney, K. D., Randell, R. A. & Rieseberg, L. H. Adaptive introgression of herbivore resistance traits in the weedy sunflower Helianthus annuus. The American Naturalist 167, 794–807 (2006).
Heiser, C. B. Hybridization in the annual sunflowers: Helianthus annuus x H. debilis var. cucumerifolius. Evolution 5, 42–51 (1951).
Rieseberg, L. H., Beckstrom-Sternberg, S. & Doan, K. Helianthus annuus ssp. texanus has chloroplast DNA and nuclear ribosomal RNA genes of Helianthus debilis ssp. cucumerifolius. Proceedings of the National Academy of Sciences 87, 593–597 (1990).
Campbell, L. G., Snow, A. A. & Sweeney, P. M. When divergent life histories hybridize: insights into adaptive life‐history traits in an annual weed. New Phytologist 184, 806–818 (2009).
Sackman, A. M. & Rokyta, D. R. The adaptive potential of hybridization demonstrated with bacteriophages. J Mol Evol 77, 221–230 (2013).
Whitney, K. D., Randell, R. A. & Rieseberg, L. H. Adaptive introgression of abiotic tolerance traits in the sunflower Helianthus annuus. New Phytologist 187, 230–239 (2010).
Holsinger, K. E. & Wallace, L. E. Bayesian approaches for the analysis of population genetic structure: an example from Platanthera leucophaea (Orchidaceae). Molecular Ecology 13, 887–894 (2004).
Gingerich, P. Rates of evolution: effects of time and temporal scaling. Science 222, 159–162 (1983).
Rieseberg, L. H. & Wendel, J. F. Introgression and its consequences in plants. Hybrid zones and the evolutionary process 70, 109 (1993).
Martin, S. H. & Jiggins, C. D. Interpreting the genomic landscape of introgression. Current Opinion in Genetics & Development 47, 69–74 (2017).
Whitney, K. D. et al. Quantitative trait locus mapping identifies candidate alleles involved in adaptive introgression and range expansion in a wild sunflower. Molecular Ecology 24, 2194–2211 (2015).
Abbott, R. J. et al. Recent hybrid origin and invasion of the British Isles by a self-incompatible species, Oxford ragwort (Senecio squalidus L., Asteraceae). Biological invasions 11, 1145 (2009).
Ingvarsson, P. K. & Whitlock, M. C. Heterosis increases the effective migration rate. Proceedings of the Royal Society of London B: Biological Sciences 267, 1321–1326 (2000).
Kim, B. Y., Huber, C. D. & Lohmueller, K. E. Deleterious variation shapes the genomic landscape of introgression. PLOS Genetics 14, e1007741 (2018).
Bone, E. & Farres, A. Trends and rates of microevolution in plants. Genetica 112–113, 165–182 (2001).
Endler, J. A. Natural selection on color patterns in Poecilia reticulata. Evolution 34, 76–91 (1980).
Reznick, D. N. & Bryga, H. Life-history evolution in guppies (Poecilia reticulata): 1. phenotypic and genetic changes in an introduction experiment. Evolution 41, 1370–1385 (1987).
Reznick, D. A., Bryga, H. & Endler, J. A. Experimentally induced life-history evolution in a natural population. Nature 346, 357–359 (1990).
Reznick, D. N. Life history evolution in guppies (Poecilia reticulata): guppies as a model for studying the evolutionary biology of aging. (Pergamon, 1997).
Pilson, D. & Decker, K. L. Compensation for herbivory in wild sunflower: response to simulated damage by the head‐clipping weevil. Ecology 83, 3097–3107 (2002).
Team, R. C. R: A language and environment for statistical computing (2016).
Plummer, M. JAGS: A program for analysis of Bayesian graphical models using Gibbs sampling. In Proc. 3rd Int. Workshop on Distributed Statistical Computing (eds Hornik, K. et al.) (2003).
Gelman, A. & Rubin, D. B. Inference from iterative simulation using multiple sequences. Statistical Science 7, 457–472 (1992).
Gabry, J. & Goodrich, B. rstanarm: Bayesian applied regression modeling via Stan. R package version 2, 0–3 (2016).
Canty, A. J. Resampling methods in R: the boot package. R News 2, 2–7 (2002).
Acknowledgements
The authors wish to thank B. Haley, D. Kent, L. Albert, S. Hammer, L. Siefert, H. Luu, H. Gilbreath, J. Ahern, P. Sun, C. Simao, L. Dugan, A. Ewbank, C. Dana, N. Wagner, C. Caseys, T. Kalynyak, A. Anderson, A. Garcia, and A. Ortiz for field and lab assistance, and the Lady Bird Johnson Wildflower Center (Austin, TX), The Brackenridge Field Laboratory, and T. Juenger and D. Bolnick for lab space and assistance. J. Rudgers provided advice and support. This work was supported by NSF DEB-1257965 to K.D.W. and L.H.R.
Author information
Authors and Affiliations
Contributions
K.D.W. and L.H.R. designed the study. N.M., S.M.H. and K.D.W. conducted fieldwork. N.M. and G.L.O. processed samples, designed data analyses, and analyzed data. N.M., G.L.O. and K.D.W. wrote the manuscript. All authors contributed to revisions.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mitchell, N., Owens, G.L., Hovick, S.M. et al. Hybridization speeds adaptive evolution in an eight-year field experiment. Sci Rep 9, 6746 (2019). https://doi.org/10.1038/s41598-019-43119-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43119-4
This article is cited by
-
Molecular evidence for natural hybridization between Rumex crispus and R. obtusifolius (Polygonaceae) in Korea
Scientific Reports (2022)
-
Reticulate evolution in the Pteris fauriei group (Pteridaceae)
Scientific Reports (2022)
-
Strong bidirectional gene flow between fish lineages separated for over 100,000 years
Conservation Genetics (2022)
-
Phenotypic shifts following admixture in recombinant offspring of Arabidopsis thaliana
Evolutionary Ecology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
