
Abstract
Gibberellin (GA) plays a controversial role in the legume-rhizobium symbiosis. Recent studies have shown that the GA level in legumes must be precisely controlled for successful rhizobial infection and nodule organogenesis. However, regulation of the GA level via catabolism in legume roots has not been reported to date. Here, we investigate a novel GA inactivating C20-GA2-oxidase gene MtGA2ox10 in Medicago truncatula. RNA sequencing analysis and quantitative polymerase chain reaction revealed that MtGA2ox10 was induced as early as 6 h post-inoculation (hpi) of rhizobia and reached peak transcript abundance at 12 hpi. Promoter::β-glucuronidase fusion showed that the promoter activity was localized in the root infection/differentiation zone during the early stage of rhizobial infection and in the vascular bundle of the mature nodule. The CRISPR/Cas9-mediated deletion mutation of MtGA2ox10 suppressed infection thread formation, which resulted in reduced development and retarded growth of nodules on the Agrobacterium rhizogenes-transformed roots. Over-expression of MtGA2ox10 in the stable transgenic plants caused dwarfism, which was rescued by GA3 application, and increased infection thread formation but inhibition of nodule development. We conclude that MtGA2ox10 plays an important role in the rhizobial infection and the development of root nodules through fine catabolic tuning of GA in M. truncatula.
Similar content being viewed by others

Introduction
Nodulation is the mutual interaction between legume plants and rhizobial bacteria that forms a symbiotic nitrogen-fixing nodule. The process is tightly controlled by the host plant via the nodulation signaling pathway, wherein plant hormones including cytokinin, auxin, ethylene, and gibberellin (GA) participate (reviewed by Oldroyd1). The roles of GA in nodulation of legume species are controversial and both positive and negative effects have been reported. Pea na, a loss-of-function mutant of the ent-kaurenoic acid oxidase gene (KAO), was characterized by a reduction in the size and number of nodules, indicating that GA is required to support nodule formation2. In contrast, other studies have indicated negative roles of GA in nodulation. In Lotus japonicus and Medicago truncatula, exogenous GA application at concentration ranges of 0.1 to 1 µM resulted in inhibition of rhizobial infection and nodule organogenesis3,4. Considering the fact that root hair deformation was also reduced by GA application, the negative effect of GA on nodulation was proposed to act at the very early stage of the Nod factor signaling3. Negative regulation of the number of nodules formed by exogenous GA was shown to be mediated by the DELLA protein, which can interact with NSP2 and NF-YA1 in vitro4. Over-expression of MtDELLA1 increased infection thread formation without changes in nodule number. However, null mutant della or RNAi knockdown plants had reduced numbers of infection thread and nodule formation2,4,5. Nodules formed in the della lines were similar in appearance to those of the wild types and still fixed the same amount of N as the wild types in pea. In addition, GA-deficient mutant plants recovered normal nodule organogenesis via knockout of DELLA5. Based on these results, a dual role of GA in two distinct stages of nodule organogenesis was proposed; the suppression of infection thread formation and promotion of nodule development6. A recent study validated this hypothesis by using various mutant pea plants with defective GA biosynthesis or signaling pathways5.
In higher plants, biosynthesis of GA occurs first in the plastid where trans-geranylgeranyl diphosphate is converted to ent-copalyl diphosphate and then to ent-kaurene by serial action of ent-copalyl diphosphate synthase (CPS) with ent-kaurene synthase (KS). A tetracyclic diterpene ent-kaurene is oxidized to ent-kaurenoic acid by ent-kaurene oxidase (KO) and further converted to GA12 by KAO on the membrane of the endoplasmic reticulum. GA12 can be oxidized to GA53 by GA13-oxidase (GA13ox). In the cytosol, GA12 and GA53 are further oxidized to bioactive GAs through the early 13-hydroxylation pathway or the non-hydroxylation pathway by a series action of GA20-oxidase (GA20ox) and GA3-oxidase (GA3ox). At each step, intermediate or bioactive GAs can be oxidized by GA2-oxidase (GA2ox), leading to the inactivation of these hormone molecules7. There are two types of GA2ox in the catabolic pathway for GAs8. Initially identified GA2ox utilized bioactive C19 GAs (GA1 and GA4) and their immediate precursor (GA20 and GA9) as substrates. Later, a novel type of GA2ox was discovered, which contained three unique conserved amino acid motifs and catalyzed only earlier intermediate C20 GAs (GA12 and GA53) (Fig. 1A). The ‘Janus face’ of GA on nodulation9 suggested that GA biosynthesis and inactivation must be precisely regulated in accordance with the progress of nodule organogenesis. Therefore, root GA concentration should be maintained at a low level at the early stage of epidermal rhizobial infection and then at a high level at the later stage of nodule organogenesis.

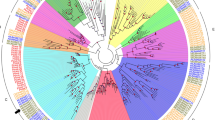
Schematic representation of gibberellin biosynthesis pathway and GA2-oxidase gene family in plant. (A) The major GA biosynthesis and metabolic pathway showing intermediate molecules, final products, and responsible enzymes for every step. GGDP, geranylgeranyl diphosphate; CPS, ent-copalyl diphosphate synthase; CDP, ent-copalyl diphosphate; KS, ent-kaurene synthase; KO, ent-kaurene oxidase; KAO, ent-kaurenoic acid oxidase; GA13ox, GA13-oxidase; GA20ox, GA20-oxidase; GA3ox, GA3-oxidase; C20 GA2ox, C20-GA2-oxidase; C19 GA2ox, C19-GA2-oxidase. (B) Phylogenetic relationship of the plant GA2-oxidase gene family. A Maximum-Likelihood tree was constructed using the deduced amino acid sequences of 9 A. thaliana, 16 B. rapa, 33 G. max, 11 L. japonicus, 14 M. truncatula, 11 S. lycopersicon, and 9 V. vinifera putative GA2-oxidase genes. Groups of genes are represented by color arcs. Bootstrap values are indicated on the branches and the branch length reflects the substitution per site. Groups I to III of GA2ox are designated according to the criteria suggested for A. thaliana and S. lycopersicon31. Group IV is comprised of dual specificity GA2-oxidase like (GAOL) genes. Closed symbols denote legume species (G. max, L. japonicus, and M. truncatula) and open symbols denote non-legume species. Genes of M. truncatula are color coded according to their group.
The cellular level of bioactive GA can be regulated in several ways, including transport of precursors or active forms of GA into the cells, inactivation of bioactive GA, or transcriptional regulation of genes involved in the biosynthesis and catabolic pathways (reviewed by Olszewski et al.10). As demonstrated in the reproductive transition of rice11 and Lolium12, regulation of GA transport via the vascular system is responsible for controlled organ development. GA12, the first GA compound produced by the GA biosynthesis pathway, is imported into the cytosol; it is then further oxidized by GA oxidases and converted to the bioactive form of GAs10. Recently, GA12 was identified as the major form of GA responsible for long-distance transport through the vascular system13,14. This finding is consistent with the expectation that GAs involved in long-distance transport should be inactive to avoid any nonspecific effects, and then converted to an active form at the location where the active GAs are required. The GA-deficient pea mutant na had dwarfism and decreased nodule formation due to disruption of production in GA12 precursor that ultimately leads to reduction in bioactive GA115. Therefore, control of GA12 metabolism is expected to be an effective means to regulate the pools of precursors of downstream GA biosynthesis. The cellular GA level can also be changed through inactivation of the bioactive forms by GA2ox13. The major GA inactivation enzyme is C19 GA2ox16 and the significance of C20 GA2ox was demonstrated by floral initiation in Arabidopsis thaliana17. Over the last decade, transcriptional regulation of genes related to the GA biosynthesis pathway in legume plants has been investigated, which has provided a comprehensive understanding of the dynamic nature of GA regulation. Gene expression studies revealed that the GA biosynthetic pathway genes are regulated in response to rhizobial inoculation or Nod factor treatment. For example, SrGA20ox1 of Sesbania rostrata was upregulated during lateral root-based nodulation and its infection-related expression pattern was dependent on Nod factors18. Similarly, several GA20ox and GA3ox genes of soybean were upregulated during the early stage of nodulation at 12 and 48 h after rhizobial inoculation19,20. Early GA precursor biosynthesis genes were also highly expressed upon rhizobium inoculation of the root hair cells of M. truncatula21.
Most of our current understanding of the roles of GA in symbiotic nodulation is based on mutant or gene studies of GA biosynthesis-related genes in pea and DELLA in L. japonicus and M. truncatula2,3,4,5,15,22,23. However, genes related to inactivation or catabolic regulation of GA during symbiotic nodulation of legume plants have not been studied to date. Previously, we investigated massive temporal transcriptome dynamics of nodulation signaling in M. truncatula wild-type cv. Jemalong A17, compared to mutants with absent (nfp24) or decreased Nod factor sensitivity (lyk325) and an ethylene-insensitive mutant (skl26) at the early symbiotic stages (0 to 48 h post-inoculation [hpi]) with rhizobia27. Among the thousands of novel candidate genes undergoing Nod factor-dependent and ethylene-regulated expression, GA biosynthesis and signaling pathway genes were enriched at 12 hpi when root hair deformation and branching occurred. We surveyed the GA-related genes in a list of symbiosis-specific genes in which transcription was activated by Nod factors, and found a partial complementary DNA (cDNA) sequence showing similarity to GA2ox that mapped to the Medtr4g074130 locus in the recent M. truncatula genome release (Mt4.0).
In this study, we first report the functional characterization of MtGA2ox10 encoding a novel C20 GA catabolic enzyme in symbiotic nodulation. We combine phylogenetic sequence comparison, expression analyses using RNA sequencing (RNA-seq) data and quantitative polymerase chain reaction (qPCR), native promoter::β-glucuronidase (GUS) fusion, CRISPR/Cas9-mediated gene deletion, and over-expression experiments. Our findings suggest that MtGA2ox10 plays important roles in both rhizobial infection at an early stage and nodule development at a late stage of symbiotic nodulation in M. truncatula.
Results
Genome-wide identification of GA2ox genes in M. truncatula
The MtGA2ox genes were identified based on a BLASTP search of all M. truncatula reference gene models against the A. thaliana GA2ox gene family, including seven GA2ox genes and two GA2-oxidase like (GAOL) genes, defined in the METACyc database28. A total of 13 MtGA2ox genes and 1 MtGAOL gene were identified from the M. truncatula genome (Mt4.0) and were named as MtGA2ox1-13 and MtGAOL15 (Fig. 1A and Supplementary Table S1). None of the M. truncatula orthologs to AtGA2ox3, AtGA2ox7, and AT3G47190.1 were identified, whereas C20 GA-specific GA2ox genes in M. truncatula were present and outnumbered A. thaliana by six genes (MtGA2ox8 to 13) to one gene (AtGA2ox8).
The phylogenetic relationship of the MtGA2ox gene family with its homologs in the sequenced plant genomes was reconstructed to investigate and characterize the phylogenetic patterns of the subgroups (Fig. 1B). A total of 113 deduced amino acid sequences of GA2ox and GAOL identified from eight sequenced plant genomes, including A. thaliana, Brassica rapa, Glycine max, L. japonicus, M. truncatula, Oryza sativa, Solanum lycopersicon, and Vitis vinifera, were multiple aligned to construct a phylogenetic tree. A Maximum-Likelihood tree using the protein sequences of the GA2ox genes showed that the plant GA2ox gene family is divided into four major clades. Groups I to III consist of GA2ox and Group IV includes only GAOL. Interestingly, Group I and II contain C19 GA-specific GA2ox (C19 GA2ox), whereas Group III comprises C20 GA-specific GA2ox (C20 GA2ox). Moreover, Group III GA2ox genes contained three unique conserved amino acid motifs that are absent in C19 GA2ox (Supplementary Fig. S1) and were relatively abundant in legume species (4–15 genes) compared to the non-legume species (1–4 genes). In each Group, legume (G. max, L. japonicus, and M. truncatula) and crucifer (A. thaliana and B. rapa) genes clustered into taxa-specific subgroups, indicating the close evolutionary relationship of genes in the same family.
MtGA2ox10 is the unique gene of the MtGA2ox gene family induced by rhizobium inoculation
To examine the expressional characteristics of each MtGA2ox gene as well as other genes related to GA biosynthesis in response to rhizobial infection, we investigated the expression pattern of the genes by searching the Medicago truncatula Gene Expression Atlas (MtGEA29) database, and by transcriptome analysis based on our large scale RNA-seq data from A17, nfp, lyk3, and skl roots that were inoculated with Sinorhizobium medicae ABS7M27. In MtGEA, none of the genes related to GA biosynthesis and inactivation exhibited nodule-specific expression (data not shown). In the transcriptome analysis using RNA-seq data, 19 out of 22 GA biosynthesis-related genes (6 GA20ox, 2 GA3ox, and 14 GAOL) and 11 out of 14 GA2ox genes were expressed in M. truncatula roots (Supplementary Table S2). Among these genes, one GA biosynthesis-related gene (MtGA3ox1) and two GA inactivation-related genes (MtGA2ox10 and MtGAOL15) showed transcriptional changes between the genotypes, which occurred between several hours to 2 days post-inoculation (dpi) with S. medicae (Fig. 2A,B). Their transcriptions responded to rhizobium inoculation in the wild type at 12 or 24 hpi and were markedly enhanced in skl (Fig. 2C). Of particular interest was that MtGA2ox10 was transcriptionally up-regulated at 6 hpi, peaked at 12 hpi where its expression level was approximately 3- to 5-fold higher than that in nfp and lyk3, and slowly declined over the rest of the time course. In contrast, MtGA3ox1 and MtGAOL15 showed similar expression patterns in A17, nfp, and lyk3 over the time course. The peak expression of these genes in A17 at 24 hpi was only 1.4- to 1.5-fold higher than those in nfp and lyk3 (Fig. 2C). Therefore, MtGA2ox10 was a unique member of the GA metabolic pathway genes in M. truncatula, which showed up-regulation in a rhizobia-dependent and ethylene-regulated manner between 6 and 48 hpi. Moreover, the rhizobia-dependent induction of MtGA2ox10 required NFP and LYK3, indicating that its transcription occurs downstream of Nod-factor recognition.

Expression of gibberellin (GA) biosynthesis and inactivation pathway genes in M. truncatula at various times after S. medicae inoculation. (A,B) Heatmaps representing the expression of GA biosynthesis (A) and inactivation (B) pathway genes in A17, nfp, lyk3, and skl based on relative log-transformed expression values (average trimmed mean of M-values [TMM] counts normalized to A17 at 0 hpi) of genes that were quantified from RNA sequencing data (NCBI BioProject accession No. PRJNA269201). The vertical axis dendrogram organizes genes according to their coexpression. The horizontal axis shows the genotype-specific time course (0–48 hpi with rhizobium). (C) Line graphs showing average TMM counts normalized to A17 at 0 hpi for MtGA3ox1, MtGA2ox10, and MtGAOL15. (D) Expression of MtGA2ox10 in various tissues verified using quantitative polymerase chain reaction. Values (the comparative cycle threshold [2−DDct]) represent the relative expression calculated using the 0 hpi sample as a reference. Error bars depict the standard error calculated from three independent biological replicates.
Transcriptional induction of MtGA2ox10 in M. truncatula root by rhizobium inoculation was evaluated by qPCR analysis of gene expression in a series of root samples and in different tissues, namely nodules, leaves, flowers, and pods (Fig. 2D). Consistent with the results of the RNA-seq data analysis, expression of MtGA2ox10 was highly specific to the early stage of S. medicae inoculation. Transcription of MtGA2ox10 was barely detected from the un-inoculated roots at 0 hpi. However, the transcript level increased from 6 hpi, peaked at 12 hpi with a ~73-fold increase in transcript abundance compared to 0 hpi, and then gradually declined until 48 hpi. Expression of MtGA2ox10 was also detected in the mature nodule and other tissues, including roots in the absence of rhizobia, leaves, flowers, and pods; however, the levels were lower than those of the rhizobium-inoculated roots at 36 hpi.
MtGA2ox10 is expressed in symbiotic tissues and nodules
Transcriptional fusion of the native MtGA2ox10 promoter and GUS reporter gene was used to examine temporal and spatial patterns of expression in transformed hairy roots of wild type A17 plants. The MtGA2ox10pro::GUS fusion construct exhibited an expression pattern nearly identical to that in the qPCR experiment, with GUS activity detected from 1 hpi, peaking at 12 hpi and declining thereafter (Supplementary Fig. S2). To characterize the tissue-level activation of the MtGA2ox10 promoter in roots and nodules, the distribution of GUS activity in symbiotic tissues was assessed by histochemical staining and microscopic analyses of the specimens.
In the absence of rhizobium inoculation, MtGA2ox10pro::GUS expression was not detected in roots (Fig. 3A). Inoculation of transgenic roots with S. medicae induced strong expression of MtGA2ox10pro::GUS at 12 hpi, with GUS activity differing between different zones; GUS activity was detected in the entire root area (epidermis, cortex, and vascular tissues), in the differentiation or maturation zone, in the vascular tissues in the elongation zone, and in the apical meristem and apices of the root cap (Fig. 3B). Interestingly, only infected or deformed root hairs in the differentiation zone exhibited GUS staining (Fig. 3C). GUS activity was reduced but localized to infected root hairs and cortex tissues, where infection thread extended at 24 hpi (Fig. 3E). At 5 dpi, strong expression of the MtGA2ox10 promoter was detected in both nascent nodules and vascular tissues (Fig. 3F). MtGA2ox10pro::GUS expression in functional nodules was observed throughout the outer layers of developing nodules at 2 wpi, and in the meristem and infection zone of mature nodules at 4 wpi, without any overlapping Magenta-Gal-stained bacterial LacZ expression in the nitrogen fixation zone (Fig. 3G,H). Root vascular bundles at 4 wpi also showed GUS activity, except in the regions basal to the nodules. Similar expression patterns persisted at 6 wpi, while GUS staining was also detected in the nitrogen fixation and senescent zones of the nodule (Fig. 3I,J).

Histochemical localization of MtGA2ox10 promoter activity in M. truncatula roots and nodules. (A–J) M. truncatula roots transformed using the MtGA2ox10pro::GUS construct and stained using X-Gluc as a substrate. (A) Uninoculated roots expressing red fluorescent protein (RFP) at 0 hpi. (B,C) Roots at 12 hpi. (D) Deformed root hairs at 12 hpi. Arrow denotes infection thread. (E) Roots at 24 hpi. (F–J) Nodules at different stages of development (5 dpi to 6 wpi). (G–J) S. medicae expressing a LacZ construct stained using Magenta-Gal as a substrate. Scale bars are 1 mm (A,B,H–J) or 200 µm (D–G).
Deletion mutation of MtGA2ox10 reduces nodule number and retards nodule development
For the loss-of-function analysis of MtGA2ox10, CRISPR/Cas9 was utilized to generate a deletion mutation of MtGA2ox10 in Agrobacterium rhizogenes-mediated transformed roots. We used the promoter of the M. truncatula U6–8 small nuclear RNA gene30 instead of A. thaliana U6–26 for efficient transcription of guide RNAs in the transformed roots of M. truncatula. To introduce a large deletion in motif 6 of the GA2ox family which functions as an oxygenase31, co-expression of two distinct guide RNAs was carried out; two single guide RNAs (sgRNAs; G851 and G907) were designed on exon 3 of MtGA2ox10 (Fig. 4A,B) and placed together in a single vector under the control of the MtU6–8 promoter, resulting in a dual sgRNA construct (G851.907; Fig. 4C). Screening of transformed roots by PCR-restriction fragment length polymorphism (RFLP) with BsrD I and Eco105 I, as well as PCR amplicon sequencing, revealed that 19% (7 out of 36) of the transformed roots expressing green fluorescent protein (GFP) harbored deletion mutations in the target region (Fig. 4D,E and Supplementary Fig. S3). Among the transformed roots with edited MtGA2ox10, three samples (G851.907 KO-1 to 3) were selected and further analyzed. G851.907 KOs were characterized by heterozygous biallelic sequences with large deletions between the G851 and G907 target regions, resulting in an in-frame deletion, frame shift, or premature stop codon in the reading frame (Fig. 4F).

CRISPR/Cas9-mediated deletion mutation of MtGA2ox10. (A) Gene structure of MtGA2ox10. Target sequences of two guide RNAs, G851 and G907, were designed on exon 3. Relative nucleotide positions of the PAM sites marked in boxes are numbered from the start codon. Restriction sites for BsrD I and Eco105 I are also presented. (B) Restriction maps of the wild type PCR products amplified with the 2347-F and 2905-R primers for genotyping by restriction fragment length polymorphism (RFLP), using BsrD I (left) or Eco105 I (right). (C) T-DNA structure of the Cas9 binary construct G851.907 for the deletion mutation of MtGA2ox10. Two single guide RNAs (sgRNAs) (G907 and G851 under MtU6–8 promoters) were tandem-assembled into the binary pGK3304 vector, which contains the GFP::BAR selection marker and Cas9::NLS under CaMV 35S promoters. (D) PCR-RFLP genotyping of A. rhizogenes-transformed roots harboring G851.907. PCR amplicons from four root samples of the pGK3304 empty vector (NULL) and seven root samples of G851.907 (G851.907) were digested independently by BsrD I (left lane) or Eco105 I (right lane). Note that all the amplicons from the NULL samples were digested to fragments of the expected sizes, as shown in the left margin of the agarose gel. In contrast, none of the amplicons from G851.907 samples were digested, indicating disruption of the restriction sites for BsrD I and Eco105 I. Sample 6 shows an increased amplicon size, presumably due to an insertion. (E) Sanger sequencing chromatograms for the PCR amplicons of the G851.907 KO-1, -2, and -3. The PAM sequence for G851 is denoted in the box and the expected cleavage site (−4 bp from PAM) is marked with an arrow, where mixed peaks appear in the sequencing chromatograms. (F) Sequences of each allele in the G851.907 KO-1, -2 and -3. The PAM sequences for sgRNAs are shown in boxes. Predicted changes in the protein structure by deletion mutations are described on the right.
Deletion of MtGA2ox10 strongly affected both nodule number and development on the transformed roots of M. truncatula (Fig. 5). Root growth over 2 months in pots of Perlite showed no significant difference of root length between G851.907 KOs and the control roots transformed with the empty vector (Fig. 5A,F). In contrast, the number and size of the nodules were significantly reduced in the G851.907 KO roots (Fig. 5B,C). Unlike the fully grown, cylindrical pink nodules, which measured ~2.5 mm in length on the control roots, G851.907 KO roots formed pale, white, immature nodules that measured <1 mm in length (p < 0.001; Fig. 5G) and were on average 3.7-fold fewer in number (p < 0.001; Fig. 5H). Interestingly, there were no significant differences in rhizobial colonization or zonal organization of similar-sized nodules between G851.907 KOs and the control, as seen by staining of LacZ activity (Fig. 5D). On the other hand, the number of infection thread per cm in the differentiation zone of G851.907 KO roots was 4.0-fold fewer than in the control (p < 0.001), indicating that epidermal infection of rhizobia was highly affected in G851.907 KO roots (Fig. 5E,H).

Root and nodule development of the MtGA2ox10 deletion plantlet. (A,B) Roots and nodules of the 2-month-old NULL and G851.907 KO plantlets. Arrowheads indicate nodules developed from 4 wpi with S. medicae. (C) Typical mature nodule of the NULL and the largest nodule of the G851.907 KO plantlets are shown. (D) S. medicae expressing LacZ in the nodules of the NULL and G851.907 KO plantlets stained using Magenta-Gal as a substrate. (E) Epidermal infection of S. medicae of the NULL and G851.907 KO roots. Scale bars are 1 cm (A,B) or 500 µm (C–E). (F–I) Root length (F), nodule length (G), nodule number per plantlet (H), and infection thread number per cm in the root differentiation zone (I) were measured. Error bars depict the standard error calculated from six NULL and seven G851.907 KO plantlets. Asterisks represent statistical significance (***p < 0.001) by t-test.
Over-expression of MtGA2ox10 causes a dwarf phenotype and inhibition of nodule formation
To assess the effect of ectopic expression of MtGA2ox10 in plant growth and nodule development, MtGA2ox10 was over-expressed under the CaMV 35S promoter in the A. tumefaciens-transformed stable transgenic plants. A total of 16 independent stable transgenic plants were selected and analyzed. As shown in Fig. 6, over-expression of MtGA2ox10 (MtGA2ox10 OE) affected plant architecture. Two-month-old transgenic plants grown in pots showed characteristics of GA-deficient phenotypes; dwarfism, small dark-green leaves, and reduced stem and root growth. Biomass of the MtGA2ox10 OE plants was only 7.8% to that of the control plants (Fig. 6A–C). Moreover, all of the T0 plants of MtGA2ox10 OE failed to yield seeds even with application of GA3. MtGA2ox10 OE in A. rhizogenes-transformed hairy roots also showed a ~1.8-fold decreased root mass compared to the control (Supplementary Fig. S4). To test whether exogenous application of GA could rescue the dwarf phenotypes of the MtGA2ox10 OE stable transgenic plants, nine independent transgenic plants were treated with GA3 at concentrations of 10 µM or 100 µM through irrigation. GA3 application resulted in a dose-dependent recovery of plant growth in two weeks after the application (Fig. 6D,E). The transgenic plants showed different sensitivity of growth response to GA3 compared with the control lines. Changes in the number of stem internode and length of stem internode were obvious in the MtGA2ox10 OE lines but not in the control lines at 10 µM GA3 (Fig. 6F to H).

Effect of MtGA2ox10 over-expression on plant architecture and growth of the stable transgenic plantlet. (A) Photograph of the 2-month-old NULL and MtGA2ox10 OE stable transgenic plantlets. (B,C) Fresh weight of overall plant (B) or leaf (C) of the transgenic plantlet. (D) Effect of GA3 application on the growth phenotype of the transgenic plantlets. Photograph was taken 2 weeks after exogenous application of 10 µM GA3 on the transgenic plants. (E) Dose-dependent growth phenotype of the MtGA2ox10 OE stable transgenic plantlets. Photograph was taken 2 weeks after exogenous application of 10 µM or 100 µM GA3 on the transgenic plants. (F–H) Shoot length (F), number of stem internode (G), and internode length (H) per plantlet were measured. Error bars depict the standard error calculated from nine NULL and nine MtGA2ox10 OE plantlets. Scale bars are 5 cm (A,D,E).
qPCR analysis of GA metabolic pathway genes in the MtGA2ox10 OE transgenic plants displayed more than a 2-fold increased expression of ent-kaurene synthesis-related genes (KS in root and KAO in leaf) and GA oxidase genes (CYP714A1 and GA3ox in root and CYP714C1 in leaf) (Supplementary Fig. S5). This result showed that the over-expression of MtGA2ox10 differentially altered the relative transcript levels of GA synthesis pathway genes in root and leaf of transgenic plants compared with the control lines. MtGA2ox10 OE also significantly affected nodulation (Fig. 7). In the control lines (n = 4), a number of nodules formed at 3 weeks post inoculation of S. medicae ABS7M (Fig. 7A). In contrast, lines over-expressing MtGA2ox10 had 23-fold increase in the number of infection threads compared with the control line (p < 0.001). However, no nodules were detected on the roots of MtGA2ox10 OE stable transgenic plants (n = 6) even after 4 weeks post rhizobium inoculation (Fig. 7B to D). Meanwhile, approximately a 1.9-fold fewer nodules formed per A. rhizogenes-transformed plant; however, no prominent difference of nodule structure or rhizobial colonization was observed in the mature nodule (Supplementary Fig. S4).

Nodule development of the MtGA2ox10 over-expression stable transgenic plantlet. (A) Roots of the 2-month-old NULL and MtGA2ox10 OE stable transgenic plantlets at 3 wpi with S. medicae. (B) S. medicae expressing LacZ in the nodules of the NULL and on the root epidermis of the MtGA2ox10 OE stable transgenic plantlets at 3 wpi with S. medicae. (C) Epidermal infection of S. medicae of the NULL and MtGA2ox10 OE roots at 4 days post inoculation with S. medicae. Scale bars are 5 mm (A) or 200 µm (B,C). (D) Infection thread number per cm in the root differentiation zone was measured. Error bars depict the standard error calculated from four NULL and six MtGA2ox10 OE plantlets. Asterisks represent statistical significance (***p < 0.001) by t-test.
Discussion
Symbiotic nodule organogenesis is a complex developmental reprograming process that requires tight regulation of the interaction between the rhizobium and the host plant. Plant hormones are important positive or negative regulators of legume-rhizobial symbiosis, as they affect the expression of symbiotic genes. Larrainzar et al.27 noted that symbiosis-specific transcriptional activation of biosynthetic pathways for multiple plant hormones, such as ethylene, cytokinin, abscisic acid, GA, and strigolactone, takes place within hours of inoculation with the rhizobium, suggesting that these hormones likely interact to regulate downstream symbiotic responses. Interestingly, this study also reported on nuanced aspects of the GA anabolic and catabolic pathways. Both GA biosynthesis and inactivation pathway genes were upregulated, with temporal differences in a Nod factor-dependent manner. Consistent with previous suggestions3,4,5, our findings provide new insights into the activity of GA during nodulation and show that spatiotemporal regulation of GA in nodule development must be considered not only in biosynthesis, but also in catabolism.
Previous studies of the roles of GA in nodulation have focused on the GA biosynthesis gene or the DELLA-mediated downstream signaling pathway. A low GA concentration is essential for the initial stage of infection, but inhibits the normal progress of nodule organogenesis. Therefore, GA levels must be regulated dynamically and differentially during the separate stages of nodulation, epidermal infection and nodule organogenesis4,5. In contrast, little attention has focused on the inactivation or transport of GA compared to biosynthesis and signaling in nodulation. In this study, we characterized the molecular function of MtGA2ox10 encoding the C20 GA-specific inactivation enzyme GA2-oxidase in symbiotic nodule organogenesis. This novel MtGA2ox gene exhibited rhizobium-dependent induction in the 6 to 36 hpi window, and negative regulation by ethylene in the M. truncatula root. Gene expression was induced as early as at 6 hpi and peaked at 12 hpi in wild type A17; it was highly enhanced in skl but was markedly low in nfp and lyk3. Native promoter::GUS fusion analysis confirmed that transcriptional activation of the MtGA2ox10 promoter was associated with rhizobium infection and nodule development. The formation of infection thread, as well as the number and size of nodules, were reduced by CRISPR/Cas9-mediated deletion of MtGA2ox10. Additionally, plant architecture and nodulation were also affected by over-expression of MtGA2ox10, whereas exogenous application of GA3 rescued the dwarf phenotype. These findings collectively suggested that MtGA2ox10 is a unique member of the MtGA2ox gene family, controlling the low concentration of GA by catabolic inactivation of C20 GA in roots during epidermal infection of the rhizobium. Therefore, it plays as a catabolic regulator of symbiotic nodule organogenesis.
MtGA2ox10 clustered into subgroup III GA2ox with substrate specificity to C20 GA, but not to active C19 GAs (Fig. 1). A number of studies have reported on the significance of C20 GA regulation for plant responses and organ development. Two C20 GA2ox genes, AtGA2ox7 and AtGA2ox8, control plant architecture and floral initiation in A. thaliana17,32. C20 GA2ox is also related to tillering and root development8, as well as to salt tolerance and root gravity responses33 in rice, and over-expression of a C20 GA2ox in switchgrass changes the plant architecture, for example through increased tillering, a short internode length, and reduced plant height34. It was interesting to note that all of the reported phenotypes of C20 GA2ox over-expression showed less severe dwarfism compared to C19 GA2ox over-expression, suggesting that C20 GA2ox does not completely deplete the pools of diverse GAs and may have a more specialized role in plant development. Meanwhile, MtGA2ox10 OE in the stable transgenic plants resulted in dwarfism with low fertility and inhibition of nodule development despite of increased root infection, presumably due to ectopic inactivation of earlier intermediate C20 GAs (GA12 and GA53) or disruption of the GA pool by altered expression of KS, KAO, GA13ox, and GA3ox. These results were consistent with the previous report from pea na-1 mutant5; therefore, clarified the role of GA on the different stages of nodulation (suppression of infection and activation of nodule formation). Of particular interest, the stable transgenic plants of MtGA2ox10 OE showed different root growth and nodulation pattern compared with the hairy root transformation lines (almost normal development of root and nodule). We anticipate that GAs transported to the A. rhizogenes-transformed roots from the aerial parts might compensate for the effect of MtGA2ox10 OE as demonstrated by grafting experiments in GA-deficient mutant pea and A. thaliana14,35.
GA biosynthesis is a complex and multistep process with diverse intermediates. Therefore, it is difficult to determine the exact spatial localization of GA biosynthesis. Other studies have suggested that GAs are mobile signaling molecules in plants. The successful completion of a number of development processes requires GAs to be mobile36. A study of pea using radiolabeled forms of GA19, GA20, and GA1 showed that GA20 was the major mobile form of GA in the pea35. In A. thaliana, the biologically inactive C20 GA12 is the major transported form of GA13,14. The membrane permeability of GA12 allows it to serve as a long-distance transport molecule36. Considering the fact that the A. rhizogenes-transformed hairy roots of MtGA2ox10 OE formed normal nodules and MtGA2ox10pro::GUS expression occurred in the vascular bundles of the roots and mature nodules but not near the base of mature nodules, GA transport through the vascular system in M. truncatula is expected to be under catabolic regulation by C20 GA-specific MtGA2ox and GA precursors are converted to active forms at the location where the nodule develops. Additionally, expression of MtGA2ox10 in the mature nodule suggests that it may inhibit nodule over-growth by quantitative regulation of GA, which is a known regulator of cell expansion and cell cycle activation. Further analysis such as grafting of wild-type scions onto rootstocks of stable transgenic over-expression and knock out lines or measurement of GA content in the transgenic plants will prove this hypothesis.
In conclusion, this study described the importance of fine catabolic tuning of GA for nodule development in M. truncatula. We clarified that MtGA2ox10 is a unique member of the MtGA2ox gene family regulating rhizobium infection and nodule organogenesis. This is the first report on the roles of the GA catabolic pathway gene in nodulation of legume plants and contributes towards a more comprehensive understanding of the dynamic nature of the GA regulatory mechanism. Research is underway to establish and characterize stable transformed plants with loss-of-function for MtGA2ox, to further understand the roles of GA and its regulation through catabolism and transport for symbiotic nodule development.
Methods
Plant growth conditions and inoculation of rhizobium bacteria
M. truncatula cv. Jemalong A17 seeds were scarified, germinated, and grown in a growth room at 22 °C under 16 h light/8 h dark conditions. For rhizobium inoculation of the seedlings, germinated 1-day-old seedlings were planted on the aeroponic caisson, a large plastic chamber with a perforated lid on top and a humidifier that sits on the bottom37, where they were misted with Lullien’s aeroponic culture medium38 containing 0.5 mM ammonium nitrate. The 2-week-old seedlings were inoculated with S. medicae ABS7M (pXLGD4) constitutively expressing the LacZ gene at an optical density at 600 nm (OD600) of 0.1. For rhizobium inoculation of A. rhizogenes-mediated transformed roots, 4-week-old transformed plantlets were transferred to Perlite in 1 L pots and maintained for 2 weeks with a supplement of half strength modified Fahraeus medium (mFM) containing 0.5 mM ammonium nitrate. Six-week-old transformed plantlets were then inoculated with S. medicae ABS7M (pXLGD4) at OD600 of 0.05.
Phylogenetic analysis of the GA2-oxidase gene family
For phylogenetic analysis of the GA2ox gene family in the sequenced plant genomes, putative GA2ox genes in the genomes of B. rapa, G. max, L. japonicus, M. truncatula, O. sativa, S. lycopersicon, and V. vinifera, were identified based on a BLASTP search (E value cutoff of E−10 and query coverage of 50%) using A. thaliana GA2ox genes as the seed queries. At the same time, the GA2ox protein sequences of tomato31, rice8, and grapevine39 were downloaded from the National Center for Biotechnology Information (NCBI) GenBank database and combined with the BLASTP search results. The deduced amino acid sequences of the GA2ox genes were aligned using the ClustalW program40 with the default parameters. The phylogenetic tree was constructed using the Maximum-Likelihood method in MEGA741, with bootstrap analysis of 1,000 replicates for stability testing of the tree nodes. Identification of other GA biosynthesis pathway genes, including CPS, KS, KO, KAO, GA13OX, and GA3ox, in the M. truncatula genome (Mt4.0) was also performed by BLASTP search (E value cutoff of E−10 and query coverage of 50%) using the previously reported GA biosynthesis genes of M. truncatula42 as the seed queries.
Transcriptional expression analyses
For the transcriptome analysis, our RNA-seq data, which were deposited to NCBI under the BioProject accession number PRJNA269201, were mapped to the very recent M. truncatula genome assembly Mt4.0, as described previously27. Read counts were normalized using the trimmed mean of M-values (TMM) method43. Average TMM values for the GA metabolic pathway genes per sample were selected and analyzed by hierarchical clustering using Cluster 344. A heat map was drawn with the log-transformed fold changes of the TMM values compared to 0 hpi of A17 as a control. For the qPCR analysis of MtGA2ox10, plant roots were harvested at 0, 6, 12, 24, 48 hpi and 2 weeks post-inoculation (wpi) with S. medicae ABS7M. Leaves and flowers were sampled from 8-week-old plants. Un-inoculated roots from 4-week-old plants were included as a control. Total RNA was extracted using the CTAB method45 combined with LiCl precipitation and DNase treatment using the TURBO DNA-free kit (Ambion, Life Technologies, Carlsbad, CA, USA). First strand cDNA was synthesized using the TOPscriptTM cDNA synthesis kit (Enzynomics, Daejeon, Korea) with oligo-dT. The cDNAs were diluted 10-fold and qPCR was performed using TOPrealTM qPCR premix (Enzynomics) and a CFX96™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The comparative cycle threshold (Ct) method, also known as the 2−DDCt method46, was employed for relative quantification using the GAPDH gene (Medtr3g085850) as a reference gene. qPCR analysis of other GA biosynthesis pathway genes (CPS, KS, KO, KAO, GA13ox, and GA3ox) was also performed using the oligonucleotide primers designed to amplify target genes from the closely related family genes (Supplementary Table S3).
Gene cloning and plasmid construction
All of the primers used in plasmid construction are listed in Supplementary Table S4. To construct the promoter::GUS reporter fusion, 2.1 kb upstream of the 5′-flanking region of the MtGA2ox10 gene (Medtr4g074130) was amplified from the genomic DNA of M. truncatula A17 using Phusion High Fidelity DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA). The resulting PCR amplicon was purified by agarose gel electrophoresis and cloned into pDONR221 using BP Clonase II (Thermo Fisher Scientific). The binary destination vector pRNGWFS7 (Kim, unpublished) was constructed by replacing NPT II in pKGWFS747 with the DsRed::NPT II translational fusion under the CaMV 35S promoter. The entry plasmid was recombined with pRNGWFS7 in the presence of LR Clonase II (Thermo Fisher Scientific) to obtain a transcriptional fusion of the MtGA2ox10 promoter to GFP and GUS (MtGA2ox10pro::GUS). To construct the over-expression vector, the full-length coding sequence (CDS) of MtGA2ox10 was amplified from the first strand cDNA which was synthesized with the total RNA isolated from the S. medicae-infected root tissues of M. truncatula A17. The amplicon was cloned into pDONR221 using BP Clonase II (Thermo Fisher Scientific) and recombined with pK7WG2D47 using LR Clonase II (Thermo Fisher Scientific) to obtain the binary construct for over-expression of the MtGA2ox10 CDS under the CaMV 35S promoter. The binary Cas9 expression vector pGK3304 and the sgRNA cloning vector pGK2223 were constructed as follows; the Cas9 expression cassette consisting of the CaMV 35S promoter, Cas9::NLS::HA and the CaMV 35S terminator was PCR-amplified from pBAtC48. A Hind III site within Cas9 was removed by overlap PCR and the Cas9 expression cassette was transferred to pKGWD47 by replacing the GFP expression cassette between the Sac I and Hind III sites. The resulting plasmid was named pKGWC. A fragment of the CaMV 35S promoter, GFP(S64T)::BAR, and the NOS terminator were amplified from pGK2720 (Kim, unpublished) and replaced the Kanamycin resistant gene in pKGWC to yield pGK3304. A sgRNA cloning vector was constructed by placing the gRNA cloning site and gRNA scaffold in pBAtC48 under the promoter of the MtU6–8 small nuclear RNA gene promoter in pENTR_MtU6.8::gus0::UT30. The resulting Gateway-compatible sgRNA cloning vector was named pGK2206 and the Aar I cloning site in pGK2206 was replaced by the Bsa I cloning site to yield pGK2223.
CRISPR/Cas9-mediated deletion
For the CRISPR/Cas9-mediated deletion of MtGA2ox10, two sgRNAs were designed on exon 3 of MtGA2ox10 gene using Cas-Designer49. The complementary oligonucleotides were annealed and cloned into the Bsa I cloning site of the entry vector pGK2223 using the Golden Gate assembly method50. Briefly, two complementary oligonucleotides were phosphorylated using T4 polynucleotide kinase (NEB, Ipswich, MA, USA) and annealed in a kinase buffer. The annealed oligonucleotides were mixed with pGK2223 plasmid, Bsa I and T4 DNA ligase (NEB). The reaction mixture was incubated at 37 °C for 30 min, and then subjected to 30 cycles of 5 min at 37 °C and 10 min at 24 °C. After a final incubation at 50 °C for 30 min, the Golden Gate assembly was transformed into E. coli TOP10 cells. Two entry plasmids with different sgRNA were tandem assembled using the restriction cloning method. One sgRNA expression cassette was cut out from the entry plasmid using Xba I and Spe I and inserted into another sgRNA entry plasmid which was digested by Xba I and dephosphorylated. The resulting dual sgRNA entry plasmid was recombined with the binary CRISPR/Cas9 vector pGK3304 using the LR clonase II (Thermo Fisher Scientific).
Plant transformation
For A. rhizogenes-mediated hairy root transformation, the binary constructs were electroporated into A. rhizogenes MSU440 and transformed roots were generated in M. truncatula A17 as previously described51. To select the plantlets, glufosinate herbicide BASTATM (Bayer Crop Science, Monheim am Rhein, Germany) was added to the medium at a concentration of 4 mg/l and the growing hairy roots were selected by detection of GFP using an IZX2-ILLB stereomicroscope equipped with a GFP filter set (Olympus, Tokyo, Japan). One transformed root was left for each plantlet while all non-transformed roots were removed. Four-week-old composite plantlets with transformed roots were transferred to Perlite in a 1 L pot and grown in a growth room as described above. For A. tumefaciens-mediated stable transformation, the binary constructs were electroporated into A. tumefaciens EHA105 and stable transgenic plants of M. truncatula A17 was generated as previously described52. Briefly, sterilized leaf explants of M. truncatula A17 were co-cultivated with A. tumefaciens on the P4 medium and callus was induced on the P4 medium containing 5 µM GA3 (Sigma-Aldrich, https://www.sigmaaldrich.com), 40 mg/L Kanamycin (Sigma-Aldrich), and 400 mg/L Cefotaxime (Sigma Aldrich). The transgenic somatic embryos were removed from the callus tissue and were plated onto the MS medium containing 10 g/L sucrose, 50 mg/L Kanamycin, and 0.25% Gelrite for development into plantlets. When sufficiently grown, plantlets were transferred to Perlite in a 1 L pot and grown in a growth room as described above.
Histochemical staining and fluorometric quantification of LacZ and GUS expression
Plant roots were harvested at 6, 12, 24, 48 hpi and 2 wpi with S. medicae ABS7M. Transformed roots were selected by detecting GFP under a fluorescence stereomicroscope as described above. The constitutive expression of LacZ in S. medicae ABS7M was detected using X-Gal as a substrate according to a standard protocol53. Dual staining of LacZ and GUS was carried out according to the protocol in the L. japonicus handbook54. The reaction was monitored overnight to avoid over-staining. Fluorometric quantification of GUS activity was conducted using 4-methylumbelliferyl b-D-glucuronide as a substrate55. The fluorescence was measured with a DynaQaunt 200TM fluorometer (Hoeffer, San Francisco, CA, USA).
Genotyping by PCR-RFLP and sequencing
Genomic DNA was extracted from the transformed hairy roots of the A. rhizogenes-transformed composite plantlets or leaves of the stable transgenic plants using the standard CTAB method56 for PCR, cloning, and sequencing. In parallel, a simple boiling method in 25 mM NaOH for genotyping by RFLP was applied. The CRISPR/Cas9-targeted region of MtGA2ox10 was amplified with the 2289-F and 2905-R primers, using Phusion High Fidelity DNA polymerase (Thermo Fisher Scientific). The amplicons were digested using the BsrD I (Thermo Fisher Scientific) or Eco105 I (Enzynomics) restriction enzymes and analyzed by agarose gel electrophoresis. Additionally, the amplicon was sequenced using the 2347-F primer after being cloned in the pLPS-TOPO Blunt vector (Elpis Biotech, Daejeon, Korea). Genotyping of the stable transgenic plants was performed by PCR amplification of the MtGA2ox10 coding sequence in the binary plasmid using G512-F and P35S-SF primers.
GA treatment and statistics test
GA3 (Sigma-Aldrich) was dissolved in ethanol at stock concentration of 10 mM. Two-month-old stable transgenic plants grown in pots were supplemented with nitrogen-free mFM medium containing either of 10 µM or 100 µM GA3 at final concentration. Changes in plant architecture were recorded for four weeks. To statistically test the difference in measurements, the independent t-test was performed using SPSS.
Data Availability
The RNA-seq data used in this study have been deposited in NCBI’s Bioproject collection under the Bioproject ID PRJNA269201.
References
Oldroyd, G. E. Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 11, 252–263 (2013).
Ferguson, B. J., Foo, E., Ross, J. J. & Reid, J. B. Relationship between gibberellin, ethylene and nodulation in Pisum sativum. New Phytol. 189, 829–842 (2011).
Maekawa, T. et al. Gibberellin controls the nodulation signaling pathway in Lotus japonicus. Plant J. 58, 183–194 (2009).
Fonouni-Farde, C. et al. DELLA-mediated gibberellin signalling regulates Nod factor signalling and rhizobial infection. Nat. Commun. 7, 12636 (2016).
McAdam, E. L., Reid, J. B. & Foo, E. Gibberellins promote nodule organogenesis but inhibit the infection stages of nodulation. J. Exp. Bot. 69, 2117–2130 (2018).
Madsen, L. H. et al. The molecular network governing nodule organogenesis and infection in the model legume Lotus japonicus. Nat. Commun. 1, 10 (2010).
Hedden, P. & Kamiya, Y. GIBBERELLIN BIOSYNTHESIS: Enzymes, Genes and Their Regulation. Annu. Rev. Plant Physiol. Plant Mol. Biol. 48, 431–460 (1997).
Lo, S. F. et al. A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. Plant Cell 20, 2603–2618 (2008).
Davies, P. J. Reflections from the Janus face of gibberellin in legume nodulation. J. Exp. Bot. 69, 1824–1828 (2018).
Olszewski, N., Sun, T. P. & Gubler, F. Gibberellin signaling: biosynthesis, catabolism, and response pathways. Plant Cell 14(Suppl), S61–80 (2002).
Sakamoto, T. et al. Expression of a gibberellin 2-oxidase gene around the shoot apex is related to phase transition in rice. Plant Physiol. 125, 1508–1516 (2001).
King, R. W. et al. Selective deactivation of gibberellins below the shoot apex is critical to flowering but not to stem elongation of Lolium. Mol. Plant 1, 295–307 (2008).
Regnault, T. et al. The gibberellin precursor GA12 acts as a long-distance growth signal in Arabidopsis. Nat. Plants 1, 15073 (2015).
Regnault, T., Davière, J.-M. & Achard, P. Long-distance transport of endogenous gibberellins in Arabidopsis. Plant Signal. Behav. 11, e1110661 (2016).
Ferguson, B. J., Ross, J. J. & Reid, J. B. Nodulation phenotypes of gibberellin and brassinosteroid mutants of pea. Plant Physiol. 138, 2396–2405 (2005).
Rieu, I. et al. Genetic analysis reveals that C19-GA 2-oxidation is a major gibberellin inactivation pathway in Arabidopsis. Plant Cell 20, 2420–2436 (2008).
Hisamatsu, T. & King, R. W. The nature of floral signals in Arabidopsis. II. Roles for FLOWERING LOCUS T (FT) and gibberellin. J. Exp. Bot. 59, 3821–3829 (2008).
Lievens, S. et al. Gibberellins are involved in nodulation of Sesbania rostrata. Plant Physiol. 139, 1366–1379 (2005).
Libault, M. et al. Complete transcriptome of the soybean root hair cell, a single-cell model, and its alteration in response to Bradyrhizobium japonicum infection. Plant Physiol. 152, 541–552 (2010).
Hayashi, S. et al. Transient Nod factor-dependent gene expression in the nodulation-competent zone of soybean (Glycine max [L.] Merr.) roots. Plant Biotechnol. J. 10, 995–1010 (2012).
Breakspear, A. et al. The root hair “infectome” of Medicago truncatula uncovers changes in cell cycle genes and reveals a requirement for auxin signaling in rhizobial infection. Plant Cell 26, 4680–4701 (2014).
Yaxley, J. R., Ross, J. J., Sherriff, L. J. & Reid, J. B. Gibberellin biosynthesis mutations and root development in pea. Plant Physiol. 125, 627–633 (2001).
Fonouni-Farde, C. et al. DELLA1-mediated gibberellin signaling regulates cytokinin-dependent symbiotic nodulation. Plant Physiol. 175, 1795–1806 (2017).
Amor, B. B. et al. The NFP locus of Medicago truncatula controls an early step of Nod factor signal transduction upstream of a rapid calcium flux and root hair deformation. Plant J. 34, 495–506 (2003).
Catoira, R. et al. The HCL gene of Medicago truncatula controls Rhizobium-induced root hair curling. Development 128, 1507–1518 (2001).
Penmetsa, R. V. & Cook, D. R. A legume ethylene-insensitive mutant hyperinfected by its rhizobial symbiont. Science 275, 527–530 (1997).
Larrainzar, E. et al. Deep sequencing of the Medicago truncatula root transcriptome reveals a massive and early interaction between nodulation factor and ethylene signals. Plant Physiol. 169, 233–265 (2015).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 42, D459–D471 (2014).
Benedito, V. A. et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 55, 504–513 (2008).
Kim, G.-B. & Nam, Y.-W. Isolation and characterization of Medicago truncatula U6 promoters for the construction of small hairpin RNA-mediated gene silencing vectors. Plant Mol. Biol. Rep. 31, 581–593 (2013).
Chen, S. et al. Identification and characterization of tomato gibberellin 2-oxidases (GA2oxs) and effects of fruit-specific SlGA2ox1 overexpression on fruit and seed growth and development. Hortic Res 3, 16059 (2016).
Schomburg, F. M., Bizzell, C. M., Lee, D. J., Zeevaart, J. A. & Amasino, R. M. Overexpression of a novel class of gibberellin 2-oxidases decreases gibberellin levels and creates dwarf plants. Plant Cell 15, 151–163 (2003).
Shan, C. et al. OsGA2ox5, a gibberellin metabolism enzyme, is involved in plant growth, the root gravity response and salt stress. PLOS One 9, e87110 (2014).
Wuddineh, W. A. et al. Identification and overexpression of gibberellin 2-oxidase (GA2ox) in switchgrass (Panicum virgatum L.) for improved plant architecture and reduced biomass recalcitrance. Plant Biotechnol. J. 13, 636–647 (2015).
Proebsting, W. M., Hedden, P., Lewis, M. J., Croker, S. J. & Proebsting, L. N. Gibberellin concentration and transport in genetic lines of pea: effects of grafting. Plant Physiol. 100, 1354–1360 (1992).
Binenbaum, J., Weinstain, R. & Shani, E. Gibberellin localization and transport in plants. Trends Plant Sci 23, 410–421 (2018).
Barker, D. et al. Growing M. truncatula: choice of substrates and growth conditions. In The Medicago truncatula Handbook (eds Mathesius, U., Journet, E. P. & Sumner, L. W.) (The Samuel Roberts Noble Foundation, Ardmore, OK, USA 2006).
Lullien, V., Barker, D. G., de Lajudie, P. & Huguet, T. Plant gene expression in effective and ineffective root nodules of alfalfa (Medicago sativa). Plant Mol. Biol. 9, 469–478 (1987).
Giacomelli, L. et al. Gibberellin metabolism in Vitis vinifera L. during bloom and fruit-set: functional characterization and evolution of grapevine gibberellin oxidases. J. Exp. Bot. 64, 4403–4419 (2013).
Thompson, J., Gibson, T. & DG, H. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics Chapter 2, Unit 2.3 (2002).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Igielski, R. & Kępczyńska, E. Gene expression and metabolite profiling of gibberellin biosynthesis during induction of somatic embryogenesis in Medicago truncatula Gaertn. PLOS One 12, e0182055 (2017).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
de Hoon, M. J., Imoto, S., Nolan, J. & Miyano, S. Open source clustering software. Bioinformatics 20, 1453–1454 (2004).
Chang, S., Puryear, J. & Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116 (1993).
Livak, K. & Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Karimi, M., Inze, D. & Depicker, A. GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 7, 193–195 (2002).
Kim, H. et al. A simple, flexible and high-throughput cloning system for plant genome editing via CRISPR-Cas system. J. Integr. Plant Biol. 58, 705–712 (2016).
Park, J., Bae, S. & Kim, J.-S. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31, 4014–4016 (2015).
Engler, C., Kandzia, R. & Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLOS One 3, e3647 (2008).
Boisson-Dernier, A. et al. Agrobacterium rhizogenes-transformed roots of Medicago truncatula for the study of nitrogen-fixing and endomycorrhizal symbiotic associations. Mol. Plant Microbe Interact. 14, 695–700 (2001).
Nolan, K. et al. An unusual abscisic acid and gibberellic acid synergism increases somatic embryogenesis, facilitates its genetic analysis and improves transformation in Medicago truncatula. PLOS One 9, e99908 (2014).
Penmetsa, R. V., Frugoli, J. A., Smith, L. S., Long, S. R. & Cook, D. R. Dual genetic pathways controlling nodule number in Medicago truncatula. Plant Physiol. 131, 998–1008 (2003).
Díaz, C. L., Schlaman, H. R. M. & Spaink, H. P. In Lotus japonicus Handbook (ed. Márquez, A. J.) 99–109 (Springer Netherlands 2005).
Jefferson, R. A., Kavanagh, T. A. & Bevan, M. W. GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907 (1987).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Acknowledgements
This work was supported by grants from Rural Development Administration (PJ012251 and PJ012421), Korea.
Author information
Authors and Affiliations
Contributions
J.H.M. planned the project, designed the research, analyzed data, and wrote the manuscript. G.B.K. performed the experiments, analyzed data, and wrote the manuscript. S.U.S. performed plant transformation. H.J.Y. analyzed data and participated in manuscript preparation.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, GB., Son, SU., Yu, HJ. et al. MtGA2ox10 encoding C20-GA2-oxidase regulates rhizobial infection and nodule development in Medicago truncatula. Sci Rep 9, 5952 (2019). https://doi.org/10.1038/s41598-019-42407-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42407-3
This article is cited by
-
Characterization of ZmPMP3g function in drought tolerance of maize
Scientific Reports (2023)
-
Comparisons within the Rice GA 2-Oxidase Gene Family Revealed Three Dominant Paralogs and a Functional Attenuated Gene that Led to the Identification of Four Amino Acid Variants Associated with GA Deactivation Capability
Rice (2021)
-
Phalaenopsis orchid miniaturization by overexpression of OsGA2ox6, a rice GA2-oxidase gene
Botanical Studies (2020)
-
Brassinosteroids play multiple roles in nodulation of pea via interactions with ethylene and auxin
Planta (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
