
Abstract
Acute leukemia (AL) patients undergoing intensive induction chemotherapy develop severe gut dysbiosis, placing them at heightened risk for infectious complications. Some AL patients will undergo “repeat therapy” (re-induction or salvage) due to persistent or relapsed disease. We hypothesized that prior injury to the microbiome during induction may influence dysbiosis patterns during repeat therapy. To test this hypothesis, we analyzed the bacterial microbiome profiles of thrice-weekly stool samples from 20 intensively treated AL patients (first induction: 13, repeat therapy: 7) by 16S rRNA sequencing. In mixed-effects modeling, repeat therapy was a significant predictor of Enterococcus expansion (P = 0.006), independently of antibiotic exposure, disease type, feeding mode, and week of chemotherapy. Bayesian analysis of longitudinal data demonstrated larger departures of microbial communities from the pre-chemotherapy baseline during repeat therapy compared to induction. This increased ecosystem instability during repeat therapy possibly impairs colonization resistance and increases vulnerability to Enterococcus outgrowth. Microbiota restoration therapies at the end of induction or before starting subsequent therapy warrant investigation.
Similar content being viewed by others


Introduction
Intensive chemotherapy for acute leukemia (AL) is typically accompanied by prolonged exposure to multiple antibiotics over a ~1-month inpatient stay, which constitutes a major ecological disruption to the intestinal microbiota1,2,3. The resulting dysbiosis leads to overgrowth of pathobionts enriched in antibiotic resistance genes and adept at translocation via the compromised gut barrier4. Despite widespread use of prophylactic antibiotics during intensive anti-leukemia chemotherapy, enteric bacteria are still responsible for >40% of all bloodstream infections (BSIs) and the causative organism is multidrug-resistant in ~20% of cases5. In addition, dysbiosis is the leading risk factor for Clostridium difficile infection (CDI), occurring in ~10% of AL patients6. Although treatment-related mortality has decreased in recent years7, infection remains a significant cause of morbidity and mortality and a barrier to success in curative-intent, anti-leukemia chemotherapy.
Patients with persistent disease after one cycle of induction and those relapsing after an initial remission commonly require intensive “repeat therapy” (re-induction or salvage). The clinical severity of gut barrier damage during intensive repeat therapy is thought to be comparable to the initial treatment, but can be higher if repeat therapy is started shortly after the initial treatment, before complete recovery from prior damage. However, although repeat therapy patients are at particularly high risk for infectious complications, current standard supportive care, including anti-microbial therapy, is largely independent of the treatment phase. We hypothesized that the experience of prior intensive chemotherapy may influence dysbiosis patterns during repeat therapy. This would have implications for supportive care optimization, including potential restorative microbiota therapies. To address this question, we compared gut dysbiosis patterns in intensively treated AL patients according to treatment phase.
Methods
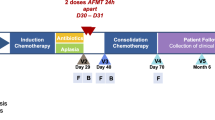
We analyzed longitudinal stool samples (n = 207) from 20 unique AL patients undergoing intensive inpatient chemotherapy. Patients with acute promyelocytic leukemia were not included. The initial treatment for medically fit adult patients with non-M3 acute myeloid leukemia (AML) is most commonly “7 + 3”, the combination of an anthracycline (3 days) and cytarabine (7 days)8. Moderate to severe mucotoxicity is common (~25% of patients) with ~10% of patients requiring parenteral nutrition8,9. Regimens used for repeat therapy differ in agents and duration of administration but are generally short-duration (5–10 days) combined regimens with comparable toxicity profile10,11. At our center, we use MEC (mitoxantrone, etoposide, and cytarabine for 5 days)12 for fit patients and Clo/Ara-C (clofarabine for 5 days and low-dose cytarabine for 10 days)13 for the less fit. The frequency and severity of mucositis with MEC or Clo/Ara-C are comparable to 7 + 314,15. There are several standard chemotherapy induction protocols for ALL, all based on multi-agent regimens with varying risks of mucotoxicty. Most of those regimens are thought to be somewhat less mucotoxic than intensive AML protocols, though formal comparisons have not been made. At our center, we use the PETHEMA16 and GRAAL17 regimens for induction in ALL patients.
We collected the first sample before or on day 1 of chemotherapy and continued thrice-weekly collections until day 30 or discharge, whichever occurred first. Samples were stored at −80 °C. Levofloxacin was administered for antibacterial prophylaxis until neutrophil recovery, and cefepime as empiric antibiotic for neutropenic fever. Variations in antibiotic choice were at the discretion of treating physicians. Parenteral nutrition was used only if oral intake was considered inadequate. The University of Minnesota Institutional Review Board approved the protocol, and the study was performed in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants.
DNA was extracted from stool using the DNeasy PowerSoil kit (Qiagen, Hilden, Germany). The V4 hypervariable region (515F/806R primer set18) of the 16S rRNA gene was amplified and paired-end sequenced (2 × 300 nucleotides) on the Illumina MiSeq platform (Illumina, Inc., San Diego, CA) at the University of Minnesota Genomics Center19. Negative (sterile water) controls were included and did not produce amplicons. Sequence data were processed in mothur20, as described previously21. Sequences were trimmed to 170 nt to remove low-quality regions at the 3′-end and paired-end joined using the fastq.join script21. Sequences were quality filtered over a window of 50 nt at a quality threshold of 35, and those with ≥2 mismatches to primer sequences, homopolymers ≥8 nt, or ambiguous bases were removed. High quality sequences were aligned against the SILVA database version 13222,23 for downstream processing and subjected to a 2% pre-cluster to remove likely sequence errors24. Chimeric sequences were identified and removed using UCHIME version 4.2.4025. Operational taxonomic units (OTUs) were classified at 97% sequence similarity using the furthest-neighbor algorithm and taxonomic classifications were made against the version 16 release from the Ribosomal Database Project26.
Samples were normalized to 9500 sequence reads/sample. Alpha diversity was calculated using the Shannon index27. Comparison of longitudinal samples to baseline (beta diversity) was done using SourceTracker (ST) version 0.9.828. For ST analysis, the baseline sample for each patient was used as “source”, and subsequent samples were used as “sink” samples. ST uses a Bayesian algorithm that leverages the information contained in taxa distributions to infer taxa attributions, thus the percentages reported reflect the estimated overlap in community composition between baseline and subsequent samples (i.e., similarity to baseline). This information cannot be derived from more conventional beta diversity indices such as the Bray-Curtis distance29. When sequences in the sink sample could not be unambiguously assigned to the baseline sample, they were assigned to an “unknown” source (newly introduced taxa or due to statistical ambiguity because of low abundances) and interpreted as divergent from baseline. Linear discriminant analysis (LDA) of effect sizes (LEfSe) was used to determine differentially abundant OTUs in the two groups; these OTUs were then classified to genera30. Species-level assignment of highly differential (LDA score ≥ 4.0) OTUs was done using BLAST31.
To evaluate the independent association between treatment phase and taxa, we applied generalized linear mixed-effects modelling using the glmmTMB package in R, where models containing fixed and random effects are fitted using maximum likelihood estimation via Template Model Builder (TMB). We defined the model in Wilkinson notation32 as Taxon ~ Treatment phase + Week of chemotherapy + Antibiotic class i (i = 1–4) + total parenteral nutrition (TPN) + Disease + (1|Patient), where taxon (relative abundance) is the dependent variable. The fixed-effects covariates (all categorical) are treatment phase (0 if induction, 1 if repeat therapy; forced in all models), week of chemotherapy (baseline or 0 vs. 1 vs. 2 vs. 3 vs. 4), TPN (1 if used within 7 days before sample, 0 otherwise), disease (ALL vs. AML), antibiotic (1 if used within 7 days before sample, 0 otherwise). The random-effects covariate is patient number. We used week, rather than day, of chemotherapy to permit categorization because the dynamics of taxa relative abundance over time was non-monotonic in many cases. We considered four classes of antibiotics, most commonly used in our patients: fluoroquinolones, third (or higher) generation cephalosporins, anti-anaerobic antibiotics (piperacillin-tazobactam, carbapenems, metronidazole, and clindamycin), and vancomycin. We applied a backward stepwise selection algorithm, starting with the complete model and removing variables with the weakest (and non-significant) association with outcome until an optimal model with the lowest Akaike and Bayesian information criterion indices was reached. We used a Beta response distribution after adding a constant (10−4) to 0’s and subtracting 10−4 from 1’s to have a non-inclusive (0,1) boundary for relative abundances. We used a false discovery rate-adjusted P value of 0.01 to define statistical significance in analyzing the 5 and 10 most abundant phyla and genera, respectively.
Significant taxa in generalized linear mixed modelling were further analyzed with the permuspliner function of SplinectomeR, a permutation-based package in R that uses weighted local polynomials (loess splines) to test for group differences in longitudinal data33. This method is less sensitive to the limitations of using aggregate data over time. We performed 999 permutations. Finally, we used the sliding_spliner function of SplinectomeR to determine whether the groups are more significantly different in specific segments of time. This non-permutation-based technique divides the time axis to 100 segments and finds segments with larger contributions to the overall intergroup difference over time for a given taxon.
To evaluate the independent association between treatment phase and alpha diversity, we applied linear mixed-effects modeling using the lmer package in R and maximum likelihood estimation. We defined the model as Shannon index ~ Treatment phase + Week of chemotherapy + Antibiotic class + TPN + Disease + (1|Patient), using similar model selection approach and definitions as those detailed above. All statistical analyses were performed in R 3.4 (R Foundation for Statistical Computing, Vienna, Austria).
Results
We studied 13 induction therapy and 7 repeat therapy patients (Table 1), who provided 133 and 74 samples, respectively. Sixteen patients had AML (9 induction therapy and 7 repeat therapy; 167 samples) and four had ALL (4 induction therapy and no repeat therapy; 40 samples). Among AML cases, 4 had myelodysplastic changes, 2 were therapy-related, and 1 was secondary to myelodysplastic syndrome. According to the 2017 European LeukemiaNet classification system34, 5 AML cases were favorable-risk, 6 were intermediate-risk, and 5 were adverse-risk (including a complex karyotype in 4 patients and complex monosomal karyotype in 1). All ALL cases were B-cell and high-risk (Ph-positive in 2 cases and Ph-like in 2). The median time from the most recent intensive chemotherapy in repeat therapy patients was 200 days. Prior intensive therapy in this group included the following: 7 + 3 (4 patients), HyperCVAD (1 patient), and 7 + 3 followed by high-dose cytarabine (2 patients). Of the 13 and 7 patients receiving induction and repeat therapy, 9 (69%) and 4 (57%) achieved a complete remission, respectively.
Antibiotic exposure was intense, with 104 antibiotic doses (all types) and 43 anti-bacterial antibiotic doses per 30 patient-days; for antibiotics given >1 dose/day, only 1 dose/day was considered (Fig. 1A). Exposure to fluoroquinolones, anti-anaerobic antibiotics, third (or higher) generation cephalosporins, and vancomycin occurred in 11, 7, 10, and 7 of the 13 induction patients, respectively. These numbers were 7, 7, 4, and 5 in the 7 repeat therapy patients, respectively. During their inpatient stay, 18 (90%), 5 (25%), and 4 (20%) patients developed neutropenic fever, BSI, and CDI, respectively, and 5 (25%; 3 patients during induction and 2 during repeat therapy) received TPN. The isolated organisms from blood cultures were Streptococcus mitis (3 patients), Strep. sanguinis (1 patient), methicillin-resistant Staphylococcus aureus (1 patient), and vancomycin-resistant Enterococcus faecium (VRE; 1 patient). Enterococcus achieved a relative abundance of >10% in the two stool samples collected before VRE BSI.

Antibacterial antibiotic use, diversity and stability of microbiota during induction and repeat therapy. Patterns of use for anti-anaerobic antibiotics, vancomycin, fluoroquinolones, and third (or higher)-generation cephalosporins are shown in (A), where the y axis shows the probability of antibiotic exposure. Panel (B) shows the Shannon diversity index. Panel (C) shows SourceTracker (ST) similarity to baseline for longitudinal samples. Spider charts in (B,C) compare samples collected during induction therapy (orange) vs. repeat therapy (blue) and include Loess splines with 95% confidence bands. Panel (D) shows a weekly comparison between the groups for ST similarity to baseline. Bar charts show mean of indices in samples collected in each week and standard error. P values are from a Mann-Whitney test.
A mean estimated Good’s coverage of 99.37 ± 0.03% was observed. Microbial diversity declined markedly with time (Fig. 1B), with Shannon indices reaching levels lower than even those we have reported in patients with multiply recurrent CDI prior to fecal microbiota transplantation35. Although repeat therapy samples had lower diversity, the difference did not reach statistical significance in linear mixed-effects modeling (P = 0.09). In contrast, recent use of TPN (P < 10−4), anti-anaerobic antibiotics (P < 10−8), or vancomycin (P < 10−6) was associated with lower diversity. Disease type (AML vs. ALL) was not independently associated with diversity.
We hypothesized that the experience of prior chemotherapy (e.g., outpatient-to-inpatient transition, chemotherapy, nutritional changes, and antibiotic exposures) may have a lasting effect on microbial ecosystems. One such detrimental effect may be diminished microbial ecosystem stability at the time of initiation of repeat therapy. As a surrogate for stability, we measured the similarity of microbiota in longitudinal samples throughout induction or repeat therapy to the baseline sample collected before initiating the corresponding treatment phase. In SourceTracker analysis, repeat therapy samples collected in weeks 1 through 3 showed less similarity to their baseline sample compared to the similarity of induction samples to their corresponding baseline sample (P < 0.05, Fig. 1C,D). Greater intestinal microbial ecosystem displacement in repeat therapy patients suggests loss of ecosystem stability relative to that seen in patients undergoing induction therapy.
One major consequence of diminished ecosystem stability is greater ease of invasion and expansion of previously rare taxa. Therefore, we evaluated whether the composition of microbial communities during intensive chemotherapy depends on treatment phase (Fig. 2A). When samples were analyzed in aggregate, the three most differentially abundant taxa were one Enterococcus OTU (LDA score: −4.95, P = 10−11) and one Veillonella OTU (P = 0.006) in repeat therapy samples, and one Parabacteroides OTU (classified as Parabacteroides distasonis; LDA score: 4.05, P = 3 × 10−4) in induction samples (Fig. 2B). In addition, repeat therapy samples experienced a progressive expansion of Enterococcus with time (Fig. 2C). Comparison of the two groups for longitudinal differences using splines fitted to Enterococcus relative abundance was significant (P = 0.027). In mixed-effects models (Table 2), Enterococcus was the only taxon independently associated with treatment phase (FDR-adjusted P = 0.006; Fig. 3). Other significant predictors of Enterococcus expansion were the use of TPN (P = 0.004) and anti-anaerobic antibiotics (P < 10−6). Finally, we evaluated whether inter-group differences during specific segments of time had a greater contribution to the overall difference in Enterococcus relative abundance between the groups. Considering day 0–14 and 15–28 intervals, the groups were different at several timepoints in both intervals. Figure 4 shows the unadjusted P values over time for the 5 most abundant phyla and 10 most abundant genera. Other taxa with significant differences between the groups in specific segments of time included the phylum Firmicutes and genera Parabacteroides, Lactobacillus, Faecalibacterium, and Veillonella, all in the interval between days 0–14, when patients tend to have highest levels of toxicity from chemotherapy.

Composition of microbial communities during induction and repeat therapy. (A) Heat map showing genera relative abundances. (B) Linear discriminant analysis (LDA) plot highlighting differentially abundant taxa in induction therapy vs. repeat therapy samples. (C) Progressive expansion of Enterococcus (more prominent in repeat therapy samples). Only the 10 most abundant genera (averaged across all samples) are shown; the remaining taxa are combined as “less abundant genera”.

Fitted splines for induction vs. repeat therapy groups. Only the 5 most abundant phyla and 10 most abundant genera are shown. The y axis shows the relative abundance of each taxon after arcsine transformation for better visualization.

Statistical significance of the comparison between induction vs. repeat therapy groups over time. Only the 5 most abundant phyla and 10 most abundant genera are shown. The y axis shows the -log(P) from Mann-Whitney tests comparing the group splines at each interval. Dotted line indicates P = 0.05. The number of patients with data at a given interval is used to scale the data point size. 100 intervals are used.
Discussion
Although the development of dysbiosis in AL patients has been reported1,2, whether the patterns of dysbiosis differ in different phases of therapy is not known. This knowledge has critical implications for potential microbiota therapeutics because AL patients often receive multiple cycles of intensive chemotherapy and are repeatedly at risk for infectious complications. Current antibiotic practice does not depend on the treatment phase; this permitted a fair comparison between induction and repeat therapy microbial communities and assessment of the independent effect of treatment phase. We also adjusted all our analyses for antibiotics. Our data suggest that the experience of prior intensive chemotherapy may lead to diminished microbiota stability at the time of subsequent chemotherapy, potentially resulting in greater vulnerability of microbiota to enterococcal outgrowths. While E. faecalis and E. faecium comprise up to 1% of the healthy adult gut microbiota36, the relative abundance of Enterococcus OTUs in some samples in our study approached 100%. Considering heavy antibiotic exposure in AL patients, many of the observed Enterococcus OTUs likely harbor antibiotic resistance genes37. Expansion of antibiotic-resistant enterococci during intensive therapy such as chemotherapy and hematopoietic cell transplantation (HCT) increases the risk of BSI38,39. Since enterococci are the third most common cause of nosocomial bacteremia in the United States with an overall mortality rate of ~30%40, preventing enterococcal blooms in the gut may decrease hospitalization, costs, morbidity, and mortality of curative-intent therapy in AL patients. Our sample size was not large enough to evaluate clinical outcomes.
Restoration of the gut microbiota to a healthy state can prevent and revert colonization by pathogens. Cooperating commensals, particularly obligate anaerobes, have a key role in colonization resistance to antibiotic-resistant enterococci and clearance of these pathogens after fecal microbiota transplantation (FMT)41,42. Parabacteroides distasonis, the most highly differentially abundant species in our induction therapy samples, was one of the four anaerobic commensals in the minimum consortium that successfully prevented and cleared vancomycin-resistant enterococci from the murine gut41. Consistent with these observations, the use of anti-anaerobic antibiotics in our cohort was associated with Enterococcus expansion. In addition, parenteral nutrition was a risk factor for Enterococcus expansion, highlighting the importance of enteral feeding, whenever possible, during intensive chemotherapy.
We suggest that microbiota restoration therapies before the initiation of repeat therapy warrant investigation. This timepoint is relevant to patients who do not achieve a remission with the first induction and those who relapse after an initial remission. Improving the stability of microbial communities before their exposure to various insults during repeat therapy may prevent pathobiont expansion and reduce infectious complications. Another timepoint where microbiota therapeutics may be beneficial is at the completion of intensive chemotherapy in patients planned to proceed to HCT. Dysbiosis has been associated with worse transplant outcomes including infections39,43, mortality44,45,46, graft-versus-host disease47,48, and relapse49. FMT has been safely applied after HCT, with resolution of dysbiosis50,51,52. We propose that correcting dysbiosis before HCT may be another approach to minimize infectious and non-infectious complications after HCT. Finally, as the first study on the subject, we enrolled both ALL and AML patients who received any form of intensive chemotherapy. Future studies in more uniform cohorts for disease and chemotherapy regimen are needed to evaluate whether our results are applicable to specific subgroups.
Data Availability
Raw sequencing data are deposited under accession number SRP141394 at the NCBI SRA.
References
Galloway-Peña, J. R. et al. Characterization of oral and gut microbiome temporal variability in hospitalized cancer patients. Genome Med. 9, 21 (2017).
Galloway-Peña, J. R. et al. The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer 122, 2186–96 (2016).
Hakim, H. et al. Gut Microbiome Composition Predicts Infection Risk During Chemotherapy in Children With Acute Lymphoblastic Leukemia. Clin. Infect. Dis., https://doi.org/10.1093/cid/ciy153 (2018).
Yu, L. C.-H. et al. Enteric dysbiosis promotes antibiotic-resistant bacterial infection: systemic dissemination of resistant and commensal bacteria through epithelial transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G824–35 (2014).
Garcia-Vidal, C. et al. Risk factors for mortality in patients with acute leukemia and bloodstream infections in the era of multiresistance. PLoS One 13, e0199531 (2018).
Vehreschild, M. J. G. T. et al. Clostridium difficile infection in patients with acute myelogenous leukemia and in patients undergoing allogeneic stem cell transplantation: epidemiology and risk factor analysis. Biol. Blood Marrow Transplant. 20, 823–8 (2014).
Othus, M. et al. Declining rates of treatment-related mortality in patients with newly diagnosed AML given ‘intense’ induction regimens: a report from SWOG and MD Anderson. Leukemia 28, 289–292 (2014).
Wiernik, P. H. et al. Cytarabine plus idarubicin or daunorubicin as induction and consolidation therapy for previously untreated adult patients with acute myeloid leukemia. Blood 79, 313–9 (1992).
Atallah, E. et al. Establishment of baseline toxicity expectations with standard frontline chemotherapy in acute myelogenous leukemia. Blood 110, 3547–51 (2007).
Mangan, J. K. & Luger, S. M. Salvage therapy for relapsed or refractory acute myeloid leukemia. Ther. Adv. Hematol. 2, 73–82 (2011).
Thol, F., Schlenk, R. F., Heuser, M. & Ganser, A. How I treat refractory and early relapsed acute myeloid leukemia. Blood 126 (2015).
Kohrt, H. E. et al. Second-line mitoxantrone, etoposide, and cytarabine for acute myeloid leukemia: a single-center experience. Am. J. Hematol. 85, 877–81 (2010).
Faderl, S. et al. Clofarabine plus low-dose cytarabine followed by clofarabine plus low-dose cytarabine alternating with decitabine in acute myeloid leukemia frontline therapy for older patients. Cancer 118, 4471–4477 (2012).
Feldman, E. J. et al. Phase III Randomized Multicenter Study of a Humanized Anti-CD33 Monoclonal Antibody, Lintuzumab, in Combination With Chemotherapy, Versus Chemotherapy Alone in Patients With Refractory or First-Relapsed Acute Myeloid Leukemia. J. Clin. Oncol. 23, 4110–4116 (2005).
He, F. et al. A real-world study of clofarabine and cytarabine combination therapy for patients with acute myeloid leukemia. Leuk. Lymphoma 59, 2352–2359 (2018).
Ribera, J.-M. et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: final results of the PETHEMA ALL-AR-03 trial. J. Clin. Oncol. 32, 1595–604 (2014).
Chalandon, Y. et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood 125, 3711–3719 (2015).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–4 (2012).
Gohl, D. M. et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 34, 942–9 (2016).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–41 (2009).
Aronesty, E. Comparison of sequencing utility programs. Open Bioinforma. J. 7, 1–8 (2013).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, (7188–96 (2007).
Schloss, P. D. & Westcott, S. L. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 77, 3219–26 (2011).
Huse, S. M., Welch, D. M., Morrison, H. G. & Sogin, M. L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–98 (2010).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–200 (2011).
Cole, J. R. et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–5 (2009).
Shannon, C. E. & Weaver, W. The Mathematical Theory of Communication. (The University of Illinois Press, 1949).
Knights, D. et al. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–3 (2011).
Bray, J. R. & Curtis, J. T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349 (1957).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Wilkinson, G. & Rogers, C. Symbolic Description of Factorial Models for Analysis of Variance. J. R. Stat. Soc. Ser. C (Applied Stat. 22, 392–399 (1973).
Shields-Cutler, R. R., Al-Ghalith, G. A., Yassour, M. & Knights, D. SplinectomeR Enables Group Comparisons in Longitudinal Microbiome Studies. Front. Microbiol. 9, 785 (2018).
Döhner, H. et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447 (2017).
Staley, C. et al. Successful Resolution of Recurrent Clostridium difficile Infection using Freeze-Dried, Encapsulated Fecal Microbiota; Pragmatic Cohort Study. Am. J. Gastroenterol. 112, 940–947 (2017).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–8 (2005).
Donskey, C. J. et al. Effect of Antibiotic Therapy on the Density of Vancomycin-Resistant Enterococci in the Stool of Colonized Patients. N. Engl. J. Med. 343, 1925–1932 (2000).
Ubeda, C. et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Invest. 120, 4332–4341 (2010).
Taur, Y. et al. Intestinal Domination and the Risk of Bacteremia in Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Clin. Infect. Dis. 55, 905–914 (2012).
Wisplinghoff, H. et al. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 39, 309–17 (2004).
Caballero, S. et al. Cooperating Commensals Restore Colonization Resistance to Vancomycin-Resistant Enterococcus faecium. Cell Host Microbe 21, 592–602.e4 (2017).
Ubeda, C. et al. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infect. Immun. 81, 965–73 (2013).
Montassier, E. et al. Pretreatment gut microbiome predicts chemotherapy-related bloodstream infection. Genome Med. 8, 49 (2016).
Weber, D. et al. Microbiota Disruption Induced by Early Use of Broad-Spectrum Antibiotics Is an Independent Risk Factor of Outcome after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 23, 845–852 (2017).
Shono, Y. et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci. Transl. Med. 8, 339ra71–339ra71 (2016).
Taur, Y. et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124, 1174–1182 (2014).
Mathewson, N. D. et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol. 17, 505–513 (2016).
Jenq, R. R. et al. Intestinal Blautia Is Associated with Reduced Death from Graft-versus-Host Disease. Biol. Blood Marrow Transplant. 21, 1373–1383 (2015).
Peled, J. U. et al. Intestinal Microbiota and Relapse After Hematopoietic-Cell Transplantation. J. Clin. Oncol. 35, 1650–1659 (2017).
DeFilipp, Z. et al. Third-party fecal microbiota transplantation following allo-HCT reconstitutes microbiome diversity. Blood Adv. 2, 745–753 (2018).
Taur, Y. et al. Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci. Transl. Med. 10, eaap9489 (2018).
Kakihana, K. et al. Fecal microbiota transplantation for patients with steroid-resistant acute graft-versus-host disease of the gut. Blood 128, 2083–2088 (2016).
Acknowledgements
A University of Minnesota Medical School Innovation Grant (to A.R.), and funding from Achieving Cures Together and Hubbard Broadcasting Foundation supported this research. We thank Andrea Hoeschen and Kevin Olson for coordinating sample collections. Sequence data were processed and analyzed using the resources of the Minnesota Supercomputing Institute.
Author information
Authors and Affiliations
Contributions
A.R. designed and led the study. C.G., T.U.R. and J.W. collaborated with A.R. in conducting the study. T.K., R.-S.C. and C.S. analyzed the data. A.R., T.K. and C.S. wrote the manuscript. A.R., T.K., C.G., S.G.H., G.D., A.K., T.U.R., J.W., D.J.W. and C.S. critically evaluated the results and enhanced the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rashidi, A., Kaiser, T., Shields-Cutler, R. et al. Dysbiosis patterns during re-induction/salvage versus induction chemotherapy for acute leukemia. Sci Rep 9, 6083 (2019). https://doi.org/10.1038/s41598-019-42652-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42652-6
This article is cited by
-
Gut diversity and the resistome as biomarkers of febrile neutropenia outcome in paediatric oncology patients undergoing hematopoietic stem cell transplantation
Scientific Reports (2024)
-
Klebsiella quasipneumoniae in intestine damages bile acid metabolism in hematopoietic stem cell transplantation patients with bloodstream infection
Journal of Translational Medicine (2023)
-
Compilation of longitudinal gut microbiome, serum metabolome, and clinical data in acute myeloid leukemia
Scientific Data (2022)
-
Loss of microbiota-derived protective metabolites after neutropenic fever
Scientific Reports (2022)
-
Gut microbiota response to antibiotics is personalized and depends on baseline microbiota
Microbiome (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
