
Abstract
Arterial/venous thrombosis is the major cardiovascular disorder accountable for substantial mortality; and the current demand for antithrombotic agents is extensive. Heparinases depolymerize unfractionated heparin (UFH) for the production of low molecular-weight heparins (LMWHs; used as anticoagulants against thrombosis). A microbial strain of Streptomyces sp. showing antithrombotic activity was isolated from the soil sample collected from north India. The strain was characterized by using 16S rRNA homology technique and identified as Streptomyces variabilis MTCC 12266 capable of producing heparinase enzyme. This is the very first communication reporting Streptomyces genus as the producer of heparinase. It was observed that the production of intracellular heparinase was [63.8 U/mg protein (specific activity)] 1.58 folds higher compared to extracellular heparinase [40.28 U/mg protein]. DEAE-Sephadex A-50 column followed by Sepharose-6B column purification of the crude protein resulted 19.18 folds purified heparinase. SDS-PAGE analysis of heparinase resulted an estimated molecular-weight of 42 kDa. It was also found that intracellular heparinase has the ability to depolymerize heparin to generate LMWHs. Further studies related to the mechanistic action, structural details, and genomics involved in heparinase production from Streptomyces variabilis are warranted for large scale production/purification optimization of heparinase for antithrombotic applications.
Similar content being viewed by others

Introduction
Heparin and its structural analogues such as heparin sulphate (HS) are acidic and linear polysaccharides, belongs to the family glycosaminoglycans1. These polysaccharides are composed of 1–4 linked repeating units of β-D-glucuronic acid, N-acetyl-glucosamine and disaccharides with different degrees of sulphation2. These moieties are present on the cell surfaces of various animal tissues as a part of extracellular matrix or integral membrane components3. Heparins, also known as unfractionated heparins (UFHs), has been used clinically in the prevention and cure of thromboembolism since 19352. The depolymerization of these polymers using various chemicals or enzymatic agents results into low molecular weight heparins (LMWHs), which possess a variety of crucial biological functions and can be used as therapeutic agents4. Due to heavy increase in thrombosis cases globally, the current demand for antithrombotic agents is very high.
Arterial/venous thrombosis is one of the principal causes of myocardial infarction, which lead to substantial mortality. Warfarin, is unfractionated and low molecular weight heparin, clinically used as anticoagulant/antithrombotic agent5. LMWHs are more efficacious on account of higher bioavailability, micro-structural differences depending upon the method of depolymerization, longer half-life and predictable pharmacokinetic profile. Heparinases are commercially used for the depolymerization of unfractionated heparin (UFH) into disaccharide and oligosaccharide products, known as low molecular weight heparins (LMWHs)4.
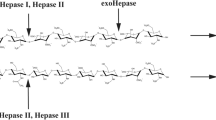
Heparinases (heparin lyases) are the enzymes that catalyze the depolymerization reactions of heparin or heparan sulphate, resulting in the generation of double bond in the uronic acid moiety at the non-reducing end. Heparinase enzymes are classified into three major types – heparinase I, heparinase II, and heparinase III, which specifically recognize and cleave at different sequences of heparin6. Heparinases/modified heparinases and LMWHs are reported to control angiogenesis and metastasis.
The production of heparinases from various microbial sources having wider clinical, pharmaceutical applications is well documented in the published literature. For example, Pedobacter heparinus is a commercial heparinase producing species. Besides this, Sphingobacterium, Bacillus circulans, Bacteroides, Acinetobacter calcoaceticus and Pseudomonas aeruginosa are some of the bacterial strains reported to produce various forms of heparinases2,7,8. Among the fungal strains, various species of Aspergillus such as Aspergillus flavus and Aspergillus oryzae are also reported to produce heparinases9,10.
To the best of information available till date no Actinomycetes is reported to produce heparinase, thus screening and investigation of novel microbial sources capable of producing different/modified heparinases is still a worth exploring area of research as heparinases have potential commercial, pharmaceutical and clinical applications. Keeping above facts in view, the present study was started with the aim of screening, isolation, purification, and characterization of heparinase from novel microbial source based on in silico screening of heparinase gene present in the genome of Streptomyces genus.
Results
In silico screening of heparinase producer strains
In order to screen the presence of heparinase producing genes in Streptomyces genera, in silico study was performed. In silico screening results suggest that some specific proteins matching with heparinase or heparinase like proteins or some hypothetical proteins are enlisted in different species of Streptomyces genus such as Streptomyces himastatinicus ATCC 53653 (Streptomyces hygroscopicus), Streptomyces olivochromogen, Streptomyces vindochromogenes, Streptomyces venezuelae, Streptomyces griseoruber, Streptomyces orinoci, Streptomyces acidiscabies 84–104, Streptomyces griseochromogenes etc (data not shown). Further, to evaluate the relationship between the isolated strain and other microbial strains having heparinase like proteins, a phylogenetic analysis was performed and the results suggested that the isolated strain showed significant similarity with a group of strains having ‘gene of interest’, i.e., heparinase producing gene. This in silico screening of heparinase producing gene step had paved the way to further explore the possibilities of identifying heparinase producing novel isolates from Streptomyces genus.
Characterization of producer strain
During the screening process, a novel heparinase producer bacterial strain was isolated from the soil sample of north India. On the basis of biochemical and morphological characteristics, the strain showed closeness to Streptomycetes. The strain was further characterized on the basis of 16S rRNA homology search as Streptomyces variabilis. A partial 16S rRNA gene sequence of 1465 base-pairs was used for BLAST and phylogenetic analysis, and showed 99% similarity with the members of Streptomyces genus. The evolutionary relationship of the isolated strain with the Streptomyces genus was further confirmed by the phylogenetic tree made, which was based on neighbor-joining method (Fig. 1). The generated phylogenetic tree can be categorized in two main clads; the first clad is further divided into two subclads, and each subclad has four strains. Whereas, in the second clad only one strain is present. The evolutionary distances among the selected species in phylogenetic tree suggested that the isolated strain has highest similarity with Streptomyces variabilis MF077022.1. The identified strain was deposited at Microbial Type Culture Collection (MTCC), Institute of Microbial Technology (IMTECH) (www.http://mtcc.imtech.res.in), Chandigarh (Punjab), India under the Accession ID: Streptomyces variabilis MTCC 12266.

16S RNA sequence based phylogenetic tree of the screened isolate by neighbor-joining method.
Heparinase production under submerged culture
Growth and production profiles of heparinase from S. variabilis MTCC 12266 were evaluated at shake flask level under submerged fermentation conditions in a defined heparin minimal medium (HMM) and complex production media (X-medium with and without heparin) (Supplementary Information: Fig. SI2). In HMM, X with and without heparin, Heparinase production was 36.41, 34.02, and 29.45 U/L after 96 hours of incubation; and with further incubation, no enhanced heparinase production was noticed. As heparinase production was high in the medium containing heparin, S. variabilis MTCC 12266 showed a heparin-dependent induction of heparinase production in the complex medium (X-medium). The total heparinase production was higher for the intracellular enzyme (41.04 U/L) compared to the extracellular enzyme (34.02 U/L). A major amount of heparin was utilized in the stationary phase during 96 to 120 hours of incubation (Fig. 2). In other words, heparin degrading enzyme, i.e., heparinase was maximally accumulated in the stationary phase. The specific activity of the intracellular heparinase production was found to be 63.8 U/mg protein (specific activity) compared to 40.28 U/mg protein of extracellular heparinase; which is 1.58 folds (140.5%) higher. In HMM, a significant lower growth rate (µmax, 0.11 h−1) was observed compared to that of X-medium (µmax, 0.23 h−1) for the culture.

Fermentation profile of S. variabilis in the production medium (dotted-line represents the microbial growth in terms of dry-cell weight and solid line represents the production of heparinase).
Purification of heparinase
Streptomyces variabilis MTCC 12266 was capable of producing intracellular heparinase enzyme, which was released by sonication; afterwards the protein was precipitated by ammonium sulphate. The enzyme was purified on DEAE-Sephadex A-50 column and the fractions collected were enriched 6.2 folds in heparinase concentration with respect to the cell extract (Table 1) [(Supplementary Information: Fig. SI2(A)]. This partially purified enzyme was further subjected to the purification step by using Sepharose-d6B column [(Supplementary Information: Fig. SI2(B)]. Heparinase was better purified by 19.18 folds with respect to the crude cell extract (Table 1). The purified enzyme was checked on SDS-PAGE, where single band of 42 kDa was appeared (Supplementary Information: Fig. SI3).
Depolymerization study
During the study of the rate of depolymerization it was found that the rate of depolymerization was depended on the biomass used in the reaction (Fig. 3). As the biomass increases, the amount of accumulated intracellular enzyme also increases. It was observed that with the increase in the biomass loading upto a critical value resulted in the depolymerization enhancement. However, further increase in the biomass loading above the critical value had no significant increase in the depolymerization. Michaelis Menten and Lineweaver Burk plot was generated to determine Km and Vmax of the strain (Figs 4 and 5). The reciprocal of the y- axis intercept is the Vmax, which was found to be 56 µM/min.mg, whereas the slope of the line gives the value of Km, which was found to be 2.3 × 10−5 mg/ml (Fig. 5). Small Km value suggests greater affinity of the substrate towards the enzyme.

Effect of Packed-cell weight on heparin depolymerization.

Michaelis Menten plot of S. variabilis MTCC 12266 heparinase reaction velocity to heparin concentration.

Lineweaver-Burk plot of S. variabilis MTCC 12266 heparinase reaction velocity to heparin concentration.
Discussion
Earlier studies have shown diverse applications of in silico analyses including screening of novel microbial strains11,12,13. During the process of in silico heparinase gene screening, the findings of some Streptomyces sp. bacteria containing heparinase like protein or hypothetical protein encourage for the experimental screening of Streptomyces sp. capable of producing heparinise enzyme from natural resources. The screening of potent heparinase producer was achieved using soil sample collected from north India. The screened bacterial isolate was able to grow on heparinase selective medium (HSM) containing heparin as a sole carbon and nitrogen source. Heparin as a sole source of carbon and nitrogen acts as an indicator of hydrolyzing activity during the microbial growth. During the secondary screening on HSM agar plates, a bacterial strain showed significant utilization of heparin, as observed by the clearance zone around the microbial growth against protamine sulfate precipitation. Protamine sulfate is a polycationic polypeptide with high arginine content readily reacts with poly-anion heparin to form protamine-heparin complexes or aggregates (PHA)14. This property was exploited for the secondary screening of heparinase producing microorganisms as described by Zimmermann et al.15. This electrostatic interaction of protamine sulfate with unconsumed heparin in the plate resulted in the formation of white color precipitate and development of a clear zone around the microbial culture, which indicates heparin breakdown by heparinase produced by the microorganism. The induction of heparinase by heparin in the bacterial culture thus appears to be a case of positively-controlled inducible gene expression, where the cells need to be exposed to an inducer for the production of the desired enzyme16,17. The genetic relatedness provides a rapid and objective identification of microbial isolates compared to the traditional phenotypic characterization18, hence the microbial isolate capable of producing heparinase was genetically identified by 16S rRNA homology analysis as Streptomyces variabilis MTCC 12266. To the best of our information, this is the very first study of the production of heparinase by Streptomyces sp. This is in line with earlier study that reports Aspergilli may produce heparinase like enzymes19. Till now, only Pedobacter heparinus is the only commercial heparinase producer known and belongs to the family Sphingobacteriaceae20. The generic status of P. heparinus has been discussed widely and the organism has been repeatedly transferred to other genera. The bacteria was first described as Flavobacterium heparinum21,22 and was later reclassified into the genus Cytophaga as Cytophaga heparina23. The confusing taxonomy of this large group of bacteria was clarified by DNA and rRNA similarity studies, which led to the reclassification of the heparinase producing bacterium as Pedobacter heparinus20. Pedobacter heparinus, represented by a single isolate, remained for many years as a sole representative of a strictly aerobic Gram-negative, heparinase producing bacterium. Gesner and Jenkin24 first isolated heparinase producing Bacteroides from the human intestinal flora24. Nakamura et al.25 isolated heparinase producing Bacteroides heparinolyticus from the dental lesions25. After more than a decade of the first report of heparinase from Bacteroides heparinolyticus, Nakamura and his co-workers26 reported the purification of 63 kDa heparinase enzyme to apparent homogeneity as detected by SDS-PAGE26. Kim et al.27 reported the first complete purification and characterization of a heparinase from Bacteroides stercoris HJ-15, an isolate from human intestine27. Bacteroides stercoris HJ-15 was reported to produce two heparinases and acharan sulphate lyase27,28. It is noteworthy that according to the Bergey’s manual of systematic bacteriology29, there are few reports of isolation of other heparinase producing microorganisms from natural resources19,20. The isolation of heparinases has been reported from Bacillus sp30,31, Prevotella heparinolyticus32, Sphingobacterium sp33,34, Eubacterium and Peptostreptococcus35.
Nakamura and his coworkers26 have reported a similar study of purification of intracellular heparinase from Bacteroides heparinolyticus, where <10% heparinase production was extracellular26. On the contrary, Pedobacter heparinus produces three intracellular heparinases (I, II, III), which have widely different structural and substrate specificities36,37,38.
The biosynthesis of anti-inflammatory, immunosuppressive metabolite by Streptomyces variabilis ASU319 recovered from the rhizosphere of Triticum vulgaris has been reported in the recent past39. From Streptomyces variabilis microbial strain, other antimicrobial compounds have been reported in the past and the production of heparinase from the same bacteria will enhance its utility for other medical applications. The production of heparinase from Streptomyces variabilis MTCC 12266 achieved in the present investigation has a potential to cater as an antithrombotic agent in the near future. Overall, this study provides a preliminary step towards the long-term future goal of technology development (i.e., optimal production/purification) and commercialization of heparinase enzyme production from this microbial source.
Conclusion
In the present study heparinase enzyme was purified and characterized with an estimated molecular weight of 42 kDa from Streptomyces variabilis MTCC 12266 isolated from the soli sample collected from north India. This is the first report showing Streptomyces genus as the producer for this life saving enzyme. Primary and secondary screenings of microbial isolates confirmed that the microbial isolate is an effective heparinase producer. Intracellularly produced heparinase has the ability to depolymerize heparin to generate LMWHs as an antithrombotic agent. Future pertinent studies related to the deciphering of the precise mode of action, structure elucidation, and involved genomics will be beneficial in elucidating the role of Streptomyces variabilis MTCC 12266 in the production of antithrombotic agent.
Materials and Methods
In silico screening for heparinase producing strain
A systematic search was performed for ‘heparinase’ protein under the section of “All Database” on the NCBI website. A search was made for ‘heparinase’ or ‘heparinase gene’. Further screening was done in order to find out whether heparinase producing genes were present in the strains of Streptomyces genus or not. Through the ENTREZ search column, the scientific published literatures regarding ‘heparinase AND Streptomyces’ were retrieved.
Isolation and characterization of heparinase producer
A novel heparinase producing strain was screen out from the soil sample collected from the basin of Gomti river (Lucknow, 26°50′27″N, 80°56′48″E), using classical primary and secondary microbial screening methods40. The collection of the soil sample from the river basin did not require any specific permission. No endangered or protected species were involved in the present study. The purified strain was maintained on YMG agar slants containing dextrose 40 g/L, mycological peptone 10 g/L and agar 20 g/L (pH 7–7.2) and stored at 4 °C.
The isolated heparinase producer bacterial was characterized morphologically and biochemically according to the protocols mentioned in the Bergey’s manual of systematic bacteriology41,42. The growth at different temperatures (4, 28, 37 and 42 °C), pH (pH 5.0, 7.0 and 10.0) and salt (NaCl) tolerance (1, 3 and 5%, w/v) was examined to find out the stability of the isolate. Further, the phenotypic characterization like haemolysis (performed on 5% human blood agar plates), antibiotic susceptibility pattern, hydrolysis of arginine, phenyl alanine, citrate, casein, gelatin, tween 20, tributyrin and urea were also performed43,44.
Finally, the microbial isolate was characterized by 16S rRNA homology analyses at Microbial Type Culture Collection (MTCC), Institute of Microbial Technology (IMTECH), Chandigarh (Punjab), India. The pair-wise sequence alignment of the partial rRNA gene sequences of the isolate was performed to identify the closely related homologs with the help of BLAST search tool available at NCBI webserver45. A phylogenetic tree was constructed by Clustal W46 phylogenetic analysis program to predict the species level characterization of the studied isolates. Through phylogenetic analysis, the evolutionary relationships among the most similar sequences selected via BLAST search were established. The distances among the sequences assist in finding the evolutionary distances among the species47. The rRNA gene sequences were deposited in EMBL nucleotide database.
Heparinase assay
Heparinase produced by the microbe during the fermentation process was assayed using the protocol mentioned by Banga and Tripathi (2010). This assay protocol is based on the estimation of uronic acid (ΔUA; absorption at 232 nm), which is the reaction product using heparin as a substrate5. The assay solution comprised of 55 µl of heparin (stock solution 20 mg/mL) in 375 µL of 20 mM Tris Buffer (pH 7.5 containing 50 mM NaCl and 4 mM CaCl2). To this assay solution, enzyme (protein concentration of ≤ 0.01 mg/mL) was added and incubated at 30 °C for 5 min. The reaction was stopped by adding 2.5 mL of 0.05 M HCl. The rate of product formation i.e., uronic acid, (ΔUA) was observed at 232 nm with respect to the control. Heparinase activity was calculated using 3,800/M/cm as a molar extinction coefficient for unsaturated oligosaccharide formed during the reaction (1 IU = 1 µmol of ΔUA containing product formed/min)38. The specific enzyme activity was calculated by dividing the number of micromoles of the product formed per minute by the quantity of the protein in µg/mL. The protein concentrations were determined using the Folin Lowry assay as described by Lowry et al.48 using bovine serum albumin as a standard.
Heparinase production in submerged culture
Submerged fermentation was performed in heparinase selective medium (HSM, g/l: Heparin, 10.0; Sodium acetate, 2.05; Calcium acetate, 0.48; pH 7.1 ± 0.2), defined minimal medium (HMM) and complex production medium (X-medium with and without heparin) at 28 °C, 180 rpm for 96 hr. The extracellular and intracellular heparinase activity of the samples withdrawn at regular intervals, was measured by azure A dye metachromasia method10. Briefly, heparin assay solution was prepared by adding heparin (25 mg/mL) in acetate solution (0.025 M sodium acetate and 0.0025 M calcium acetate; pH 7). In an Eppendorf tube, 200 µL of the enzyme extract was incubated at 30 °C with 100 µL of heparin assay mix. At 10 min intervals, 10 µL samples were withdrawn from the assay tube and added to 10 mL azure A dye solution. The optical density (OD) was measured within 1 hr at 620 nm and compared with a standard curve of 0 to 8 µg/mL heparin in azure A solution. One unit of heparinase activity is defined as the amount of the enzyme required for the degradation of 1 mg of heparin in one hr5.
Purification of heparinase
The purification of heparinase was carried out in three steps at 4 °C.
Step 1. Ammonium sulphate precipitation
The bacterial cell biomass was harvested by the centrifugation of the fermented culture at 4,500 × g for 30 min at 4 °C. The biomass was washed and stored in 10 mM Tris buffer (pH 7.0). The bacterial cell suspension was disrupted by sonication for 10 min at 30 seconds interval in an ultrasonic processor with continuous cooling. The cell debris was removed by centrifugation at 6,000 × g for 15 min at 4 °C. The proteins in the cell extract were precipitated with ammonium sulphate (of 75% saturation overnight) at 4 °C. The precipitated proteins were collected by the centrifugation (10,000 × g for 20 min). The pellet was dissolved in 0.01 M phosphate buffer, pH 7 and dialyzed against the same buffer overnight at 4 °C.
Step 2. Anion exchange chromatography
The dialyzed enzyme was loaded on a pre-equilibrated (in a 0.01 M phosphate buffer) DEAE- Sephadex A-50 column (5 × 30 cm) (Sigma, USA) and left for 20 min at 4 °C for binding. The unbound excess proteins were washed with the double volume of 0.1 M phosphate buffer (pH 7), afterwards the enzyme was eluted with step wise gradient of NaCl (0.1–1.0 M) in 0.01 M potassium phosphate buffer at a flow-rate of 0.5 ml/min. The fractions (3 ml each) were collected, and tested for heparinase activity. Active fractions were pooled, concentrated and purified by gel filtration chromatography.
Step 3. Gel-filtration chromatography
The above concentrated fraction was loaded on a Shepharose-6B column pre-equilibrated with a 0.01 M phosphate buffer (pH 7) and the column was washed with the same buffer followed by isocratic elution with NaCl (0.2 M in 0.01 M phosphate buffer) at a flow rate of 0.5 ml/min. The fractions thus collected were tested for heparinase activity. The active fractions were pooled, concentrated and analyzed by SDS- PAGE.
Molecular weight determination
The active fractions were subjected to molecular weight determination by SDS-PAGE using 12.0% polyacrylamide gel by using the method of Laemmli49 with the standard marker (Bio-Rad). The samples were compared with a standard protein marker after separation was done on gel for 90 min at 120 V; and finally, the gels were stained with Coomassie Brilliant Blue 250.
Michaelis and rate constant determination
The effect of heparin on heparinase activity was investigated by ranging the heparin concentration from 0.1 to 1.0 mg/ml (w/v). Lineweaver-Burk’s plot was plotted between the reciprocal of the velocity (1/v) against the reciprocal of the substrate concentration (1/[S]) to determine Km and Vmax values.
The Michaelis constant or Km represents the substrate concentration, which results in half-maximal velocity.
Depolymerization of heparin using microbial heparinase
The depolymerization of heparin was performed by the enzymatic degradation of heparin using bacterial heparinase to produce low molecular weight heparins (LMWHs) having industrial importance. Forty-eight hour grown bacterial culture was centrifuged and the concentrated suspension (20% w/v) of microbial biomass prepared in 0.01 M phosphate buffer was used for depolymerization assay of heparin. Briefly, varying concentration of microbial biomass was incubated at 120 rpm for 6 hours at 30 °C in a reaction mixture containing fix amount of heparin. The efficiency of the microbial cells against heparin degradation was monitored by analyzing the remaining heparin (concentration) in the supernatant using Azure A dye and uronic acid formation at 620 nm.
References
Shriver, Z., Capila, I., Venkataraman, G. & Sasisekharan, R. Heparin and heparan sulfate: analyzing structure and microheterogeneity. Handb. Exp. Pharmacol. 207, 159–176, https://doi.org/10.1007/978-3-642-23056-1_8 (2012).
Pulsawat, W. & Khanitchaidecha, P. Screening and environmental factors effecting on growth of heparinase-producing bacteria. Asia-Pac. J. Sci. Technol. 17, 593–606 (2012).
Merilahti, P., Karelehto, E. & Susi, P. Role of heparan sulfate in cellular infection of integrin-binding Coxsackievirus A9 and Human Parechovirus 1 isolates. PLoS ONE. 19, e0147168 (2016).
Tripathi, C. K. M., Banga, J. & Mishra, V. Microbial Heparin/Heparan sulphate lyases: Potential and Applications. Appl. Microbiol. Biotechnol. 94, 307–321 (2012).
Banga, J. & Tripathi, C. K. M. Purification and characterization of novel heparinase from Aspergillus flavus (MTCC 8653). Appl. Biochem. Biotechnol. 160, 1004–1016 (2010).
Dhanalakshmi, R., Manasa, A. & Babu, R. N. G. Effects of micronutrients on the production of heparinase from Bacillus lentus. Inter. J. Sci. Res. 4, 426–430 (2015).
Banga, J. & Tripathi, C. K. M. Rapid Purification and Characterization of a novel heparin degrading enzyme from Acinetobacter calcoaceticus. New Biotechnol. 26, 99–104 (2009).
Dzvova, N., Colmer-Hamood, J. A., Griswold, J. A. & Hamood, A. N. Isolation and characterization of HepP: a virulence-related Pseudomonas aeruginosa heparinase. BMC Microbiol. 17, 233, https://doi.org/10.1186/s12866-017-1141-0 (2017).
Banga, J. & Tripathi, C. K. M. Response surface methodology for optimization of medium components in submerged culture of Aspergillus flavus for enhanced heparinase production. Lett. Appl. Microbiol. 49, 204–209 (2009).
Banga, J., Tripathi, C. K. M. & Bihari, V. Growth and enzyme production kinetics of a Heparinase producing fungal isolate. Med. Chem. Res. 17, 85–93 (2008).
Singh, V. et al. Isolation, characterization and antifungal docking studies of wortmannin isolated from Penicillium radicum. Sci. Rep. 5, 11948 (2015).
Singh, V. & Somvanshi, P. Computational modeling of RNA secondary structures and phylogenetic inference of evolutionary conserved 5S rDNA of Prokaryotes. J. Mol. Graph. Model. 27, 770–776 (2009).
Somvanshi, P., Singh, V. & Seth, P. K. Phylogenetic and computational proteome analysis of Influenza A virus subtype H5N1. The Internet J. Genomics Proteomics 3, https://print.ispub.com/api/0/ispub-article/11067 (2007).
Rossmann, P., Matousovic, K. & Horacek, V. Protamine-heparin aggregates. Their fine structure, histochemistry, and renalde position. Virchows Arch., B, Cell Pathol. 40, 81–98 (1982).
Zimmermann, J. J., Langer, R. & Cooney, C. L. Specific plate assay for bacteria heparinase. Appl. Environ. Microbiol. 56, 3593–3594 (1990).
Galliher, P., Linhardt, R., Conway, L., Langer, R. & Cooney, C. Regulation of heparinise synthesis in Flavobacterium heparinum. Appl. Microbiol. Biotechnol. 15, 252–257 (1982).
Ernst, S., Langer, R., Cooney, C. L. & Sasisekharan, R. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 30, 387–444 (1995).
Gevers, D. et al. Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733 (2005).
Joubert, J. J. & Pitout, M. J. A constitutive heparinase in a Flavobacterium sp. Cell. Mol. Life Sci. 41, 1541–1541 (1985).
Steyn, P. L. et al. Classification of heparinolytic bacteria into a new genus, Pedobacter, comprising four species: Pedobacter heparinus comb. nov., Pedobacter piscium comb. nov., Pedobacter africanus sp. nov. and Pedobacter saltans sp. nov. Proposal of the family Sphingobacteriaceae fam.nov. Int. J. Syst. Bacteriol. 48, 165–177 (1998).
Payza, A. N. & Korn, E. D. The degradation of heparin by bacterial enzymes. J. Biol. Chem. 223, 853–864 (1956).
Payza, A. N. & Korn, E. D. Bacterial degradation of heparin. Nature 177, 88–89 (1956).
Christensen, P. Description and taxonomic status of Cytophaga heparina (Payza and Korn) comb. nov. (Basionym: Flavobacterium heparinum Payza and Korn 1956). Int. J. Syst. Evol. Microbiol. 30, 473–475 (1980).
Gesner, B. M. & Jenkin, C. R. Production of heparinase by Bacteroides. J. Bacteriol. 81, 595–604 (1961).
Nakamura, T., Suginaka, Y. & Takazoe, I. Heparinase activity in lesion of periodontal diseases. Bull. Tokyo Dent. Coll. 17, 147–155 (1976).
Nakamura, T., Shibata, Y. & Fujimura, S. Purification and properties of Bacteroides heparinolyticus heparinise (heparinlyase, EC 4.2.2.7). J. Clin. Microbiol. 26, 1070–1071 (1988).
Kim, D. H. et al. Degradation of acharan sulfate and heparin by Bacteroides stercoris HJ-15, a human intestinal bacterium. Arch. Pharm. Res. 21, 576–580 (1998).
Kim, W. S., Kim, B. T., Kim, D. H. & Kim, Y. S. Purification and characterization of heparin lyase I from Bacteroides stercoris HJ-15. J. Biochem. Mol. Biol. 37, 684–690 (2004).
Garrity, G. M., Bell, J. A. & Lilburn T. G. The revised road map to the manual. In D. J. Brenner, N. R. Krieg, J. T. Staley and G. M. Garrity (Eds.), Bergey’s manual of systematic bacteriology, second edition (The Proteobacteria), Part A (An introductory assay), Springer, New York, 2, 159–220 (2005).
Yoshida, E. et al. Purification and characterization of heparinase that degrades both heparin and heparan sulfate from Bacillus circulans. Biosci. Biotechnol. Biochem. 66, 1181–1184 (2002).
Bellamy, R. W. & Horikoshi, K. Heparinase produced by microorganism belonging to the genus Bacillus, US Patent 5145778A (1992).
Watanabe, M. et al. Characterization of heparinase from an oral bacterium Prevotella heparinolytica. J. Biochem. 123, 283–288 (1998).
Yapeng, C. et al. Rapid purification, characterization and substrate specificity of heparinase from a novel species of Sphingobacterium. J. Biochem. 134, 365–71 (2003).
Gao, N., Cheng, X., Yang, J. & Zhang, S. Strain screening and fermentation conditions of a novel heparinase-producing strain. Wei Sheng Wu Xue Bao = Acta Microbiologica Sinica 39, 64–67 (1999).
Joubert, J. J., vanRensburg, E. J. & Pitout, M. J. A plate method for demonstrating the breakdown of heparin and chrondroitin sulphate by bacteria. J. Microbiol. Meth. 2, 197–202 (1984).
Han, Y. H. et al. Structural snap shots of heparin depolymerisation by heparin lyase I. J. Biol. Chem. 284, 34019–34027 (2009).
Shaya, D. et al. Crystal structure of heparinise II from Pedobacter heparinus and its complex with a disaccharide product. J. Biol. Chem. 281, 15525–15535 (2006).
Lohse, D. & Linhardt, R. Purification and characterization of heparin lyases from Flavobacterium heparinum. J. Biol. Chem. 267, 24347–24355 (1992).
Shubha, M., Ushashi, B., Veena, S. & Bhaskara Rao, K. V. Antimicrobial activity of Streptomyces variabilis strain-VITUMVB03 isolated from Kanyakumari marine sediments. Asian J. Pharm. Clin. Res. 10, 112–116 (2017).
Srivastava, A. et al. Screening of biologically active microbial strains having therapeutic applications. Ind. J. Exp. Biol. 56, 244–251 (2018).
Williams, S. T., Goodfellow, M. & Alderson, G. Genus Streptomyces, Waksman and Henrici 1943, 339AL. In: Williams S. T., Sharpe, M. E. & Holt, J. G., Eds, Bergey’s Manual of Systematic Bacteriology, Vol. 4, Williams and Wilkins, Baltimore, 2452–2492 (1989).
Holt, J. G., Krieg, N. R., Sneath, P. H. A., Staley, J. T. & Williams, S. T. Bergey’s Manual of Determinative Bacteriology, 9 th Edn. Baltimore, MD; Philadelphia; Hong Kong; London; Munich; Sydney; Tokyo: William & Wilkins (1994).
Bridge, P. D. et al. A reappraisal of the terverticillate penicillia using biochemical, physiological and morphological features I. Numerical taxonomy. Microbiol. 135, 2941–2966 (1989).
Collee, J. G., Miles, R. S. & Watt, B. Tests for identification of bacteria. In: Colle J G, Duguid JP, Frase AG, Marmion BP Mackie, McCartney (Edt). Practical Medical Microbiology. 14th ed., Vol 2 Churchill Livingstone, London 131–148 (1996).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25, 3389–3402 (1997).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4, 406–425 (1987).
Singh, V., Praveen, V., Khan, F. & Tripathi, C. K. M. Phylogenetics of an antibiotic producing Streptomyces strain isolated from soil. Bioinformation 4, 53–58 (2009).
Lowry, O. H., Rosebrough, N. J., Farr, A. L. & Randall, R. J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 (1951).
Laemmli, U. K. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 224, 680–685 (1970).
Acknowledgements
The authors are grateful to the Council of Scientific and Industrial Research (CSIR)- Central Drug Research Institute and Shri Ramswaroop Memorial University, Lucknow (UP), India, for providing the necessary laboratory facility for this study. The author, Dr. Shafiul Haque sincerely acknowledges Jazan University for providing the access of the Saudi Digital Library facility for this study.
Author information
Authors and Affiliations
Contributions
Conceived and designed the study and experiments: V.S., S.H., V.K., H.A.E., P.S., B.N.M. and C.K.M.T. Performed the experiments: V.S., V.K., P.S. and C.K.M.T. Analyzed the data: V.S., S.H., V.K. and P.S. Contributed reagents/materials/analysis tools: V.S., S.H., H.A.E., B.N.M. and C.K.M.T. Wrote the paper: V.S., S.H. and V.K. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, V., Haque, S., Kumari, V. et al. Isolation, Purification, and Characterization of Heparinase from Streptomyces variabilis MTCC 12266. Sci Rep 9, 6482 (2019). https://doi.org/10.1038/s41598-019-42740-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42740-7
This article is cited by
-
Identification and characterization of a novel heparinase PCHepII from marine bacterium Puteibacter caeruleilacunae
Scientific Reports (2023)
-
Cloning and Expression of Heparinase Gene from a Novel Strain Raoultella NX-TZ-3–15
Applied Biochemistry and Biotechnology (2022)
-
Plant-derived bioactive compounds produced by Streptomyces variabilis LCP18 associated with Litsea cubeba (Lour.) Pers as potential target to combat human pathogenic bacteria and human cancer cell lines
Brazilian Journal of Microbiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
