
Abstract
Development of antiviral drug resistance is a continuous concern for viruses with high mutation rates such as influenza. The use of antiviral drugs targeting host proteins required for viral replication is less likely to result in the selection of resistant viruses than treating with direct-acting antivirals. The iminosugar UV-4B is a host-targeted glucomimetic that inhibits endoplasmic reticulum α-glucosidase I and II enzymes resulting in improper glycosylation and misfolding of viral glycoproteins. UV-4B has broad-spectrum antiviral activity against diverse viruses including dengue and influenza. To examine the ability of influenza virus to develop resistance against UV-4B, mouse-adapted influenza virus was passaged in mice in the presence or absence of UV-4B and virus isolated from lungs was used to infect the next cohort of mice, for five successive passages. Deep sequencing was performed to identify changes in the viral genome during passaging in the presence or absence of UV-4B. Relatively few minor variants were identified within each virus and the ratio of nonsynonymous to synonymous (dN/dS) substitutions of minor variants confirmed no apparent positive selection following sustained exposure to UV-4B. Three substitutions (one synonymous in PB2, one nonsynonymous in M and PA each) were specifically enriched (>3%) in UV-4B-treated groups at passage five. Recombinant viruses containing each individual or combinations of these nonsynonymous mutations remained sensitive to UV-4B treatment in mice. Overall, these data provide evidence that there is a high genetic barrier to the generation and selection of escape mutants following exposure to host-targeted iminosugar antivirals.
Similar content being viewed by others


Introduction
Rapidly acquired genetic variability due to error-prone polymerases has made rational drug development against RNA viruses, such as influenza virus, arduous. The fundamental problem underlying this difficulty is the inevitable development of drug resistance, where changes in a very small number of amino acid residues in the targeted viral protein is sufficient to reduce or completely block efficacy of a drug. For example, a single amino acid substitution (H274Y) in influenza A virus (IAV) isolates confers resistance to the neuraminidase inhibitor, oseltamivir1. Rapid development of resistance necessitates new approaches to the development of antiviral drugs. Since viruses are obligate intracellular parasites, they are critically dependent upon host factors for infection, replication, and spread. Identification and targeting of host factors that are critical for the viral replication cycle provides an opportunity for the development of novel classes of antiviral drugs2,3. Due to the improbability of changes to the host genome during an acute viral infection cycle, evasion of host-directed antiviral drugs is much less likely to occur.
IAV is a good target for the development of a host-directed antiviral therapy as the host molecular pathways that interact with the immunodominant viral proteins, hemagglutinin (HA) and neuraminidase (NA), have been well described. HA and NA are the two major surface glycoproteins of IAV that play essential roles in virion attachment and budding4,5. HA attaches the incoming virions to target cells by binding terminal sialic acid residues on cell-surface glycans5, whereas NA is a sialidase that assists with the budding process by cleaving sialic acid on the cell surface6. As a prototypical homotrimeric type I integral membrane protein, HA is synthesized in the endoplasmic reticulum (ER) of infected cells and transported through the Golgi complex to the plasma membrane, where it is incorporated into budding virions7,8,9. A variable number (dependent on the strain) of N-linked oligosaccharides are added co-translationally to HA as it is extruded into the ER through the translocon, and are subsequently trimmed and modified extensively during transport to the cell surface8,10,11,12. HA folding begins co-translationally, as demonstrated by the acquisition of intrachain disulfide bonds and the binding of monoclonal antibodies (mAbs) specific for discontinuous epitopes within HA to nascent chains8,11,12,13,14,15. N-terminal glycosylation at the globular head of HA helps the virus escape the immune response, while glycosylation of some sites at the stem of HA are critical for protein folding and stability11,13,14,15. Similarly, NA is a transmembrane tetrameric protein that uses the ER-Golgi route to the cell surface and is glycosylated in the process16. As both HA and NA rely heavily on glycosylation and other processing in the ER, a host-directed drug that inhibits this process has high potential for antiviral activity.
Key targets for inhibiting the host ER glycosylation pathway include the α-glucosidase I and II enzymes. These enzymes are responsible for making modifications to the co-translationally attached N-linked oligosaccharides, which are necessary for the proper folding of many glycoproteins17,18. Enveloped viruses that express surface glycoproteins, such as HA and NA of IAV, are dependent on the host cell α-glucosidase I and II enzymes for their replication. If viral proteins are not properly glycosylated, protein folding, stability, functionality, and immune evasion are impaired, and may result in reduced viral secretion or the production of defective virions8,9,10,11.
Iminosugars are glucomimetics with structural similarity to sugar molecules that can competitively inhibit glucosidase enzymes. Some iminosugars specifically target the α-glucosidases in the ER19. As a result, their therapeutic potential has been investigated against a range of viruses both in vitro and in vivo20,21,22,23. The iminosugar UV-4B has demonstrated in vitro and in vivo activity against a phylogenetically diverse set of glycosylated, enveloped viruses, including dengue (DENV) and influenza viruses24,25,26,27,28. It was previously demonstrated that DENV has a high genetic barrier for development of resistance against UV-4B27.
Here, we assessed the development of viral resistance to UV-4B treatment in vivo using a murine model of IAV infection. Mouse-adapted influenza A/Texas/36/91 (H1N1) was passaged in mice treated with UV-4B or vehicle for five successive passages. The sensitivity of the 5-times passaged viruses (P5) to treatment with UV-4B or the unrelated antiviral oseltamivir, which is currently approved for use to treat IAV infections, was confirmed in mice. The passaged viruses were deep-sequenced and relatively few minor variants were identified within each virus. Three substitutions (one synonymous in PB2 and two nonsynonymous in M and PA) were specifically enriched in P5 viruses passaged in the presence of UV-4B. However, these substitutions did not impact the efficacy of UV-4B or oseltamivir against the P5 viruses as evident from in vivo efficacy studies. Recombinant viruses containing each individual or combinations of these mutations remained susceptible to UV-4B treatment, showing no increased replication in vitro or enhanced disease severity in vivo.
Results
Passaging IAV in vivo in the presence of the host-targeted antiviral UV-4B does not decrease susceptibility to the drug
A murine model of IAV infection was used to test for the development of viral resistance to the iminosugar UV-4B. The dosing route and regimen were selected based on available data from tolerability and pharmacokinetic studies in uninfected mice25,28 and previous efficacy studies using IAV murine models of disease24,25. Groups of mice were challenged intranasally (i.n.) with influenza A/Texas/36/91 (H1N1) (passage 0; P0) and treated by intragastric administration of UV-4B or vehicle three times a day (TID) for seven days (Fig. 1). A portion of mice (ten) in each group were observed for morbidity and mortality for 14 days. The remaining five mice in each group were sacrificed on Day 4 post-infection (p.i.) and their lungs were harvested and homogenized for virus titration and deep sequencing. Following virus titration of the lung homogenates, a portion of the homogenates were pooled by group and used as the challenge virus for the next passage (P1) in mice. Virus passaging continued for a total of five successive passages. As expected, and similar to our previously published work24,25, viral titers in the lungs of UV-4B-treated mice were significantly lower (P ≤ 0.05) than that of vehicle-treated mice after each passage except for passage 4 (P = 0.052) (Fig. 2A).

Schematic diagram of the study design. Two groups of 15 female BALB/c mice were challenged i.n. with ~1 LD90 (~52 PFU) of mouse adapted A/Texas/36/91 (H1N1) and treated by intragastric administration with 100 mg/kg UV-4B or vehicle (water) TID for 7 days, starting 1 h after infection. Mice from each group (n = 5) were sacrificed on day 4 post-infection and their lungs were isolated and homogenized. A portion of the lung homogenates were pooled by group and used as the challenge virus (~1 LD90 or ~52 PFU/mouse) for the next passage in mice, successively for a total of 5 passages. The remaining portion of the lung homogenates were used to measure viral titer and isolate RNA for amplification by multi-segment RT-PCR. Sequencing libraries were prepared and sequenced on either the Illumina HiSeq 2000 or Illumina MiSeq v2 instruments (with repeat sequencing on the Ion Torrent PMG). Virus sequence assembly and identification of SNPs were performed using the CLC Genomics Workbench. Mutant viruses recapitulating the corresponding nucleotide changes of 3 SNPs identified (individually and in combination) were generated using site directed mutagenesis. Lethality and susceptibility to UV-4B of the mutant viruses was measured in vivo using similar experimental conditions.

Properties of IAV after in vivo passaging in the presence or absence of UV-4B. (A) Mouse-adapted INFV A/Texas/36/91 (H1N1) was passaged five times in the presence or absence of UV-4B. BALB/c mice were infected i.n. with the P0 parent virus or virus isolated from lungs of infected mice after 1 (P1) to 4 (P4) passages and treated by intragastric administration with 100 mg/kg UV-4B or vehicle (water) TID beginning 1 h after infection. Lungs were isolated from mice (n = 5/group) on day 4 post-infection and viral titers were measured by plaque assay after each passage in mice. UV-4B-treated groups in passages (P)0–4 were significantly lower (p < 0.05) than vehicle-treated groups at each passage, except for P4 (p = 0.052). (B,C) Survival outcome and relative weight of mice infected with IAV passaged 5 times in vivo in the presence or absence of UV-4B. BALB/c mice (n = 10/group) were exposed i.n. to virus (~1 LD90) that was passaged 5 times in mice in the presence of UV-4B (P5A) or water (P5B), and treated via intragastric administration with UV-4B (100 mg/kg, TID), water (TID), or oseltamivir (20 mg/kg, twice daily) beginning 1 h after challenge. Survival (B) and relative average weight compared to day 0 (C) were measured daily through the end of the study (Day 14).
An efficacy study was performed to determine whether IAV isolated after 5 passages (P5) from animals treated with UV-4B (P5A) or vehicle (P5B) were still sensitive to treatment with UV-4B or the influenza virus (INFV) antiviral oseltamivir. Mice were challenged i.n. with P5A or P5B IAV and then treated with UV-4B (100 mg/kg, TID), vehicle (water), or oseltamivir (20 mg/kg, twice daily). The UV-4B treatment dose selected was based on previous mouse efficacy studies25,26. Mice were observed daily for changes in health, weight and mortality for 14 days. All mice treated with UV-4B or oseltamivir survived infection with P5A and P5B viruses (Fig. 2B). All mice infected with P5A or P5B IAV and treated with vehicle succumbed to disease with a median survival time of 13 or 9 days, respectively (Supplemental Table 1). There were no differences in the average decrease in body mass of similarly treated groups infected with either P5A or P5B viruses (Fig. 2C). The onset and severity of clinical signs of disease (health score) were comparable between groups receiving similar treatments whether they were infected with the P5A or P5B viruses. Overall, this suggests that IAV passaged in mice five times in the presence of UV-4B did not acquire mutations that could potentially cause resistance to UV-4B or oseltamivir treatment or increase in vivo pathogenicity.
Sequencing of IAV identified limited selective mutations acquired during in vivo passaging in the presence of UV-4B
IAVs from the five individual mice at each passage that were treated with UV-4B (n = 25 samples, 5 per passage) or vehicle (n = 25 samples, 5 per passage), along with the input challenge virus, were deep-sequenced to achieve at least 200-fold coverage at each base of the coding sequence. Variant analysis of deep-sequence data was performed to identify selection of variants in UV-4B-treated animals versus vehicle-treated animals. The consensus sequence for the parental input mouse-adapted influenza A/Texas/36/91 (H1N1) challenge virus used for this study was previously unpublished. Therefore, we compared it against 2 deposited consensus sequences in the National Center for Biotechnology Information (NCBI) database for the A/Texas/36/91 (H1N1) strain. Five nonsynonymous nucleotide differences were found in the HA and NA sequences for the input parental virus compared with both NCBI sequences, only one of which (HA residue N104D) removes a potential N-linked glycosylation sequon (Supplemental Table 2).
Deep sequencing analysis of the in vivo-passaged IAV was performed to determine the consensus sequence and analyze consensus differences and low frequency nucleotide variations (quasi species/ minor variants) observed in the IAV samples to identify potential genomic population variation in UV-4B-treated and vehicle-treated mice. The sequence analysis identified that the genomic population in the virus samples is fairly uniform for an RNA virus (relatively few minor variants) and most were not specifically associated with UV-4B treatment. There were 31 selectively enriched coding sequence variants with >3% Single Nucleotide Polymorphisms (SNPs) identified at P5 which were unequally divided among 7 of the 8 RNA segments (Table 1). A cut off of >3% was considered true polymorphism (with minimum of 200X coverage at each base) to account for errors we observed from systematic sequencing (as high as 1%) with the Illumina platform, in addition to errors due to reverse transcription and library preparation29,30.
The summary of results for P5 sequence changes are shown in Table 2. Fourteen SNPs did not result in an amino acid substitution (i.e., synonymous). Of the 17 nonsynonymous substitutions, 6 were enriched in samples from UV-4B-treated mice. The six nonsynonymous substitutions associated with UV-4B treatment were limited to the polymerase acidic (PA), nucleoprotein (NP), and matrix (M) proteins. Other investigators have observed mutations in PA, polymerase basic 2 (PB2), and NP, which were attributed to mouse adaptation; however, not specifically at these amino acid positions identified here31. Only two of the six nonsynonymous substitutions (one each in M and PA) and one synonymous substitution in PB2 were specific to UV-4B treatment and were not also present in viruses from the vehicle-treated samples.
Plotting of the substitution rate (minor variant >3%) for each passage shows no significant change in the rate of substitution over passaging in UV-4B-treated mice (linear regression analysis of non-zero slope p-value = 0.2795). In contrast, linear regression analysis shows a significant decrease in the number of substitutions in vehicle-treated mice (p-value < 0.0001) (Fig. 3A). Unlike what has been observed with direct antivirals (e.g., oseltamivir)32, we found neutral selection in viruses from UV-4B treated mice based on the ratio of nonsynonymous to synonymous substitutions (dN/dS), despite the constant number of substitutions over time (linear regression of non-zero slope p-value = 0.65) (Fig. 3B), and negative selection was observed in vehicle-treated mice over successive passages (linear regression of non-zero slope p-value = 0.0063). Regardless of being under pressure, as evident from neutral (UV-4B group) vs. negative (vehicle group) selection, the lack of any positive selection in the presence of UV-4B after 5-pasages further confirms the high genetic barrier for the emergence of drug resistance in the presence of host-targeted antivirals (e.g. UV-4B)

Regression lines plotting the relationship between passage number and either (A) number of substitutions per genome (>3%) or (B) the dN/dS ratio (number of nonsynonymous substitutions divided by the number of synonymous substitutions). Analysis was performed using the R package ggplot2 with method “lm” (linear model).
Recombinant IAV with substitutions enriched in P5 show no increased replication in MDCK cells
All 8 gene segments of A/Texas/36/91 (H1N1) virus were synthesized and cloned to develop reverse genetics systems for further evaluation33. M, PA, and PB2 gene segments were mutagenized to generate the three specifically enriched substitutions found in P5A viruses to determine whether these acquired substitutions have any significant selective advantage for the virus to escape UV-4B treatment. The nonsynonymous substitution in the M gene segment results in an amino acid substitution in both M1 (M248I) and M2 (C19Y), whereas the nonsynonymous substitution in PA results in the amino acid change K626R. We also generated a synonymous nucleotide substitution in PB2 (A726G). A total of 8 recombinant (r) mouse-adapted influenza A/Texas/36/91 (H1N1) viruses were rescued (Fig. 4A) using an established reverse genetics rescue system34,35.

In vitro and in vivo replication efficiency of recombinant wild-type and mutant viruses. (A) Schematic of recombinant viruses generated. (B) Viral titers for wild-type and recombinant mutant viruses were determined by TCID50 assay in MDCK cells at Day 4. (C) Mice were infected i.n. with decreasing challenge doses of wild-type or mutant viruses. Survival was monitored to the end of the study (Day 14) and the LD90 was determined for each virus.
No selective advantage of the substitutions enriched in P5 over the input mouse-adapted wild-type virus
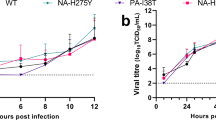
Titration of viruses in Madin-Darby Canine Kidney (MDCK) cells after rescue showed very similar titers for each of the recombinant mutant viruses, demonstrating that these substitutions were not detrimental to viral replication/fitness in vitro (Fig. 4B). Next, we compared the in vivo lethality of recombinant IAVs containing substitutions that were enriched in the P5A virus to that of recombinant wild-type mouse-adapted influenza A/Texas/36/91 (H1N1) virus in BALB/c mice. Mice were infected i.n. with decreasing doses of recombinant viruses, and changes in weight loss and survival were monitored. Less pronounced weight loss and a higher survival rate were observed with decreasing challenge doses in a largely dose-dependent manner (Supplemental Figs 1 and 2). Overall, the LD90 titers (converted as TCID50 titer) were similar among wild-type and mutant viruses in BALB/c mice (Fig. 4C and Table 3), suggesting that no increase in lethality was acquired by viruses with these specific mutations.
Substitutions enriched in P5A are sensitive to both UV-4B and oseltamivir
UV-4B treatment (100 mg/kg TID for 5 days beginning 8 h after infection) protected mice against lethal infection with all the recombinant wild-type and mutant IAV (p < 0.01 in all cases) (Fig. 5). Increased survival (60–100%) was observed with UV-4B treatment when mice were infected either with the wild-type or recombinant mutant viruses as compared to those treated with water alone (0–10%). Protection from weight loss was also apparent during the period of UV-4B treatment (day 4 p.i.) (Supplemental Fig. 3). Oseltamivir was included as a positive control compound and performed as expected against both recombinant wild-type virus and the recombinant virus with all three mutations (3 muts, M_PA_PB2) (Fig. 5A,H).

Survival outcome of mice infected with recombinant influenza A (H1N1) mutants and treated with UV-4B, oseltamivir, or vehicle. (A–H) BALB/c mice (n = 10/group) were infected i.n. (~1 LD90) with a wild-type recombinant virus or mutant recombinant viruses containing substitutions specific to UV-B treatment. Mice were treated via intragastric administration with UV-4B (100 mg/kg) or vehicle (water) three times daily or oseltamivir (20 mg/kg) twice daily for 5 days beginning 8 h post-challenge. **P < 0.01, ***P < 0.001 when compared to the vehicle group.
Discussion
In this study, we evaluated the ability of mouse-adapted IAV (A/Texas/36/91 (H1N1)) to mutate in vivo in the presence of the host-targeted iminosugar UV-4B. Viruses passaged 5 times in mice in the presence or absence of UV-4B remained sensitive to UV-4B treatment in in vivo efficacy studies, providing evidence that UV-4B treatment does not promote the generation of viral escape mutants. Amongst the passaged viruses, relatively few minor variants were identified, and only three substitutions (one synonymous in PB2 and two nonsynonymous in M and PA) were specifically enriched in UV-4B-treated viruses at passage five. Recombinant viruses containing these mutations showed no detectable selective advantage and maintained their sensitivity to UV-4B treatment in vivo, corroborating a predicted high genetic barrier to escape mutations with host-targeted iminosugar antivirals36.
We identified minimal mutations in IAV after multiple passages in mice, and most of those mutations were not specifically associated with UV-4B treatment, indicating nonspecific selective pressure. Six of the 17 nonsynonymous substitutions identified were enriched in UV-4B-treated samples, which were limited to proteins that are not glycosylated by the host-cell machinery in the ER. Mice infected with recombinant viruses containing mutations in M, PA and/or PB2 were still susceptible to treatment with UV-4B; however, a slightly lower survival benefit (60–90%) was observed for some groups compared to the control group infected with the wild-type recombinant virus (100% survival). These differences could be due, in part, to challenges during the treatment procedure, possibly resulting in early deaths, or because UV-4B doesn’t consistently protect every animal from lethal infection, which would be more evident in a larger population. However, it is also plausible that these mutations could alter the HA/NA balance on the virions themselves or through some unknown mechanisms in the context of recombinant viruses. Although, it is important to note that all three of these unique substitutions were at sub-consensus levels in the passaged viruses, even after 5 rounds of passaging, suggesting limited to no selective advantage of these substitutions.
Based on the mechanism of action of UV-4B, it was expected that mutations unique to UV-4B treatment may be identified in viral glycoproteins, especially since this has been shown for selection of IAV in presence of mAbs10,37. The lack of variants in these proteins indicates that the virus did not acquire mutations necessary to overcome the selection pressure elicited by UV-4B, confirming a high genetic barrier for development of resistance. Unlike previous studies that showed concentration dependent positive selection of the IAV in the presence of oseltamivir and rapid emergence of oseltamivir resistant mutation after only one or two passages in mice32, our dN/dS analysis showed neutral selection during five successive passages in mice for four days in the presence of UV-4B, which accounts for a total of ~25–50 replication cycles of the virus (calculated based on ~5 to 10 replication cycles over the course of 4 days in mice/passaging38), further confirming the genetic barriers to escape UV-4B treatment. At the same time, as expected, passaging without drug showed negative selection (purifying selection) over time as this virus is already well adapted to the host (mice) and only purged changes that are deleterious on viral fitness in mice.
The generation of resistant mutants following UV-4B treatment was previously evaluated in mice following infection with DENV, a mosquito-borne pathogen that can cause severe and potentially life-threatening illnesses27. Like IAV, DENV requires the host’s ER α-glucosidase I and II enzymes for replication. Plummer et al., evaluated the evolution of two populations of passaged virus in the presence or absence of UV-4B in mice: DENV passaged in individual mice after a single passage, and pooled virus over the course of 4 serial passages. Similar to the results reported here for IAV, passaging of DENV in mice in the presence of UV-4B did not provide evidence for generation of viral escape mutants. Only 13 nonsynonymous SNPs in DENV were identified in pooled samples from at least one time point, and 12 of those 13 mutations were found in virus isolated from both the vehicle-treated and UV-4B-treated animals. However, the authors noted that by pooling samples for sequencing, some of the individual mouse-specific responses may have been diluted below detection. For this reason, combined with consideration of the normal passaging of DENV from invertebrate (mosquito) to vertebrate hosts rather than from vertebrate to vertebrate, they evaluated mutations acquired in DENV following a single passage in mice in the presence or absence of UV-4B. Synonymous and nonsynonymous mutations were identified in all DENV proteins after a single passage in mice. Unlike the current study, the dN/dS was higher when passaged in the presence of UV-4B compared to vehicle. Looking specifically at DENV glycosylated proteins (membrane, M; envelope, E; and NS1), nineteen nonsynonymous mutations were present at significant levels only in UV-4B-treated mice, possibly related to the mechanism of the drug. This was not observed for IAV in the present study, where no mutations specific to UV-4B treatment were identified in any of the viral glycoproteins.
Prior to this study, the development of drug resistant IAV strains was primarily evaluated for direct-acting antivirals. Unlike host-targeted iminosugars, approved anti-influenza virus drugs that are currently available directly target viral proteins (M2: amantadine and rimantadine; NA: oseltamivir, zanamivir, peramivir; PA: Xofluza39). The potential for influenza viruses to develop resistance to direct-acting antivirals is well established. Amantadine and rimantadine directly block the M2 proton channel to prevent the delivery of the viral genome into the cytoplasm of cells infected with IAV40. The Centers for Disease Control and Prevention (CDC) no longer recommends the use of amantadine or rimantadine to treat IAV infections in the United States because of the prevalence of resistant IAV strains that are no longer sensitive to treatment with these drugs41. Since the early 1970s, multiple groups have reported instances of resistance to amantadine and rimantadine42,43,44,45. As a result, neuraminidase inhibitors (NAI) are currently recommended by the CDC for treatment of influenza virus infections. NAIs, which inhibit the enzymatic activity of the viral NA protein, prevent virus release and spread and are effective against both IAV and IBV viruses. The active site of NA is highly conserved among all influenza viruses, making NA an ideal target for antiviral therapeutics directed against influenza virus32,46. However, mutations in any of the viruses that alter the shape/charge of the NA catalytic site can reduce binding and result in decreased sensitivity or resistance to treatment47 In addition, evidence shows that in the absence of NA-specific drugs, antibody escape in HA results in epistatic compensatory mutations in NA that could also result in drug resistance1,48.
Of the approved NAIs, oseltamivir is the only drug with sufficient bioavailability to be administered orally49,50,51,52,53,54,55. Oseltamivir-resistant strains can arise from a single mutation that results in a decrease in the binding affinity of oseltamivir to NA56,57. Additionally, combinations of multiple mutations can result in a synergistic or enhanced effect on oseltamivir antiviral resistance32,58,59,60,61 and can potentially restore mild reduced viral fitness that is observed with some single mutations1,59. As the potential risk for the transmission of drug resistant strains is a continuous concern, the impact on the efficacy of other antivirals against such resistant strains must be considered. Stavale et al., demonstrated that UV-4B maintains efficacy against an oseltamivir-resistant strain in a murine model of disease25. Here, we report that IAV passaged in mice five times in the presence of UV-4B did not acquire mutations that cause resistance to the commonly used anti-influenza drug oseltamivir. While the proposed mechanism for the antiviral activity of UV-4B is through perturbation of N-linked glycan processing, further studies need to be performed to determine which, if any, glycosylation sites on viral glycoproteins are affected and whether other mechanisms contribute to the anti-IAV activity of UV-4B. Using influenza virus reassortants, Hussain et al. demonstrated that changes in glycosylation of HA are the likely antiviral mechanism for iminosugars but more detailed analysis of which glycosylation sites are most sensitive has not yet been determined62.
It is interesting to note that UV-4B protects against fatal influenza infection in vivo (Fig. 2B,C); however, treatment with UV-4B does not robustly reduce viral replication in the lung (Fig. 2A) to a level that would be biologically significant. Based on these data, it appears that UV-4B may prevent influenza-induced morbidity and mortality through multiple biologic effects that may include mechanisms other than inhibition of viral replication. One such mechanism could be reduction in localized inflammation due to blunted cytokine responses. The freebase form of UV-4B (UV-4) itself does not induce a cytokine response in the blood of naïve, UV-4-treated mice28 or in vitro using mouse splenocytes, or human PBMC (Warfield et al., unpublished data) nor does it alter cytokine responses in vitro following mitogen stimulation (ex. PHA or LPS) (Warfield, et al., unpublished data). However, UV-4B treatment does result in reduced systemic cytokine responses in dengue virus infection mice28 and could also result in a reduced local (lung) cytokine responses following influenza infection. Alternately, other novel mechanisms that limit influenza pathogenesis by host-targeted antiviral iminosugars in addition to established mechanisms (i.e., direct impact on the hosts glycosylation pathway or indirectly by protein mis-folding due to ER stress) may be discovered22. The limited reduction in virus titer observed in the mouse lung following UV-4B treatment supports the potential use of UV-4B in a combination therapy. The effect of UV-4B treatment when co-administered with an NA inhibitor, and the potential for the development of drug resistant strains following combined treatment, should be considered and evaluated in future studies.
Taken together, our data suggests that the iminosugar UV-4B, which is effective against a broad range of influenza A and B viruses in vitro and in vivo, is at a low risk for selecting for mutant, drug-resistant influenza viruses. Iminosugars have positive drug-like properties and a history of safe use in humans, including five iminosugar compounds in clinical use: miglustat (Zavesca®) for the treatment of Gaucher’s disease and Niemann-Pick Type C; migalastat (Galafold™) for the treatment Fabry disease; and miglitol (Glyset®), acarbose (Precose®), and voglibose (Basen®) for the treatment or prevention of type II diabetes mellitus. Therefore, iminosugars remain a promising class of compounds for the future development of an antiviral therapeutic targeting a range of diverse viral pathogens, including DENV and influenza viruses.
Materials and Methods
Animal welfare, husbandry and observations
All procedures of the study were performed in accordance with the guidelines and protocols set and approved by the Noble Life Sciences or Utah State University Institutional Animal Care and Use Committee (IACUC). Both institutions are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Laboratory animals were observed and veterinary care was provided for all laboratory animals as required on a 24 hr basis, including weekends and holidays, in accordance with the Public Health Service Policy, U.S. Dept. of Agriculture (USDA) and AAALAC International requirements.
Female BALB/c mice (6–8 weeks of age) maintained under specific pathogen-free conditions were procured from Charles River Laboratories and were quarantined for >48 h prior to study initiation. During in-life study duration, all animals were observed carefully 2–3 times daily for changes in clinical signs, including morbidity and mortality. Survival and health were evaluated daily using a scoring system to assess signs of clinical disease (e.g. appearance of fur coat, posture, mobility, activity level and attitude) and provide a numerical value (1–7) directly related to disease severity as previously described25. Mice were euthanized when they met a 30% weight loss cut-off or scored at ≥6 in the standard scoring system. Animals were euthanized by administration of inhaled CO2 followed immediately by cervical dislocation and in accordance with the 2013 American Veterinary Medical Association (AVMA) Guidelines on Euthanasia.
Passaging description
The original virus stock of the mouse-adapted influenza A/Texas/36/91 (H1N1) was obtained from the Baylor School of Medicine (kind gift of P. Wyde and B. Gilbert). Using 6-8-week-old female BALB/c mice, a new working virus stock (P0) was prepared using infected-lung homogenates and the LD90 was determined to be 52 PFU/mouse. Influenza A/Texas/36/91 (H1N1) was passaged successively five times in mice before determining the efficacy of UV-4B against the P5 virus. One group of 30 mice (P0) was challenged with influenza A/Texas/36/91 (H1N1) (~1 LD90 or ~52 PFU) intranasally and were treated via intragastric administration with either 100 mg/kg UV-4B or its vehicle (water) control starting one hour after infection. This treatment continued TID for seven days. A portion of these mice (n = 10) were observed daily for weight changes, morbidity (assessed using standard health scores) and mortality for 14 days. The remaining mice (n = 5) were sacrificed on Day 4 post-infection and their lungs were bisected. Half of each lung was immersed in RNALater for RNA extraction and subsequent virus sequencing. The other half of each lung was snap-frozen in liquid nitrogen and stored frozen until homogenization in PBS on wet ice. Homogenate samples from individual mice were maintained for titration with 0.2 mL from each mouse combined into a pool homogenate to be used in the next passage. Following virus titration of the lung homogenate pool, these viruses were then used as the challenge agent (P1-4) in the next round of virus challenge at ~52 PFU/mouse (~1 LD90) of input virus. Virus passaging continued for a total of five times.
Lethal dose 90% (LD90) and efficacy determination of passage 5 viruses
The LD90 was determined for influenza viruses isolated from lungs after 5 passages in mice treated with UV-4B (P5A) or vehicle (P5B). Groups of ten BALB/c mice were used to test the lethality of six different challenge doses (1, 5, 10, 25, 50, or 100 PFU) of each virus following i.n. exposure. All mice were orally administered 100 µL of water TID starting 1 h p.i. for a total of ten days to mimic treatment. Mice were monitored daily for changes in health (assessed using standard scoring system), weight, and mortality for 14 days.
Groups of 30 mice were challenged with either P5 virus isolated from animals treated with UV-4B (P5A, ~1 LD90 = 25 PFU) or from animals treated with the vehicle control (P5B, ~1 LD90 = 10 PFU). One hour following infection, the animals in these groups were treated via intragastric administration with either 100 mg/kg of UV-4B (N = 10), vehicle control (N = 10), or 20 mg/kg of the positive control, oseltamivir. The treatments with UV-4B and vehicle continued TID for seven days, while the treatment with oseltamivir continued twice a day for five days. Mice were observed daily for 14 days for changes in health, weight, and mortality.
Influenza virus next-generation sequencing
Viral RNA from the infected lung lysates were individually evaluated using multi-segment reverse transcriptase-polymerase chain reaction (M-RT-PCR)63, followed by library preparation using the Nextera DNA library preparation kit (Illumina) and the Ion Xpress™ Plus Fragment Library Kit (Thermo Fisher Scientific) for sequencing using Illumina MiSeq and Ion Torrent PGM, respectively, to overcome platform specific errors. The IAV genomic RNA segments were simultaneously amplified from 3 µl of purified RNA using M-RT-PCR34. Illumina libraries were prepared from these M-RT-PCR products using the Nextera DNA Sample Preparation Kit (Illumina, Inc., San Diego, CA, USA) with half-reaction volumes. PCR products were quantified using QIAxcel (Qiagen, Hilden, Germany), and 25 ng of DNA amplicons for each sample were tagmented (fragmented and tagged) at 55 °C for 5 min. Tagmented DNA amplicons were cleaned with the ZR-96 DNA Clean & Concentrator Kit (Zymo Research Corporation, Irvine, CA, USA) and eluted in 25 µl resuspension buffer. Illumina sequencing adapters and barcodes were added to tagmented DNA via PCR amplification by combining 20 µl tagmented DNA with 7.5 ul Nextera PCR Master Mix, 2.5 ul Nextera PCR Primer Cocktail, and 2.5 µl of each index primer (Integrated DNA Technologies, Coralville, IA, USA) for a total volume of 35 µl per reaction. Five cycles of PCR were performed as per the Nextera DNA Sample Preparation Kit protocol (3 min at 72 °C, denaturation for 10 sec at 98 °C, annealing for 30 sec at 63 °C, and extension for 3 min at 72 °C) to create a dual-indexed library for each sample. After PCR amplification, 10 µl of each library derived from M-RT-PCR products were pooled into a 1.5-ml tube; separately, 10 µl of each library derived from HA-specific amplicons were pooled into a 1.5-ml tube. Each pool was cleaned two times with Ampure XP Reagent (Beckman Coulter, Inc., Brea, CA, USA) to remove all leftover primers and small DNA fragments. The first and second cleanings used 1.2x and 0.6x volumes of Ampure XP Reagent, respectively. The cleaned pool derived from M-RTPCR products was sequenced on the Illumina HiSeq 2000 instrument (Illumina, Inc.) with 100-bp paired-end reads, while the cleaned pool derived from HA-specific amplicons was sequenced on the Illumina MiSeq v2 instrument with 300-bp paired-end reads.
Separately, influenza M-RT-PCR products were randomly amplified and prepared for NGS using a sequence-independent single-primer amplification (SISPA) method as previously described64. The methodology used 100 ng of amplified viral DNA that was denatured in the presence of DMSO and a chimeric oligonucleotide containing a known 22-nt barcode sequence followed by a 3′ random hexamer. A Klenow reaction was prepared with the denatured DNA template by adding NEB buffer II, 3′-5′ exo- Klenow (New England Biolabs, Ipswich, MA, USA), and dNTPs (Thermo Fisher Scientific, Waltham, MA, USA). The Klenow reaction was incubated at 37 °C for 60 min, followed by incubation at 75 °C for 10 min. The resulting cDNA was randomly amplified by PCR using the Promega GoTaq Hot Start Polymerase (Promega Corporation, Madison, WI, USA) for 35 cycles (denaturation: 30 sec, 94 °C; annealing: 30 sec, 55 °C; extension: 48 sec, 68 °C). PCR reactions contained primers corresponding to the known 22-nt barcode sequence from the oligonucleotide utilized in the previous Klenow step. The resulting cDNA was then treated with Exonuclease I at 37 °C for 60 min, followed by incubation at 72 °C for 15 min. SISPA products were normalized and pooled into a single reaction that was purified using the QIAquick PCR purification kit (Qiagen). Pooled samples were further purified to select for SISPA products 300–500 bp in size for Illumina Miseq paired-end (2 × 300) sequencing.
For additional sequencing coverage, samples were re-sequenced using the Ion Torrent platform. M-RT-PCR products were sheared for 7 min, and Ion-Torrent-compatible barcoded adapters were ligated to the sheared DNA using the Ion Xpress Plus Fragment Library Kit (Thermo Fisher Scientific) to create 400-bp libraries. Libraries were pooled in equal volumes and cleaned with the Ampure XP Reagent. Quantitative PCR was performed on the pooled, barcoded libraries to assess the quality of the pool and to determine the template dilution factor for emulsion PCR. The pool was diluted appropriately and amplified on Ion Sphere Particles (ISPs) during emulsion PCR on the Ion One Touch 2 instrument (Thermo Fisher Scientific). The emulsion was broken, and the pool was cleaned and enriched for template-positive ISPs on the Ion One Touch ES instrument (Thermo Fisher Scientific). Sequencing was performed on the Ion Torrent PGM using a 318v2 chip (Thermo Fisher Scientific).
Virus genome assembly and variant analysis
For virus sequence assembly, all sequence reads were sorted by barcode, trimmed, and de novo assembled using CLC Bio’s clc_novo_assemble program (Qiagen). The resulting contigs were searched against custom full-length influenza segment nucleotide databases to find the closest reference sequence for each segment. All sequence reads were then mapped to the selected reference influenza A virus segments using CLC Bio’s clc_ref_assemble_long program.
Minor allele variants (i.e., nucleotide sequences that differ from the reference sequence and are supported by sequencing reads at or above a pre-defined threshold) were identified using FindStatisticallySignificantVariants (FSSV) software (http://sourceforge.net/projects/elvira/). The FSSV software applied statistical tests to minimize false-positive Single Nucleotide Polymorphism (SNP) calls generated by Illumina sequence-specific errors (SSEs)65. SSEs usually result in false SNP calls if sequences are read in one sequencing direction. The FSSV analysis tool requires observing the same SNP at a statistically significant level in both sequencing directions. Once a minimum minor allele frequency threshold and significance level are established, the number of minor allele observations and major allele observations in each direction and the minimum minor allele frequency threshold are used to calculate p-values based on the binomial distribution cumulative probability. If the p-values calculated in both sequencing directions are less than the Bonferroni-corrected significance level, then the SNP calls are accepted. A significance level of 0.05 (Bonferroni-corrected for tests in each direction to 0.025) and a minimum minor allele frequency threshold of 3% were applied for this analysis, and the consensus sequence from the input parental virus was used as the reference sequence for each sample.
Differences in the consensus sequence compared to the reference sequence were identified using CLC Bio’s find_variations software. The identified consensus and minor allele variations were analyzed by assessing the functional impact on coding sequences or other regions based on overlap with identified features of the genome. For each sample, the reference sequence was annotated using VIGOR software31, and then the variant data and genome annotation were combined using VariantClassifier software66 to produce records describing the impacts of the identified variations.
Recombinant viruses
To generate the recombinant A/Texas/36/1991 (H1N1) viruses, the appropriate gene segments were cloned into reverse genetics plasmids as previously described33. Substitutions in PA, PB and M were performed using site-directed mutagenesis. Wild-type and recombinant mutant viruses A/Texas/36/1991 (H1N1) were rescued using reverse genetics systems with 293 T and MDCK co-culture as previously described35,63,67.
Virus titration
The 50% tissue culture infectious dose (TCID50) assay was performed for in vitro titrations of IAV. Briefly, the supernatants from the lung homogenates were evaluated using 10-fold serial dilutions in 96-well plates (six dilutions total, 10−2 to 10−7) to determine the viral titers. MDCK cells were seeded at a density of 1 × 104 cells per well in 100 µL of medium into 96-well plates and incubated overnight (20–24 h). Cells were infected with the dilutions in 9 replicates, three plates per yield dilution, with one column serving as the negative control. Cells were incubated at 35 °C until the cytopathic effect (CPE) in wells containing infected cells remained constant (6–10 days). The cells were then fixed with 5% glutaraldehyde and stained with 0.1% crystal violet to measure CPE. The number of infected wells at each dilution was used to calculate the TCID50 value based on the Reed-Muench method68. The calculation of PFU from TCID50 is based on the ratio PFU/TCID50 of the limiting dilution which would infect 50% of the challenged cell layers69.
LD90 and efficacy determinations of recombinant IAV
To determine the LD90 of each recombinant virus (wild-type or substituted), groups of five mice were exposed intranasally (75 µl) to an individual virus using dilutions of 1:100, 1:320, 1:1000, and 1:3200. A group of 5 mice were not infected and served as weight/survival controls. Mice in all groups were treated with water via intragastric administration 3 times daily for 7 days, beginning 8 hours after infection. Mice were monitored for weight loss, morbidity and mortality as described above. LD90 values were graphically estimated and used to set challenge inoculum dilutions for each individual recombinant virus (ranging from 1:320-1:1000).
Groups of mice were exposed intranasally (75 µl) to the wild-type or recombinant viruses at ~1LD90. Groups of mice were treated with UV-4B (100 mg/kg/dose or vehicle (water) via intragastric administration TID for 5 days beginning 8 hours after infection. For two of the viruses, a group treated with oseltamivir was also included as a positive control, where mice were treated with oseltamivir (10 mg/kg/day) twice daily for 5 days beginning four hours after infection. Mice were monitored for weight loss, morbidity and mortality as described above.
Statistical analyses
Statistical analyses were performed using GraphPad Prism. Virologic titers were log10‐transformed and statistical analysis between vehicle- and UV‐4B‐treated groups at each passage performed using an unpaired parametric t‐test. Survival analysis was performed using the Kaplan-Meier graphing method and log-rank test. Linear regression analysis was performed to test if slopes of the number of substitutions or the dN/dS ratio over passage number were significantly different than zero.
Accession number of sequences
All the sequences are submitted to GenBank with access no MK615188-MK615595.
References
Bloom, J. D., Gong, L. I. & Baltimore, D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 328, 1272–1275, https://doi.org/10.1126/science.1187816 (2010).
Lee, S. M. & Yen, H. L. Targeting the host or the virus: current and novel concepts for antiviral approaches against influenza virus infection. Antiviral Res 96, 391–404, https://doi.org/10.1016/j.antiviral.2012.09.013 (2012).
Min, J. Y. & Subbarao, K. Cellular targets for influenza drugs. Nat Biotechnol 28, 239–240, https://doi.org/10.1038/nbt0310-239 (2010).
Hers, J. F. Disturbances of the ciliated epithelium due to influenza virus. Am Rev Respir Dis 93(Suppl), 162–177, https://doi.org/10.1164/arrd.1966.93.3P2.162 (1966).
Skehel, J. J. & Wiley, D. C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69, 531–569, https://doi.org/10.1146/annurev.biochem.69.1.531 (2000).
Shtyrya, Y. A., Mochalova, L. V. & Bovin, N. V. Influenza virus neuraminidase: structure and function. Acta Naturae 1, 26–32 (2009).
Russ, G., Bennink, J. R., Bachi, T. & Yewdell, J. W. Influenza virus hemagglutinin trimers and monomers maintain distinct biochemical modifications and intracellular distribution in brefeldin A-treated cells. Cell Regul 2, 549–563, https://doi.org/10.1091/mbc.2.7.549 (1991).
Yewdell, J. W., Yellen, A. & Bachi, T. Monoclonal antibodies localize events in the folding, assembly, and intracellular transport of the influenza virus hemagglutinin glycoprotein. Cell 52, 843–852 (1988).
Daniels, R., Kurowski, B., Johnson, A. E. & Hebert, D. N. N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell 11, 79–90 (2003).
Das, S. R. et al. Fitness costs limit influenza A virus hemagglutinin glycosylation as an immune evasion strategy. Proc Natl Acad Sci USA 108, E1417–1422, https://doi.org/10.1073/pnas.1108754108 (2011).
Das, S. R. et al. Glycosylation focuses sequence variation in the influenza A virus H1 hemagglutinin globular domain. PLoS Pathog 6, e1001211, https://doi.org/10.1371/journal.ppat.1001211 (2010).
Han, Y. et al. Monitoring cotranslational protein folding in mammalian cells at codon resolution. Proc Natl Acad Sci USA 109, 12467–12472, https://doi.org/10.1073/pnas.1208138109 (2012).
Gething, M. J., McCammon, K. & Sambrook, J. Expression of wild-type and mutant forms of influenza hemagglutinin: the role of folding in intracellular transport. Cell 46, 939–950 (1986).
Copeland, C. S. et al. Folding, trimerization, and transport are sequential events in the biogenesis of influenza virus hemagglutinin. Cell 53, 197–209 (1988).
Copeland, C. S., Doms, R. W., Bolzau, E. M., Webster, R. G. & Helenius, A. Assembly of influenza hemagglutinin trimers and its role in intracellular transport. J Cell Biol 103, 1179–1191 (1986).
Air, G. M. Influenza neuraminidase. Influenza Other Respir Viruses 6, 245–256, https://doi.org/10.1111/j.1750-2659.2011.00304.x (2012).
Pieren, M., Galli, C., Denzel, A. & Molinari, M. The use of calnexin and calreticulin by cellular and viral glycoproteins. J Biol Chem 280, 28265–28271, https://doi.org/10.1074/jbc.M501020200 (2005).
Hammond, C., Braakman, I. & Helenius, A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci USA 91, 913–917 (1994).
Caputo, A. T. et al. Structures of mammalian ER alpha-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proc Natl Acad Sci USA 113, E4630–4638, https://doi.org/10.1073/pnas.1604463113 (2016).
Dalziel, M., Crispin, M., Scanlan, C. N., Zitzmann, N. & Dwek, R. A. Emerging principles for the therapeutic exploitation of glycosylation. Science 343, 1235681, https://doi.org/10.1126/science.1235681 (2014).
Miller, J. L., Tyrrell, B. E. & Zitzmann, N. Mechanisms of Antiviral Activity of Iminosugars Against Dengue Virus. Adv Exp Med Biol 1062, 277–301, https://doi.org/10.1007/978-981-10-8727-1_20 (2018).
Tyrrell, B. E., Sayce, A. C., Warfield, K. L., Miller, J. L. & Zitzmann, N. Iminosugars: Promising therapeutics for influenza infection. Crit Rev Microbiol 43, 521–545, https://doi.org/10.1080/1040841X.2016.1242868 (2017).
Alonzi, D. S., Scott, K. A., Dwek, R. A. & Zitzmann, N. Iminosugar antivirals: the therapeutic sweet spot. Biochem Soc Trans 45, 571–582, https://doi.org/10.1042/BST20160182 (2017).
Warfield, K. L. et al. The Iminosugar UV-4 is a Broad Inhibitor of Influenza A and B Viruses ex Vivo and in Mice. Viruses 8, 71, https://doi.org/10.3390/v8030071 (2016).
Stavale, E. J., Vu, H., Sampath, A., Ramstedt, U. & Warfield, K. L. In vivo therapeutic protection against influenza A (H1N1) oseltamivir-sensitive and resistant viruses by the iminosugar UV-4. PLoS One 10, e0121662, https://doi.org/10.1371/journal.pone.0121662 (2015).
Warfield, K. L. et al. Inhibition of endoplasmic reticulum glucosidases is required for in vitro and in vivo dengue antiviral activity by the iminosugar UV-4. Antiviral Res 129, 93–98, https://doi.org/10.1016/j.antiviral.2016.03.001 (2016).
Plummer, E. et al. Dengue Virus Evolution under a Host-Targeted Antiviral. J Virol 89, 5592–5601, https://doi.org/10.1128/JVI.00028-15 (2015).
Perry, S. T. et al. An iminosugar with potent inhibition of dengue virus infection in vivo. Antiviral Res 98, 35–43, https://doi.org/10.1016/j.antiviral.2013.01.004 (2013).
Lakdawala, S. S. et al. The soft palate is an important site of adaptation for transmissible influenza viruses. Nature 526, 122–125, https://doi.org/10.1038/nature15379 (2015).
Poon, L. L. et al. Quantifying influenza virus diversity and transmission in humans. Nat Genet 48, 195–200, https://doi.org/10.1038/ng.3479 (2016).
Wang, S., Sundaram, J. P. & Stockwell, T. B. VIGOR extended to annotate genomes for additional 12 different viruses. Nucleic Acids Res 40, W186–192, https://doi.org/10.1093/nar/gks528 (2012).
Pizzorno, A. et al. Evolution of oseltamivir resistance mutations in Influenza A(H1N1) and A(H3N2) viruses during selection in experimentally infected mice. Antimicrob Agents Chemother 58, 6398–6405, https://doi.org/10.1128/AAC.02956-14 (2014).
Dormitzer, P. R. et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci Transl Med 5, 185ra168, https://doi.org/10.1126/scitranslmed.3006368 (2013).
Zhou, B. et al. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol 83, 10309–10313, https://doi.org/10.1128/JVI.01109-09 (2009).
Zhou, B. et al. Reverse genetics plasmid for cloning unstable influenza A virus gene segments. J Virol Methods 173, 378–383, https://doi.org/10.1016/j.jviromet.2011.01.021 (2011).
Gotte, M. The distinct contributions of fitness and genetic barrier to the development of antiviral drug resistance. Curr Opin Virol 2, 644–650, https://doi.org/10.1016/j.coviro.2012.08.004 (2012).
Kosik, I. et al. Influenza A virus hemagglutinin glycosylation compensates for antibody escape fitness costs. PLoS Pathog 14, e1006796, https://doi.org/10.1371/journal.ppat.1006796 (2018).
Baccam, P., Beauchemin, C., Macken, C. A., Hayden, F. G. & Perelson, A. S. Kinetics of influenza A virus infection in humans. J Virol 80, 7590–7599, https://doi.org/10.1128/JVI.01623-05 (2006).
Lackenby, A. et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors and status of novel antivirals, 2016–2017. Antiviral Res 157, 38–46, https://doi.org/10.1016/j.antiviral.2018.07.001 (2018).
Sidwell, R. W. & Smee, D. F. In vitro and in vivo assay systems for study of influenza virus inhibitors. Antiviral Res 48, 1–16 (2000).
Centers for Disease Control and Prevention, N. C. f. I. a. R. D. N. Influenza Antiviral Drug Resistance.
Oxford, J. S., Logan, I. S. & Potter, C. W. In vivo selection of an influenza A2 strain resistant to amantadine. Nature 226, 82–83 (1970).
Monto, A. S. & Arden, N. H. Implications of viral resistance to amantadine in control of influenza A. Clin Infect Dis 15, 362–367, discussion 368–369 (1992).
Hayden, F. G. & Hay, A. J. Emergence and transmission of influenza A viruses resistant to amantadine and rimantadine. Curr Top Microbiol Immunol 176, 119–130 (1992).
Hayden, F. G. et al. Emergence and apparent transmission of rimantadine-resistant influenza A virus in families. N Engl J Med 321, 1696–1702, https://doi.org/10.1056/NEJM198912213212502 (1989).
Colman, P. M., Hoyne, P. A. & Lawrence, M. C. Sequence and structure alignment of paramyxovirus hemagglutinin-neuraminidase with influenza virus neuraminidase. J Virol 67, 2972–2980 (1993).
Abed, Y. & Boivin, G. A Review of Clinical Influenza A and B Infections With Reduced Susceptibility to Both Oseltamivir and Zanamivir. Open Forum Infect Dis 4, ofx105, https://doi.org/10.1093/ofid/ofx105 (2017).
Hensley, S. E. et al. Influenza A virus hemagglutinin antibody escape promotes neuraminidase antigenic variation and drug resistance. PLoS One 6, e15190, https://doi.org/10.1371/journal.pone.0015190 (2011).
Barroso, L., Treanor, J., Gubareva, L. & Hayden, F. G. Efficacy and tolerability of the oral neuraminidase inhibitor peramivir in experimental human influenza: randomized, controlled trials for prophylaxis and treatment. Antivir Ther 10, 901–910 (2005).
Cass, L. M., Efthymiopoulos, C. & Bye, A. Pharmacokinetics of zanamivir after intravenous, oral, inhaled or intranasal administration to healthy volunteers. Clin Pharmacokinet 36(Suppl 1), 1–11 (1999).
Elliott, M. Zanamivir: from drug design to the clinic. Philos Trans R Soc Lond B Biol Sci 356, 1885–1893, https://doi.org/10.1098/rstb.2001.1021 (2001).
Davies, B. E. Pharmacokinetics of oseltamivir: an oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J Antimicrob Chemother 65(Suppl 2), ii5–ii10, https://doi.org/10.1093/jac/dkq015 (2010).
Dutkowski, R. et al. Safety and pharmacology of oseltamivir in clinical use. Drug Saf 26, 787–801 (2003).
He, G., Massarella, J. & Ward, P. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin Pharmacokinet 37, 471–484, https://doi.org/10.2165/00003088-199937060-00003 (1999).
Kim, C. U. et al. Structure-activity relationship studies of novel carbocyclic influenza neuraminidase inhibitors. J Med Chem 41, 2451–2460, https://doi.org/10.1021/jm980162u (1998).
Pollara, C. P. et al. Nosocomial outbreak of the pandemic Influenza A (H1N1) 2009 in critical hematologic patients during seasonal influenza 2010–2011: detection of oseltamivir resistant variant viruses. BMC Infect Dis 13, 127, https://doi.org/10.1186/1471-2334-13-127 (2013).
Sheu, T. G. et al. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob Agents Chemother 52, 3284–3292, https://doi.org/10.1128/AAC.00555-08 (2008).
LeGoff, J. et al. I223R mutation in influenza A(H1N1)pdm09 neuraminidase confers reduced susceptibility to oseltamivir and zanamivir and enhanced resistance with H275Y. PLoS One 7, e37095, https://doi.org/10.1371/journal.pone.0037095 (2012).
Pizzorno, A. et al. Impact of mutations at residue I223 of the neuraminidase protein on the resistance profile, replication level, and virulence of the 2009 pandemic influenza virus. Antimicrob Agents Chemother 56, 1208–1214, https://doi.org/10.1128/AAC.05994-11 (2012).
Hurt, A. C., Leang, S. K., Speers, D. J., Barr, I. G. & Maurer-Stroh, S. Mutations I117V and I117M and oseltamivir sensitivity of pandemic (H1N1) 2009 viruses. Emerg Infect Dis 18, 109–112, https://doi.org/10.3201/eid1801.111079 (2012).
Hurt, A. C. et al. Increased detection in Australia and Singapore of a novel influenza A(H1N1)2009 variant with reduced oseltamivir and zanamivir sensitivity due to a S247N neuraminidase mutation. Euro Surveill 16 (2011).
Hussain, S. et al. Strain-specific antiviral activity of iminosugars against human influenza A viruses. J Antimicrob Chemother 70, 136–152, https://doi.org/10.1093/jac/dku349 (2015).
Das, S. R. et al. Defining influenza A virus hemagglutinin antigenic drift by sequential monoclonal antibody selection. Cell Host Microbe 13, 314–323, https://doi.org/10.1016/j.chom.2013.02.008 (2013).
Djikeng, A. et al. Viral genome sequencing by random priming methods. BMC Genomics 9, 5, https://doi.org/10.1186/1471-2164-9-5 (2008).
Nakamura, K. et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res 39, e90, https://doi.org/10.1093/nar/gkr344 (2011).
Doceul, V., Hollinshead, M., van der Linden, L. & Smith, G. L. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327, 873–876, https://doi.org/10.1126/science.1183173 (2010).
Zhou, B. & Wentworth, D. E. Influenza A virus molecular virology techniques. Methods Mol Biol 865, 175–192, https://doi.org/10.1007/978-1-61779-621-0_11 (2012).
Reed, L. J. & Muench, H. A simple method of estimating fifty percent endpoints. The American Journal of Hygiene 27, 493–497 (1938).
Bryan, W. R. Interpretation of host response in quantitative studies on animal viruses. Ann N Y Acad Sci 69, 698–728 (1957).
Acknowledgements
We thank the laboratory and veterinary staff at Utah State University and Integrated Biotherapeutics for their contributions to the work. Aruna Sampath, Mansoora Khaliq, Anthony Treston, Christina Hedberg and Michael Callahan, as well as Amy Krafft, Helen Schiltz from National Institute of Allergy and Infectious Diseases (NIAID), engaged in helpful discussions. We thank Mr. Suman Pakala for sequence data submission to GenBank. We thank Reed Shabman for the initial discussion on sequencing approaches. The work here was supported by Emergent BioSolutions (formerly Unither Virology, LLC). UV-4B material used for these studies was generated under NIAID contract HHSN272201100030C awarded to Emergent BioSolutions (formerly Unither Virology, LLC). The viral sequencing infrastructure used for the study was funded in whole or part with federal funds from the NIAID, National Institutes of Health (NIH), Department of Health and Human Services (HHS) under award number U19AI110819 to SRD. SRD is also supported by NIH-funded Tennessee Center for AIDS Research (P30 AI110527) and U19AI095227. Content is solely the responsibility of the authors.
Author information
Authors and Affiliations
Contributions
K.L.W., W.W., E.S., M.M., M.H.S., T.B.S., D.L.B., U.R. and S.R.D. designed and conducted work described herein. All authors analyzed and interpreted data described here. K.L.W., K.R.S., L.E.D., K.B.S. and S.R.D. generated the first draft of the manuscript and all authors have reviewed and approved the content.
Corresponding authors
Ethics declarations
Competing Interests
W.W., E.S., M.M., K.R.S., M.H.S., T.B.S. and D.L.B. declare no competing interests. K.L.W., L.E.D. and K.B.S. are employees and shareholders of Emergent BioSolutions. K.L.W. and U.R. are authors on pending and issued patents related to the use of iminosugars as treatments for viral disease. S.R.D. has been funded by Emergent BioSolutions. Other authors have no competing financial and/or non-financial interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Warfield, K.L., Schaaf, K.R., DeWald, L.E. et al. Lack of selective resistance of influenza A virus in presence of host-targeted antiviral, UV-4B. Sci Rep 9, 7484 (2019). https://doi.org/10.1038/s41598-019-43030-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43030-y
This article is cited by
-
Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
