
Abstract
Primordial follicles, consisting of granulosa cell (GC)-enveloped oocytes are maintained in a state of developmental arrest until activated to grow. The mechanism that operates to maintain this arrested state in GCs is currently unknown. Here, we show the TGFβ-activated transcription factor SMAD3 is expressed in primordial GC nuclei alongside the cell cycle proteins, cyclin D2 (CCND2) and P27. Using neonatal C57/Bl6 mouse ovaries densely populated with primordial follicles, CCND2 protein co-localised and was detected in complex with P27 by immunofluorescence and co-immunoprecipitation, respectively. In the same tissue, SMAD3 co-precipitated with DNA sequences upstream of Ccnd2 and Myc transcription start sites implicating both as direct SMAD3 targets. In older ovaries follicle growth was associated with nuclear exclusion of SMAD3 and reduced P27 and CCND2 in GCs, alongside elevated Myc expression. Brief (2 H) exposure of neonatal ovaries to TGFβ1 (10 ng/ml) in vitro led to immediate dissociation of SMAD3 from the Ccnd2 and Myc promoters. This coincided with elevated Myc and phospho-S6, an indicator of mTOR signalling, followed by a small increase in mean primordial GC number after 48 H. These findings highlight a concentration-dependent role for TGFβ signalling in the maintenance and activation of primordial follicles, through SMAD-dependent and independent signalling pathways, respectively.
Similar content being viewed by others

Introduction
Primordial follicles, each consisting of a central oocyte surrounded by a single layer of supporting granulosa cells (GCs) are held in a relative state of developmental arrest until activated to grow1,2,3. Maintenance and regulation of the arrested state is fundamental for ensuring that a steady supply of oocytes is available for ovulation throughout the reproductive life time in most mammals4,5; however, the molecular mechanisms that operate to maintain this quiescent phenotype are currently unclear.
We recently showed that in the mouse ovary, the TGFβ-mediated transcription factors SMAD2/3 are detectable in nuclei of primordial GCs, suggesting that this pathway is active in these cells6. It is well established that TGFβ signalling is important for maintaining growth arrest in a range of cell types by directly inhibiting Myc7,8,9, as well as regulating the expression of other cell cycle proteins10,11. Cell proliferation initially involves cell cycle progression from G0-G112. Typically, this occurs through growth factor stimulated expression of cyclin D, which then binds to cyclin-dependent kinase (CDK) proteins, precipitating release of E2F transcription factors to drive cell cycle progression12,13,14. In many cell types, cyclin D-CDK complexes can also be inhibited by P27, creating a stable trimeric complex that maintains cell cycle arrest15. In the ovary, in contrast to other D-type cyclins, cyclin D2 (CCND2) mRNA is detectable in GCs of follicles across a range of stages and knockout of both cyclin D2 (Ccnd2−/−) and p27−/− exhibit dis-regulated early follicle development with major effects on fertility16,17,18.
We therefore carried out detailed analysis of CCND2 and P27 in the context of arrested primordial and early developing follicles, focussing on the relationship of these factors with TGFβ signalling in GCs. Using the C57/Bl6 mouse as a model, we show that SMAD3 specifically localises to GCs of small follicles where it directly promotes the expression of Ccnd2 and represses Myc. The CCND2 protein is detectable in complex with the inhibitory factor P27 and together highlights a mechanism that potentially maintains primordial GC arrest, whilst ensuring they are poised ready to proliferate. Interestingly, the loss of SMAD3 in GCs of growing follicles is associated with elevated mTOR signalling, potentially explaining a dual mechanism of TGFβ signalling in follicle growth and arrest.
Results and Discussion
SMAD3 is predominantly expressed in GCs of primordial follicles
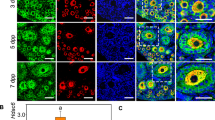
Both SMAD2 and SMAD3 mediate TGFβ signalling and regulate gene expression10,11,19; however, whether both or either SMAD is important in this context is not known. In this study, we have used ovaries from C57/Bl6 mice at postnatal day 4, 8 and 16 as a natural developmental paradigm to analyse gene expression in primordial, early activated (transitional), and growing follicles (primary, primary+ , secondary), respectively20,21 (Fig. 1A). Immunofluorescent localisation of SMAD2 was very weak in ovaries at each age, while SMAD3 was specifically expressed in GCs of small follicles, notably in the earliest primordial stages as exemplified in sections from day 4 ovaries (Fig. 1B). Consistent with this, the level of Smad2 mRNA, although detectable, did not vary between the different ovary ages (Fig. 1C), whereas Smad3 mRNA expression was reduced in day 8 and 16 ovaries in which the relative proportion of primordial follicles is reduced (Fig. 1D). Based on these findings we focused on SMAD3 for further analyses.

SMAD3 is predominantly expressed in GCs of primordial follicles. (A) Diagram showing the follicle composition of the prepubertal mouse ovary at 4, 8 and 12 days of age (d4, d8, d12). (B) Immunofluorescent localisation of SMAD2 and SMAD3 protein in the prepubertal mouse ovary. SMAD2 (green) was weak/undetectable in prepubertal ovaries, including primordial (yellow arrow) and early growing primary follicles (yellow asterisk), but was strong in corporal lutea from adult ovary (d16 inset). SMAD3 (red) localised to the nuclei and cytoplasm of GCs of primordial and transitional follicles (arrows), with nuclear expression appearing weaker in GCs of growing primary-plus follicles (arrowheads). Cell nuclei are counterstained with DAPI (blue). Rabbit IgG is shown as a negative control. (C,D) Relative expression of Smad2 and Smad3 mRNA by qPCR in d4, d8 and d16 ovaries. (E–G) Quantitative analyses of SMAD3 immunofluorescence. (E) Data represents total SMAD3 staining intensity in GCs by follicle stage. Each point represents the % positive pixels in the GC compartment for an individual follicle. (F) Nuclear SMAD3 intensity in GCs by follicle stage. Each point represents the % positive pixels in individual GC nuclei within each follicle stage. Points in red and blue represent measurements from d4 and d8 ovaries, respectively. (G) Nuclear/cytoplasmic ratio of SMAD3 in GCs by follicle stage. Each point represents the % positive pixels in all GC nuclei relative to % positive pixels in the GC compartment for an individual follicle obtained from d8 ovaries. Dashed lines (E,F) demarcate lower and upper quartiles to represent proportions of samples that were considered low, medium and high intensity staining, respectively. (H) Subcellular expression of SMAD3 by western blotting in protein lysates from d4 mouse ovaries. Samples were separated into cytosolic, membrane-bound organelle, and nuclear fractions. The cytoplasmic protein GAPDH is included as a control. Gel images have been cropped from originals provided in Fig. S1. PF, primordial; T, transitional; P, primary; P + , primary plus, S, secondary follicle. Data in C, D are means ± SEM (n = 5 ovaries/age) and differences are relative to d4. Data in E, F, G are medians ± interquartile ranges and differences are relative to the follicle stage in parentheses. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
When SMAD3 immunofluorescence intensity values were plotted by follicle stage, a bi-phasic pattern was evident, with total GC-expression being highest in transitional follicles that are poised to initiate growth, and lowest in more advanced secondary-staged growing follicles (Fig. 1E). Since SMAD3 is a cytoplasmic signalling mediator and nuclear transcription factor19,22, we analysed GC expression in both cellular compartments. In GC nuclei SMAD3 was highly variable; however, the median expression was similar between primordial, transitional and primary staged follicles, and was lower from the primary plus stage (Fig. 1F). Nuclear SMAD3 in each follicle was then normalised against cytoplasmic expression to determine if the ratio was equal throughout follicle development. The results, however, show that SMAD3 was similarly located in the nuclei and cytoplasm of GCs in primordial and transitional follicles, with a relative reduction in nuclear expression from the primary stage, indicating that nuclear exclusion occurs prior to the general loss of SMAD3 expression in GCs (Fig. 1G). Sub-cellular fractionation of protein lysates from day 4 ovaries further confirmed that SMAD3 was detectable in both nuclear and non-nuclear fractions (Fig. 1H). Since SMAD3 exhibits a DNA binding domain22, our findings support a role for SMAD3 as a potential transcriptional regulator in GCs of primordial follicles.
CCND2 and P27 are maintained in GCs of primordial follicles but decrease as follicles initiate growth
The expression of D-type cyclins occurs early in the cell cycle12,15; therefore, the localisation and regulation of CCND2, as well as the inhibitory partner P27 in quiescent primordial follicles were investigated. CCND2 was detectable in GC and oocyte nuclei, with relatively strong staining evident in GCs of small, single layered follicles (Fig. 2A). Strong expression (intensity values >75%) of CCND2 was detectable in GCs of over 50% of primordial follicles analysed, with a relative reduction in expression from the primary stage onwards (Fig. 2B). Consistent with this, Ccnd2 mRNA levels in whole ovaries were significantly reduced in older ovaries containing proportionately more growing follicles, relative to day 4 ovaries (Fig. 2C). As with CCND2, P27 protein localised to GC nuclei of small follicles (Fig. 2D). Staining was highly variable between primordial follicles; however, the nuclear expression of P27 significantly declined in the majority of growing follicles from the primary stage onwards (Fig. 2E). This loss of expression in developing follicles reflected p27 mRNA expression in whole ovaries, which was significantly reduced in day 8 and 16 ovaries, relative to day 4 (Fig. 2F).

CCND2 and P27 are maintained in GCs of primordial follicles but decrease as follicles initiate growth. (A) Immunofluorescent localisation of CCND2 (red) in the prepubertal mouse ovary. High power images show strong (persistent) nuclear CCND2 staining in GCs of primordial follicles (PF; arrows). Transitional follicles (T) show nuclei with variable CCND2 (arrow heads) whereas larger primary plus staged follicles (P+) show weak CCND2 expression. (B) Nuclear CCND2 protein expression in GCs by follicle stage. Each point represents the % positive pixels for all GC nuclei in an individual follicle. (C) Relative expression of Ccnd2 mRNA by qPCR in d4, d8 and d16 ovaries. (D) Immunofluorescent localisation of P27 (green) in the prepubertal mouse ovary. High power images (lower panels) show strong nuclear P27 staining in GCs of primordial follicles (PF; arrows). Transitional follicles (T) show nuclei with variable P27 whereas larger secondary staged follicles (S) show weak P27 expression. (E) Nuclear P27 protein expression in GCs by follicle stage. Each point represents the % positive pixels for all GC nuclei in an individual follicle. (F) Relative expression of p27 mRNA by qPCR in d4, d8 and d16 ovaries. (G) Co-localisation of CCND2 and P27 in the prepubertal mouse ovary. After image processing (d4 and d8; lower panels), only positive pixels in the green and red channels are shown, highlighting co-localisation in white. A negative IgG control (d16; lower right) is shown. (H) P27/CCND2 ratio in individual GCs by follicle stage. Each point represents the % positive pixels for P27 relative to % positive pixels for CCND2 an individual nucleus. Numbers in parentheses refer to the GC number range for each follicle stage. (I) Western blot of P27 and CCND2 following co-immunoprecipitation from d4 and d16 ovaries. Protein samples (12.5 µg) were immunoprecipitated using anti- P27 (IP) or non-specific rabbit IgG (IgG; control). Non-immunoprecipitated protein (5 µg) was used as positive control (IN). Gel images have been cropped from originals provided in Fig. S2. PF, primordial; T, transitional; P, primary; P+, primary plus, S, secondary follicle. Nuclei in A, D, G are labelled with DAPI (blue). Data in B, E, H are medians ± interquartile ranges and differences are relative to the follicle stage in parentheses. Points in red and blue represent measurements from d4 and d8 ovaries, respectively. Data in C, F are means ± SEM (n = 5 ovaries/age) and differences are relative to d4. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Both proteins co-localised in GC nuclei of small follicles regardless of age (Fig. 2G). We then focussed on d4 and d8 ovaries due to the preponderance of primordial and small activated follicles. Although the degree of co-localisation in each cell varied widely within follicles and between follicle stages, the ratio of P27 to CCND2 declined from the transitional stage onwards. Thus, as follicles activate, P27 is initially reduced while CCND2 persists (Fig. 2H). In support of this, both CCND2 and P27 co-immunoprecipitated in protein lysates from day 4 ovaries. However, in day 16 ovaries, although CCND2 was detectable by western blot, the same protein was beyond the limits of detection by western blotting following immunoprecipitation with P27 (Fig. 2I). Together these data indicate that CCND2 and P27 exist in molecular complexes in GCs of primordial follicles. Consistent with a role for P27 as a cell-cycle inhibitory factor15, it is likely that P27 is inhibiting the activity of CCND2 in primordial GCs, thereby participating in maintaining the quiescent phenotype of primordial follicles.
SMAD3 directly regulates Ccnd2 and Myc expression in primordial follicles
Since the transcription factor SMAD3 is expressed in GC nuclei of primordial/transitional follicles alongside CCND2 and P27, we then investigated whether SMAD3 has the ability to directly bind and regulate the expression of Ccnd2 and p27 in this context. The immediate early gene Myc was also used as a positive control, since it is a well-known target of SMAD3 and is involved in cell-cycle progression7,8,9. In day 4 and day 16 ovaries, SMAD3 was detected on the promoter of Ccnd2 using ChIP-PCR, which was significant when quantified in relation to non-immune control IgG (Fig. 3A). This is consistent with the observed reduction in Ccnd2 gene expression in ovaries of Smad3−/− mice23,24. SMAD3 was also detectable on the promoter of the Myc gene in day 4 ovary samples; however, this binding was not significantly different from control IgG in day 16 ovary samples (Fig. 3B). By comparison, SMAD3 binding to the p27 promoter was undetectable by ChIP-PCR in day 4 ovaries, and barely detectable in day 16 ovaries (Fig. 3C). Therefore, it is unlikely that p27 is a direct target of SMAD3 in small follicles.

SMAD3 directly regulates Ccnd2 and Myc expression in primordial follicles. (A–C) Amplification of target binding sites upstream of the Ccnd2, Myc and p27 genes following ChIP. Representative gel images showing amplification of DNA after immuno-precipitation with anti-SMAD3, anti-FOXL2 (GC expressed transcription factor – control), or non-specific antibodies (IgG) in d4 and d16 mouse ovaries. A positive sample of chromatin (Input) prior to immunoprecipitation is also shown (upper panels). Relative quantification of anti-SMAD3 IgG binding to Ccnd2 (lower panel A) and Myc (lower panel B) gene promoters in d4 and d16 ovaries by qPCR. % input was calculated by normalizing anti-SMAD3 IgG and non-specific IgG (control) against input DNA as described in the materials and methods. Each group (d4, d16) contained 5 repeats (n = 5 ovaries). Data are medians ± interquartile range and statistical differences are relative to IgG control. *P < 0.05, NS: Not significant. Note the Ccnd2 gel is a composite as indicated with divisional space and all gel images have been cropped from originals provided in Fig. S3. (D) Relative expression of Myc mRNA by qPCR in d4, d8 and d16 ovaries. Means ± SEM (n = 5 ovaries/age) and differences are relative to d4. *P < 0.05, ****P < 0.0001.
Since Myc has a well-ascribed role in promoting cell proliferation25,26, we then quantified expression in immature ovaries. The increase in Myc mRNA expression in day 8 and day 16 ovaries relative to day 4 ovaries (Fig. 3D) suggests that SMAD3 acts to repress Myc transcription in primordial follicles. Conversely, the decrease in Ccnd2 mRNA expression in day 16 ovaries relative to day 4 ovaries observed previously (Fig. 2A–C) shows that SMAD3 likely promotes cyclin D2 expression in primordial follicles. These findings imply that TGFβ-SMAD3 signalling acts directly on GCs of primordial follicles to differentially regulate genes involved in cell cycle regulation.
Short term exposure to TGFβ1 promotes mTOR signalling in neonatal mouse ovaries
Considering the above observations, we then investigated the effect of an altered TGFβ environment on small follicle development in vitro. Previous studies have shown that long-term culture of neonatal rodent ovaries ( > 7 days) with a TGFβ ligand can influence primordial follicle viability27,28,29. In cell culture models, changes in SMAD3-regulated gene expression have been observed as little as 30 minutes after stimulation with TGFβ ligands30,31. Furthermore, a 60–90 min exposure of kit ligand or the pathway inhibitor imatinib to post-natal day 7 mouse ovaries in culture was sufficient to cause a significant shift in FOXO3a translocation in oocytes32. We therefore evaluated effects on gene expression and signalling following a brief 2-hour exposure to TGFβ1 ligand and A83-01, a Type I receptor inhibitor, in day 4 ovaries in vitro. Consistent with in vivo findings, SMAD3 bound to the promoter of Ccnd2 and Myc in unstimulated (control) ovaries. By comparison, binding to Ccnd2 and Myc was no longer evident in ovaries exposed to either TGFβ1, A83-01, or a combination of TGFβ1 and A83-01 (Fig. 4A), indicating that modulation of TGFβ signalling causes dissociation of SMAD3 from the promoters of these genes. Moreover, dissociation of SMAD3 to target gene promoters was not able to be over-ridden by exposure to TGFβ1. Interestingly, this change in binding was associated with elevated expression of the pro-proliferation factor Myc in ovaries exposed to TGFβ1. Myc is an early response gene and in other studies Myc mRNA reaches maximum levels after 2–3 hours after growth factor stimulation, whereas induction of Ccnd2 mRNA peaks around 4 hours33,34. The latter point may explain why the loss of SMAD3 occupancy on the Ccnd2 promoter did not effect Ccnd2 mRNA expression in the same way as Myc (Fig. 4B). Elevated transcript levels of Smad7 – a negative regulator of TGFβ signalling35,36 were also observed, suggesting one effect of increased exposure to TGFβ1 is to inhibit the action of SMAD3 (Fig. 4B). In ovaries exposed to A83-01 or a combination of TGFβ1 and A83-01, Myc expression was unchanged. As Myc is normally repressed in cells with low turnover, it is possible that other transcriptional repressors independent of TGFβ signalling, such as WT137,38, are active in primordial GCs, although further studies would be required to confirm this.

Short term exposure to TGFβ1 promotes mTOR signalling in neonatal mouse ovaries. Whole neonatal mouse ovaries (d4) were maintained in vitro and exposed for 2 hours in either 1 µM DMSO (control; C), 10 ng/ml of TGFβ1 ligand (T), or 1 µM A83-01 inhibitor (I). An additional group was exposed to 1 µM A83-01 inhibitor followed by 10 ng/ml of TGFβ1 ligand (IT). Ovaries were analysed for gene and protein analysis immediately following treatment. (A) Relative quantification of SMAD3 binding to Ccnd2 and Myc gene promoters in cultured ovaries by ChIP-qPCR. Each group contained 5 ovaries (n = 5). Data are medians ± interquartile range and statistical differences are relative to IgG control (*P < 0.05). (B) Relative expression of Myc, Smad7 and Ccnd2 mRNA in cultured ovaries. Each group contained 5 ovaries (n = 5). Data are means ± SEM; *P < 0.05 vs group indicated in parentheses. (C) Expression of phospho-intermediates in the canonical TGFβ (p-SMAD3), PI3 kinase/Akt (p-AKT), p44/42 MAPK (p-ERK1/2) and mTOR (p-S6) pathways in cultured ovaries by western blot. Example blots are shown (left-hand panels; originals provided in Fig. S4) and levels quantified by normalization against beta-actin (loading control). Data represents four samples from each group, each from an independent blot. *P < 0.05 vs group indicated in parentheses.
Since exposure to A83-01 and TGFβ1 led to a similar loss of SMAD3 promoter binding, but with differential effects on Myc and Smad7 expression, we sought to determine if this was associated with elevated SMAD3 signalling, or activation of SMAD-independent pathways. Levels of p-SMAD3, p-AKT and p-ERK1/2 proteins did not vary between TGFβ1, A83-01 or control groups. By comparison, the mTOR mediator p-S6 was significantly increased in ovaries exposed to TGFβ1 in comparison to ovaries exposed to A83-01 or control (Fig. 4C), indicating that elevated TGFβ signalling in neonatal ovaries leads to early effects on the mTOR pathway.
Acute modulation of TGFβ signalling causes stage-specific effects on follicle development
Finally, in order to evaluate the consequence of an acutely altered TGFβ environment, ovaries were returned to control media for an additional two days. Processed ovaries were then stained with SMAD3 and DDX4 as markers of the GC compartment and oocytes, respectively, for morphological assessment and quantification. The overall distribution of follicles was similar between groups, although GCs of growing primary follicles in ovaries exposed to A83-01 had failed to adequately cuboidalise (Fig. 5A). When follicles were classified according to oocyte size, TGFβ1 or A83-01 had no effect on stage of follicle development (Fig. 5B). Likewise, exposure to TGFβ1 or A83-01 had no effect on oocyte size (Fig. 5C). Interestingly, brief exposure to TGFβ1 ligand promoted a small, but measurable increase in the mean GC number of follicles classified as primordial, suggesting that 2 hours of elevated TGFβ1 was sufficient to promote GC proliferation but not sufficient enough to promote significant oocyte growth. By comparison, exposure to A83-01 had no effect on GCs of primordial follicles but caused a significant reduction in mean GC number in follicles classified as growing, indicating that inhibition of TGFβ receptors inhibits normal GC proliferation in activated follicles (Fig. 5D). It should also be noted that very few apoptotic cells were detectable in any of the groups as determined by TUNEL labelling (Fig. S6). The reduction in GC number along with inadequate cuboidalisation indicates that inhibition of TGFβ receptors likely impairs normal GC proliferation in activated follicles. Thus, although SMAD3 is reduced in GCs of growing follicles (Fig. 1), TGFβ signalling is still essential, which is consistent with the well-ascribed role of other TGFβ ligands that signal through the same Type I receptors, such as GDF939,40,41,42 and activin43,44,45,46 in supporting early follicle development.

Acute modulation of TGFβ signalling causes stage-specific effects on follicle development. Whole neonatal mouse ovaries (d4) were maintained in vitro and exposed for 2 hours with either 10 ng/ml of TGFβ1 ligand (T), 1 µM A83-01 inhibitor (I), or 1 µM DMSO (C). Ovaries were then placed in basic culture media for two additional days prior to morphological analysis. (A) Confocal images of sections of cultured ovaries immunolabelled with DDX4 (red) and SMAD3 (green) to highlight oocytes and GCs, respectively. Cell nuclei are counterstained with DAPI (blue). Low power images (upper panels) showing general distribution of follicles in each group. Scale bars = 100 µm. High power images (lower panels) showing examples of early growing primary (P) and primary plus (P+) follicles in each group as classified by oocyte size. Note the smaller size of the GC-layer in follicles exposed to the inhibitor. Scale bars = 20 µm. (B) Proportion of follicles classified by follicle stage. (C) Distribution of oocyte size by treatment. (D) Mean GC number per follicle. Quantitative data from cultured ovaries (B–D) was obtained from six different ovary sections per treatment group (n = 6 ovaries/group). Classification was based on oocyte size (Fig. S5). Data show means ± 95% CI. *P < 0.05 and ****P < 0.0001 vs control. PF, primordial; T, transitional; P, primary; P+, primary plus follicle.
Conclusions
The ovarian reserve consists of a limited pool of primordial follicles held in a state of developmental arrest1,3,4,5. The molecular programme that maintains this arrest, and conversely regulates activation and growth is largely unknown. We recently presented a detailed analysis highlighting an inverse relationship between nuclear SMAD2/3 expression and cell proliferation in GCs of mouse ovaries, suggesting a role for TGFβ signalling in primordial follicle maintenance and activation6. Here, we show that SMAD3 recapitulates the pattern of SMAD2/3 and is associated with expression of the cell cycle regulators cyclin D2 and P27. The specific localisation of these proteins was similar in ovaries derived from d4 and d8 mice, each with different populations of developing follicles, further highlighting a stage-specific, rather than age-dependent role in in GCs of non-growing and early growing follicles (Fig. S7). In non-growing follicles, it is unlikely that SMAD3 directly regulates P27 in this context; instead, SMAD3 binds directly to Ccnd2 and Myc to differentially regulate these genes, which may be important for ensuring GCs are ‘poised’ in a state ready to progress through the cell cycle. The presence and accumulation of CCND2 in complex with P27 suggests a mechanism for primordial follicle maintenance, where P27 may prevent GC cycle progression by inhibiting cyclin-CDK activity15. This hypothesis is consistent with other studies reporting an inhibitory effect of P27 on early follicle activation16,47. Although the mechanism leading to a reduction in P27 activity in this context is currently unknown, it is clear that follicle growth is also associated with a reduction in accumulated CCND2 and SMAD3 – the latter also allowing de-repression of Myc. CCND2 and SMAD3 are important regulators of the cell cycle and although their steady-state expression is reduced in growing follicles, both proteins are still expressed at a moderate level in GCs (Figs 1 and 2). Furthermore, briefly exposing ovaries to an elevated environment of TGFβ led to a slight increase in GC proliferation. This occurred following an immediate uncoupling of SMAD3 from Ccnd2 and Myc gene promoters alongside elevated mTOR signalling. Activation of mTOR signalling in primordial GCs is a key early event in follicle initiation48, has been implicated in TGFβ-driven models of epithelilal-mesenchymal transition49, and plays an important role more generally in cellular growth and proliferation50.
Based on these findings, we propose a multi-step mechanism of follicle activation that initially involves a basal level of SMAD3 expression and nuclear activity to prepare GCs for proliferation. It remains to be determined whether this activity in primordial GCs is dependent on receptor-mediated signalling, and if so, which is the principle ligand involved; however, increased TGFβ1 ligand exposure promotes a shift to a SMAD-independent pathway, which drives cell proliferation and eventual follicle activation (Fig. 6). The role of other TGFβ superfamily members and the cross-talk involved in regulating SMAD signalling in this context will also be important to establish. Although this study has used the mouse as a model organism, revelation of the molecular framework that underscores the phenotype of pre-granulosa cells in other species is essential for understanding how the ovarian reserve is maintained throughout the reproductive lifetime of the female.

Model of cell cycle regulation in GCs of small follicles. Arrested GCs of primordial follicles express the transcription factor SMAD3, which directly promotes expression of CCND2 and represses Myc. In this ‘poised’ state, CCND2 is bound by the inhibitory factor P27. In transitional GCs, P27 dissociates from CCND2 and SMAD3 expression transiently increases, possibly via elevated TGFβ signalling, allowing maintenance of CCND2 expression for cell cycle progression. Increased SMAD3 promotes expression of factors such as Smad7, which act to inhibit SMAD3 in a self-regulatory manner. The eventual loss of SMAD3 leads to de-repression of Myc and activation of SMAD-independent TGFβ signalling involving mTOR to drive GC proliferation in growing follicles. Solid black line arrows indicate predominant signalling pathway. Coloured block arrows indicate moderate (unfilled arrows) or substantial (filled arrows) changes in expression relative to primordial stage. Note in growing follicles, although SMAD3 and CCND2 are reduced, they are still detectable and play an essential role in regulation of the GC cycle. PF, primordial; T, transitional; P, primary follicle.
Materials and Methods
Animals and tissue collection
All ovary tissues used in this study were obtained from wild-type C57Bl6 mice housed in the Biological Services Unit at the University of Sheffield in compliance with the Animals (Scientific Procedures) Act, 1986. Mice were killed by a registered practitioner (Schedule 1 procedures) with approval from the University of Sheffield Training and Competency Officer. For total RNA, protein extraction and ChIP experiments, ovaries from immature mice (day 4, 8 and 16) were dissected and either placed in culture (day 4 only) or snap frozen in liquid nitrogen and stored at −80 °C. For immunostaining, ovaries were immersed in 10% neutral buffered formalin solution (Sigma-Aldrich) for 24 hours and processed in paraffin blocks and sectioned at 5 μm.
Neonatal mouse ovary culture
Ovaries were placed in 24 mm Transwell plates containing membrane inserts (Corning; Sigma). Culture medium consisted of Waymouth medium 752/1 (Life Technologies) supplemented with 10% fetal bovine serum (FBS; Sigma), 0.23 mM pyruvic acid (Sigma), 10 µg/ml streptomycin sulfate (Sigma), 75 µg/ml penicillin G (PENK; Sigma) and 0.3 mg/mL BSA. Ovaries were maintained in a 37 °C incubator (5% CO2) for 24 h prior to any treatment. For the assessment of the molecular changes under different TGFβ conditions (SMAD3 binding and gene regulation) ovaries were exposed for 2 h in serum free culture media, supplemented with either TGFβ1 ligand (10 ng/ml; R&D systems, 240-B-002), A83-01 (1 µM; MedChem Press, 2939), a selective inhibitor of Type 1 TGFβ receptors ALK4, ALK5 and ALK751, a combination of TGFβ1 (10 ng/ml) and A83-01 (1 µM), or DMSO (control; same volume as for A83-01; Sigma). A single time point (2 hours) and concentration for TGFβ1 ligand (10 ng/ml) and A 83-01 inhibitor (1 µM) were selected based on previous studies27,28,29,51,52. Immediately following treatment, ovaries were collected and either fixed and processed for ChIP-qPCR or flash frozen in liquid nitrogen for gene expression analysis. For the assessment of the morphological changes, ovaries were exposed for 2 h in serum free culture media followed by supplementation as above. In the TGFβ1 treatment group, the addition of A83-01 inhibitor for an extra 1 hour was used to block further TGFβ1 signalling as previously described53. Treatments were then washed out and ovaries were left in culture for two days before processing.
Immunofluorescence staining
Sections were dewaxed before microwaving in 0.1 M citrate buffer (pH6.0). Sections were blocked with CAS universal blocking reagent (ThermoFisher) for 20 minutes at room temperature to reduce non-specific binding. Specific antibodies against SMAD2 (0.59 µg/ml, #5339, Cell Signaling Technology, MA, USA), SMAD3 (0.75 µg/ml, #9523, Cell Signaling), SMAD2/3 (0.25 µg/ml, 133098, Santa Cruz Biotechnology, TX, USA), CCND2 (0.5 µg/ml, sc-593, Santa Cruz Biotechnology, TX, USA), DDX4 (2.2 µg/ml, ab13840, Abcam, Cambridge, UK) and P27 (1 µg/ml, sc-528, Santa Cruz or 0.5 µg/ml, sc-1641 for co-localisation) were diluted in blocking solution and incubated overnight at 4 °C. A species-specific isotype control IgG antibody at the same concentration (normal rabbit IgG I-1000 and normal mouse IgG I-200, Vector Laboratories) were included in each experiment. Sections were washed in PBS and incubated with secondary antibodies (either donkey anti-rabbit AlexaFluor® 488 or AlexaFluor® 555; donkey anti-mouse AlexaFluor® 488 and AlexaFluor® 555, each 1:400; Invitrogen). Sections were mounted with a drop of Prolong® Gold anti-fade reagent with DAPI (ThermoFisher) and were imaged using an inverted Leica SP5 confocal laser-scanning microscope (Leica Microsystems, Wetzlar, Germany).
Image analysis and quantification of imunofluorescence
Quantification of GC factors in situ was performed from a total of 7 ovaries (four day 4 and three day 8 ovaries) using the largest cross section per ovary. Confocal images were stitched together using DoubleTake (http://echoone.com/doubletake/) to create a high-resolution composite of each section. Images were obtained as an 8-bit RGB stack file and the threshold for each channel in every composite section was set up with the same criteria across the different sections of the same staining. Pixels above the threshold were considered as ‘positive pixels” while the remaining pixels were registered as negative. The oocyte cytoplasm was used as a baseline setting (due to lack of specific staining) and threshold settings were normalised against this to account for variation between slides. Follicles with a visible oocyte nucleus were selected with the region of interest (ROI) manager tool of ImageJ software (https://imagej.nih.gov/ij/). In order to quantify each of the candidate proteins in different ROIs, the area of positive pixels “Area fraction” was calculated and referred as “Intensity” for simplicity. ROIs from each follicle were selected manually using the DAPI channel and the “measure and label” tool, which allowed the intensity of each ROI in either the green or red channel to be quantified. Follicles were classified by the number of GCs and oocyte area (Fig. S5). For each follicle, measurements were recorded for the intensities of individual GC nuclei. For SMAD3 analysis, the intensity of the entire GC compartment was also measured and the ratio between the geometric mean of the intensities for the GC nuclei, and the geometric mean for the entire GC compartment were calculated and plotted as a measure of nuclear:cytoplasm expression. CCND2 and P27 co-localisation was analysed with the “Colocalization threshold” plugin from ImageJ. A minimum of 20 individual GC nuclei from primordial and transitional follicles were selected from a single day 4 and day 8 ovary section. The ratio of intensities between CCND2 and p27 was represented as (P27/CCND2). For cultured ovaries, sections were stained with antibodies against DDX4 and SMAD3 and image analysis was performed as described above. When more than one section from the same ovary was used, a minimum separation of 2 sections (10 µm) from the largest cross-section was considered to avoid double counting.
Gene expression analysis
Total RNA was isolated from ovaries using RNeasy® Micro Kits (QIAGEN, Crawley, UK). Concentration and purity of all RNA samples was assessed using the Agilent 2100 Bioanalyzer. Samples with an RNA integrity number (RIN) between 8 and 10 were used for gene expression analysis. Fifty ng of RNA from each sample was converted to cDNA using the SuperScript III Reverse Transcription kit (Invitrogen; ThermoFisher). Quantivative PCR (qPCR) using 500 nM of primers for Smad2, Smad3, Smad7, Myc, Ccnd2 and p27 (Table S1) were used with the SensiFAST™ SYBR® Hi-ROX kit (Bioline). Efficiencies were 90–130% for all primers. Samples (1 µl per reaction) and the same volume of distilled water (negative control) were run in duplicate in 384 well-plates. qPCR conditions were as follows: activation at 95 °C for 2 minutes and then 40 cycles of amplification (3 seconds at 95 °C for DNA denaturation, 20 seconds at 60 °C for primer annealing and 10 seconds at 72 °C for primer extension) using a 7900HT Fast Real-Time PCR System (Applied Biosystems). Ct values were normalised against Atpb5 (PrimerDesign, Southampton, UK) as previously described6,21 and fold changes (relative to control sample) were calculated for each gene according to the 2−ΔΔCt method54.
Protein extraction from mouse ovaries
Protein extraction from whole ovaries was performed after initial disruption with 25 G needles with Pierce IP lysis buffer (Thermo Fisher Scientific) containing protease inhibitors (cOmpleteTM Mini ULTRA tablets; Roche) and phosphatase inhibitors (PhosSTOPTM, Roche). Pooled ovaries (3) from day 4 and 16 mice were used for co-immunoprecipitation or (2) for processing cultured ovaries. Samples were vortexed, centrifuged and concentration of supernatant was measured using the Pierce bicinchoninic acid (BCA) protein assay kit (ThermoFisher). For subcellular fractionation, proteins were extracted from six day 4 mouse ovaries using a sequential lysis protocol as described previously55. This protocol utilises a gradient of detergent lysis buffers to sequentially yield a cytosolic fraction, a membrane-bound organelle fraction and a nuclear fraction, respectively. Equal amounts (3–5 µg) of the cytosolic, organelle and nuclear protein fractions were used for analysis by western blotting.
Co-immunoprecipitation and Western blotting
Twenty-five µg of total protein was diluted with Pierce IP lysis buffer (ThermoFisher Scientific) and 2 µg of the immunoprecipitating antibody P27 (sc-528, Santa Cruz) were incubated for 1.5 hours at 4 °C with rotation. The same amount of protein was used to incubate with a non-specific IgG (Normal rabbit IgG I-1000, Vector Laboratories). 20 µl of Protein G magnetic beads (Merck-Millipore, MA, USA) were added to each aliquot and incubated with rotation for an additional hour. Bead/protein complexes were washed with IP buffer and gentle centrifugation. After the last wash, 35 µl of 5x loading buffer (EC-887, National Diagnostics, GA, USA) was used to elute the beads. Half of the volume of the IP samples was loaded on the western blot, and input samples (control for the IP) contained 20% of the initial amount of protein used for the IPs. Proteins were separated in 12% separating gel and 4% stacking PAGE gels. Proteins were transferred to a nitrocellulose membrane using a semi-dry transfer. Membranes were washed with TBS-Tween and blocked in either 5% (w/v) BSA (Sigma) for phospho-protein detection or 5% (w/v) non-fat dried milk at room temperature for 2 hours. Blots were washed in TBS-Tween before applying primary antibodies for CCND2 (0.4 µg/ml; sc-593, Santa Cruz), P27 (0.6 µg/ml; sc-528, Santa Cruz), p-SMAD3 (1:1000; ab52903, Abcam), p-AKT (1:1000; 4060, Cell Signaling), p-ERK1/2 (1:1000; 4370, Cell Signaling), p-S6 (1:1000; 2211, Cell Signaling) overnight at 4 °C. Antibodies were detected using HRP-conjugated secondary antibodies (1:20,000; P0448, DAKO) and ECL reagents (Westar Supernova enhanced chemiluminescent HRP substrate; Cyanagen, Bologna, Italy).
Chromatin immunoprecipitation (ChIP) - qPCR
Whole ovaries were fixed with 1% formaldehyde (Sigma) in Leibovitz’s L-15 medium (Life Technologies) for 30 minutes. An EZ- Magna ChIPTM G kit (Millipore) was used according to manufacturer’s instructions for the preparation of nuclear extracts and subsequent ChIP. 2% of the initial volume was taken as control for the IPs (Input). Protein G magnetic beads (Merck-Millipore) were added to each aliquot along with 1 µg of immunoprecipitating antibody against SMAD2/3 (sc-133098, Santa Cruz) and non-immune mouse IgG (I-200, Vector Laboratories). As a positive control, an antibody against the protein FOXL2 was used (0.2 µg/ml; sc-55655, Santa Cruz), since Ccnd2 and p27 are known targets of this protein56,57. An equal volume (4 µl) of IP sample, control or RNAse/DNAse free water (additional negative control) were analysed by qPCR in duplicate using primers designed to flank predicted FOXL2, SMAD3 and MYC binding sites (TRANSFAC; Table S1) in 384 well-plates as described above. Input, IPs and IgG control samples were analysed using the ‘% input’ method where the mean Ct value of each IP and IgG sample was normalised against the input sample. Since the starting input sample was 2% of the initial chromatin, a dilution factor of 50 or 5.64 cycles (log 2 of 50) was subtracted and calculations were performed as follows: i) Mean Ct INPUT – 5.64, ii) Mean Ct IP (or IgG) - Mean Ct INPUT = ΔCt, iii)% INPUT = (2 (−ΔCt)) × 100.
Statistical analysis
For immunofluorescent quantification of SMAD3, CCND2 and P27, follicle stage-specific differences in expression were analysed using a non-parametric Kruskal-Wallis test with a post-hoc Dunn’s multiple comparisons test. For all gene expression analyses by qPCR, fold changes between groups were evaluated using one-way ANOVAs with post-hoc Bonferroni multiple comparisons tests. For the ChIP-qPCR data, non-parametric Mann-Whitney U tests were used to compare % input values in SMAD3 vs IgG control groups. Western blot data was analysed using one-way ANOVAs with post-hoc Tukey’s multiple comparisons tests. Effect of different culture conditions on oocyte size was evaluated using a Kruskal-Wallis test with post-hoc Dunn’s multiple comparisons. All analyses were carried out using Prism (v7.03; Graphpad). Differences were considered significant when P < 0.05.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Adhikari, D. & Liu, K. Molecular Mechanisms Underlying the Activation of Mammalian Primordial Follicles. Endocr Rev 30, 438–464, https://doi.org/10.1210/er.2008-0048 (2009).
Da Silva-Buttkus, P. et al. Effect of cell shape and packing density on granulosa cell proliferation and formation of multiple layers during early follicle development in the ovary. J Cell Sci 121, 3890–3900, https://doi.org/10.1242/jcs.036400 (2008).
McLaughlin, E. A. & McIver, S. C. Awakening the oocyte: controlling primordial follicle development. Reproduction 137, 1–11, https://doi.org/10.1530/Rep-08-0118 (2009).
Monniaux, D. et al. The ovarian reserve of primordial follicles and the dynamic reserve of antral growing follicles: what is the link? Biol Reprod 90, 85, https://doi.org/10.1095/biolreprod.113.117077 (2014).
Skinner, M. K. Regulation of primordial follicle assembly and development. Hum Reprod Update 11, 461–471, https://doi.org/10.1093/humupd/dmi020 (2005).
Hardy, K. et al. Nuclear exclusion of SMAD2/3 in granulosa cells is associated with primordial follicle activation in the mouse ovary. J Cell Sci 131, https://doi.org/10.1242/jcs.218123 (2018).
Pietenpol, J. A. et al. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 61, 777–785 (1990).
Seoane, J. et al. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol 3, 400–408, https://doi.org/10.1038/35070086 (2001).
Yagi, K. et al. c-myc is a downstream target of the Smad pathway. J Biol Chem 277, 854–861, https://doi.org/10.1074/jbc.M104170200 (2002).
Massague, J. TGFbeta signalling in context. Nat Rev Mol Cell Biol 13, 616–630, https://doi.org/10.1038/nrm3434 (2012).
Morikawa, M., Derynck, R. & Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb Perspect Biol 8, https://doi.org/10.1101/cshperspect.a021873 (2016).
Sherr, C. J. & Roberts, J. M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13, 1501–1512 (1999).
Haberichter, T. et al. A systems biology dynamical model of mammalian G1 cell cycle progression. Mol Syst Biol 3, 84, https://doi.org/10.1038/msb4100126 (2007).
Knudsen, E. S. & Wang, J. Y. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol 17, 5771–5783 (1997).
Susaki, E., Nakayama, K. & Nakayama, K. I. Cyclin D2 translocates p27 out of the nucleus and promotes its degradation at the G0-G1 transition. Mol Cell Biol 27, 4626–4640, https://doi.org/10.1128/MCB.00862-06 (2007).
Rajareddy, S. et al. p27kip1 (cyclin-dependent kinase inhibitor 1B) controls ovarian development by suppressing follicle endowment and activation and promoting follicle atresia in mice. Mol Endocrinol 21, 2189–2202, https://doi.org/10.1210/me.2007-0172 (2007).
Sicinski, P. et al. Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature 384, 470–474, https://doi.org/10.1038/384470a0 (1996).
Robker, R. L. & Richards, J. S. Hormone-induced proliferation and differentiation of granulosa cells: a coordinated balance of the cell cycle regulators cyclin D2 and p27Kip1. Mol Endocrinol 12, 924–940, https://doi.org/10.1210/mend.12.7.0138 (1998).
Moustakas, A. & Heldin, C. H. The regulation of TGFbeta signal transduction. Development 136, 3699–3714, https://doi.org/10.1242/dev.030338 (2009).
Fenwick, M. A., Mansour, Y. T., Franks, S. & Hardy, K. Identification and regulation of bone morphogenetic protein antagonists associated with preantral follicle development in the ovary. Endocrinology 152, 3515–3526, https://doi.org/10.1210/en.2011-0229 (2011).
Sharum, I. B., Granados-Aparici, S., Warrander, F. C., Tournant, F. P. & Fenwick, M. A. Serine threonine kinase receptor associated protein regulates early follicle development in the mouse ovary. Reproduction 153, 221–231, https://doi.org/10.1530/REP-16-0612 (2017).
Hill, C. S. Transcriptional Control by the SMADs. Cold Spring Harb Perspect Biol 8, https://doi.org/10.1101/cshperspect.a022079 (2016).
Looyenga, B. D. & Hammer, G. D. Genetic removal of Smad3 from inhibin-null mice attenuates tumor progression by uncoupling extracellular mitogenic signals from the cell cycle machinery. Mol Endocrinol 21, 2440–2457, https://doi.org/10.1210/me.2006-0402 (2007).
Tomic, D. et al. Ovarian follicle development requires Smad3. Mol Endocrinol 18, 2224–2240, https://doi.org/10.1210/me.2003-0414 (2004).
Adams, M. R., Sears, R., Nuckolls, F., Leone, G. & Nevins, J. R. Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol Cell Biol 20, 3633–3639 (2000).
Leone, G., DeGregori, J., Sears, R., Jakoi, L. & Nevins, J. R. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 387, 422–426, https://doi.org/10.1038/387422a0 (1997).
Rosairo, D., Kuyznierewicz, I., Findlay, J. & Drummond, A. Transforming growth factor-beta: its role in ovarian follicle development. Reproduction 136, 799–809, https://doi.org/10.1530/Rep-08-0310 (2008).
Schindler, R., Nilsson, E. & Skinner, M. K. Induction of ovarian primordial follicle assembly by connective tissue growth factor CTGF. PLoS One 5, e12979, https://doi.org/10.1371/journal.pone.0012979 (2010).
Wang, Z. P. et al. Transforming growth factor-beta signaling participates in the maintenance of the primordial follicle pool in the mouse ovary. J Biol Chem 289, 8299–8311, https://doi.org/10.1074/jbc.M113.532952 (2014).
Du, D. et al. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep 19, 135–155, https://doi.org/10.15252/embr.201744250 (2018).
Zhang, Y. et al. High throughput determination of TGFbeta1/SMAD3 targets in A549 lung epithelial cells. PLoS One 6, e20319, https://doi.org/10.1371/journal.pone.0020319 (2011).
Ezzati, M. M. et al. Regulation of FOXO3 subcellular localization by Kit ligand in the neonatal mouse ovary. J Assist Reprod Genet 32, 1741–1747, https://doi.org/10.1007/s10815-015-0589-9 (2015).
Lau, L. F. & Nathans, D. Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: coordinate regulation with c-fos or c-myc. Proc Natl Acad Sci USA 84, 1182–1186 (1987).
Park, Y. et al. Induction of cyclin D2 in rat granulosa cells requires FSH-dependent relief from FOXO1 repression coupled with positive signals from Smad. J Biol Chem 280, 9135–9148, https://doi.org/10.1074/jbc.M409486200 (2005).
Nakao, A. et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 389, 631–635, https://doi.org/10.1038/39369 (1997).
Yan, X. et al. Smad7 Protein Interacts with Receptor-regulated Smads (R-Smads) to Inhibit Transforming Growth Factor-beta (TGF-beta)/Smad Signaling. J Biol Chem 291, 382–392, https://doi.org/10.1074/jbc.M115.694281 (2016).
Gao, F. et al. Wt1 functions in ovarian follicle development by regulating granulosa cell differentiation. Hum Mol Genet 23, 333–341, https://doi.org/10.1093/hmg/ddt423 (2014).
Hartkamp, J., Carpenter, B. & Roberts, S. G. The Wilms’ tumor suppressor protein WT1 is processed by the serine protease HtrA2/Omi. Mol Cell 37, 159–171, https://doi.org/10.1016/j.molcel.2009.12.023 (2010).
Elvin, J. A., Yan, C., Wang, P., Nishimori, K. & Matzuk, M. M. Molecular characterization of the follicle defects in the growth differentiation factor 9-deficient ovary. Mol Endocrinol 13, 1018–1034, https://doi.org/10.1210/mend.13.6.0309 (1999).
Hayashi, M. et al. Recombinant growth differentiation factor-9 (GDF-9) enhances growth and differentiation of cultured early ovarian follicles. Endocrinology 140, 1236–1244, https://doi.org/10.1210/endo.140.3.6548 (1999).
Vitt, U. A., Hayashi, M., Klein, C. & Hsueh, A. J. Growth differentiation factor-9 stimulates proliferation but suppresses the follicle-stimulating hormone-induced differentiation of cultured granulosa cells from small antral and preovulatory rat follicles. Biol Reprod 62, 370–377, https://doi.org/10.1095/biolreprod62.2.370 (2000).
Wang, C. & Roy, S. K. Expression of growth differentiation factor 9 in the oocytes is essential for the development of primordial follicles in the hamster ovary. Endocrinology 147, 1725–1734, https://doi.org/10.1210/en.2005-1208 (2006).
Ding, C. C., Thong, K. J., Krishna, A. & Telfer, E. E. Activin A inhibits activation of human primordial follicles in vitro. J Assist Reprod Genet 27, 141–147, https://doi.org/10.1007/s10815-010-9395-6 (2010).
Liu, X. et al. A comparative study on transforming growth factor-beta and activin A for preantral follicles from adult, immature, and diethylstilbestrol-primed immature mice. Endocrinology 140, 2480–2485, https://doi.org/10.1210/endo.140.6.6827 (1999).
Mizunuma, H. et al. Activin from secondary follicles causes small preantral follicles to remain dormant at the resting stage. Endocrinology 140, 37–42, https://doi.org/10.1210/endo.140.1.6409 (1999).
Smitz, J., Cortvrindt, R., Hu, Y. & Vanderstichele, H. Effects of recombinant activin A on in vitro culture of mouse preantral follicles. Mol Reprod Dev 50, 294–304, https://doi.org/10.1002/(SICI)1098-2795(199807)50:3<294::AID-MRD5>3.0.CO;2-E (1998).
Hirashima, Y., Moniruzzaman, M. & Miyano, T. p27(Kip1) negatively regulates the activation of murine primordial oocytes. J Reprod Dev 57, 217–222 (2011).
Zhang, H. et al. Somatic cells initiate primordial follicle activation and govern the development of dormant oocytes in mice. Curr Biol 24, 2501–2508, https://doi.org/10.1016/j.cub.2014.09.023 (2014).
Lamouille, S., Connolly, E., Smyth, J. W., Akhurst, R. J. & Derynck, R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci 125, 1259–1273, https://doi.org/10.1242/jcs.095299 (2012).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293, https://doi.org/10.1016/j.cell.2012.03.017 (2012).
Tojo, M. et al. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta. Cancer Sci 96, 791–800, https://doi.org/10.1111/j.1349-7006.2005.00103.x (2005).
Yamamura, S. et al. The activated transforming growth factor-beta signaling pathway in peritoneal metastases is a potential therapeutic target in ovarian cancer. Int J Cancer 130, 20–28, https://doi.org/10.1002/ijc.25961 (2012).
Clarke, D. C., Brown, M. L., Erickson, R. A., Shi, Y. & Liu, X. Transforming growth factor beta depletion is the primary determinant of Smad signaling kinetics. Mol Cell Biol 29, 2443–2455, https://doi.org/10.1128/MCB.01443-08 (2009).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Baghirova, S., Hughes, B. G., Hendzel, M. J. & Schulz, R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2, 440–445, https://doi.org/10.1016/j.mex.2015.11.001 (2015).
Bentsi-Barnes, I. K., Kuo, F. T., Barlow, G. M. & Pisarska, M. D. Human forkhead L2 represses key genes in granulosa cell differentiation including aromatase, P450scc, and cyclin D2. Fertil Steril 94, 353–356, https://doi.org/10.1016/j.fertnstert.2009.09.050 (2010).
Garcia-Ortiz, J. E. et al. Foxl2 functions in sex determination and histogenesis throughout mouse ovary development. BMC Dev Biol 9, 36, https://doi.org/10.1186/1471-213X-9-36 (2009).
Acknowledgements
The authors would like to thank Dr Paul Heath, Dr Neil Chapman and Ms Orla Gallagher (University of Sheffield) for helpful technical advice. This work was supported by funds received from The Royal Society UK awarded to M.F. (RG130193), the Infertility Research Trust awarded to M.F. and a University of Sheffield postgraduate scholarship awarded to S.G.-A.
Author information
Authors and Affiliations
Contributions
The study was conceived by S.G.-A., K.H., S.F., and M.A.F. Experimental work was carried out by S.G.-A., I.B.S., S.L.W., M.A.F.; Formal analysis and validation S.G.-A., M.A.F.; Writing original draft S.G.-A., M.A.F.; All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Granados-Aparici, S., Hardy, K., Franks, S. et al. SMAD3 directly regulates cell cycle genes to maintain arrest in granulosa cells of mouse primordial follicles. Sci Rep 9, 6513 (2019). https://doi.org/10.1038/s41598-019-42878-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42878-4
This article is cited by
-
Integrated analysis reveals the regulatory mechanism of the neddylation inhibitor MLN4924 on the metabolic dysregulation in rabbit granulosa cells
BMC Genomics (2024)
-
Effect of aging on the formation and growth of colonic epithelial organoids by changes in cell cycle arrest through TGF-β-Smad3 signaling
Inflammation and Regeneration (2023)
-
A role for orphan nuclear receptor liver receptor homolog-1 (LRH-1, NR5A2) in primordial follicle activation
Scientific Reports (2021)
-
Human placenta-derived mesenchymal stem cells stimulate ovarian function via miR-145 and bone morphogenetic protein signaling in aged rats
Stem Cell Research & Therapy (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
