
Abstract
G-protein signaling components have been attributed many biological roles in plants, but the extent of involvement of G-protein coupled receptor 1 (GCR1) with the Gα (GPA1) remained unknown. To address this, we have performed transcriptomic analyses on Arabidopsis gpa1-5gcr1-5 double mutant and identified 656 differentially expressed genes (DEGs). MapMan and Gene Ontology analyses revealed global transcriptional changes associated with external stimulus, cell wall organization/biogenesis and secondary metabolite process among others. Comparative transcriptomic analyses using the single and double mutants of gcr1-5 and gpa1-5 identified 194, 139 and 391 exclusive DEGs respectively, whereas 64 DEGs were common to all three mutants. Further, pair wise comparison of DEGs of double mutant with single mutants of gcr1-5 or gpa1-5 showed about one-third and over half common DEGs, respectively. Further analysis of the DEGs exclusive to the double mutant using protein-protein interaction networks revealed molecular complexes associated with nitrate and light signaling and plant-pathogen interactions among others. Physiological and molecular validation of nitrate-response revealed the sensitivity of germination to low N in the double mutant and differential expression of nitrate transporter (and nitrate reductase in all three mutants). Taken together, GCR1 and GPA1 work in partnership as well as independently to regulate different pathways.
Similar content being viewed by others


Introduction
Heterotrimeric G-proteins regulate diverse signaling events in plants, following the dissociation of heterotrimer into GTP-bound Gα subunit and Gβγ dimers, which further activate the various downstream effectors for the coordinated regulation of plant responses. The model dicot Arabidopsis has been so far found to have only one α (GPA1), one β (AGB1), three γ subunits (AGG1-3), and three extra-large Gα proteins (XLG1-3)1,2. It has been shown that heterotrimeric G-proteins regulate cell growth and development, hormonal signaling, nitrate reductase gene expression and response to both abiotic and biotic stresses3,4,5,6,7. The upstream components of plant G-protein signalling and their interactions with G-proteins have been studied8,9,10 but still poorly understood. The best-considered GPCR candidate, GCR1 in Arabidopsis, has been implicated in the regulation of DNA synthesis11, abolishing seed dormancy, reducing flowering time12, brassinosteroid and gibberellin-regulation of seed germination13, drought stress, ABA response, regulation of stomatal apperture14, blue light response15 and most recently in biotic stress, flavonoid biosynthesis, cytokinin biosynthesis, salicylic acid and ethylene response, and phosphate starvation16. Transcriptome analyses of gpa1-5 has also identified DEGs involved in similar pathways including flavonoid biosynthesis, transcription factors, transporters and nutrient responses to nitrate and phosphate17.
The demonstration of self-activation of GPA118, lack of a confirmed GPCR and its ligand or guanine nucleotide exchange factor (GEF) activity in plant GCR so far19 and the disagreement20 over the reported interaction between GCR1 and GPA1 in Arabidopsis14,21 were used to question the existence and the role of GPCRs in plant G-protein signalling20. Instead, it has been shown with the help of crystal structure and in vitro experiments that plant Gα-proteins are self-activating and spontaneously exchange GDP with GTP without the need of GEF activity18,22. The sustained activation of G-protein signaling occurs by endocytosis of the regulator of G-protein signalling (RGS1) in Arabidopsis23. The seven transmembrane RGS proteins were initially thought to be absent in most studied grasses and monocots24 but later it was found that RGS proteins are present in many grasses with frequent losses in different species like rice25. Moreover, the argument regarding the lack of heterotrimeric G-proteins in green algae (which are predicted to have GCRs) has been countered recently by the discovery of a complete G-protein complex in a green alga, Chara braunii26. Most recently, transcriptome analyses on gcr1-5 mutant revealed differentially expressed genes belonging to known G-protein regulated processes16 suggesting the need to revisit the role of GCR1 in plant signalling in general and G-proteins in particular.
Till the GEF activity for GCR1 and its GPCR properties are proven, an overlap between the genes/processes/responses between the single mutants of GCR116 and GPA117 remains the best genetic evidence in favour of their functional association. This can be best validated by transcriptomic analyses of a double mutant, in comparison with either of the single mutants. The above two single mutant studies were done in the WS ecotype, whereas the double mutant isolated elsewhere was in the Col-0 ecotype11 and therefore, a double mutant in WS ecotype was necessary to validate the predictions made using the single mutants16,17. Accordingly, in this study, we used the GPA1 and GCR1 double mutant generated in WS background for whole transcriptome microarray analysis and comparison with single mutant data to demonstrate their combinatorial roles for various cellular responses and sensitivity of its seed germination to low nitrate.
Results
Characterization of the gpa1-5gcr1-5 double mutant
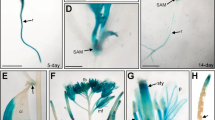
A double mutant of gpa1-5gcr1-5 was generated by crossing their confirmed single null mutants16,17 but its characterization was not reported earlier6. The null double mutant, devoid of expression of both GPA1 and GCR1, was confirmed by qPCR (Fig. 1A). The mutant plants were phenotypically characterized for root length, plant height, leaf shape and other phenotypic traits. It was found that gpa1-5gcr1-5 is similar to the only other known gpa1gcr1 double mutant in Col-0 background11 with longer roots, less plant height, longer siliques, and rounded leaves and smaller rosette (Fig. 1B–E). Overall, the double mutant gpa1-5gcr1-5 was found to be phenotypically closer to the gpa1-5 single mutant17 than the gcr1-5 single mutant16, though in most cases, the phenotype is somewhere between the two single mutants.

Characterization of the gpa1-5gcr1-5 double mutant. (A) The mutants and WT were grown for 23 days and subjected to total RNA isolation and qRT-PCR to confirm the lack of expression of GPA1 or GCR1 in the single as well as double mutants. The data represent averages of three independent replicates ± SE. (B–E) Phenotypic characterization of the gpa1-5gcr1-5 mutants. The double mutant and the WT were grown for 5 days on agar plates for root length comparison and were subsequently transferred to pots and grown to complete their life cycle to evaluate other phenotypic parameters shown. Each experiment was performed twice independently and the data represent averages of 10 individual plants ± SE (*P < 0.05, **P < 0.01 according to unpaired t-test using GraphPad Prism). The photographic strip of the Ws2 control have been reproduced from our previous paper16 under creative commons attribution license for ready reference. Scale bar = 1.0 cm.
Microarray analysis and validation
The MIAME compliant microarray replicates had high correlation coefficient (>0.9), clearly indicating the robustness and a high level of reproducibility of the data (Table S1). The Benjamini Hochberg FDR procedure at a cut-off value of p ≤ 0.05 was used for multiple testing corrections. A stringent cut-off value of 1.0 (geometric mean log2) with a p-value of ≤0.05 was used to identify 829 differentially regulated transcripts in the double mutant (422 up-regulated and 407 down-regulated). These transcripts corresponded to 656 unique differentially expressed genes (DEGs), 306 up-regulated and 350 down-regulated. A list of 10 most up- and down-regulated genes is shown in Table 1 and the heat map of all the DEGs and their GO classification is shown in Fig. 2. In order to validate the microarray results, 19 DEGs (10 up- and 9 down-regulated) were selected spanning each of the important functional categories and subjected to RT-qPCR using gene specific primers tested for efficiency (100 ± 10%). The list of these genes and the primer sequences used are given in the Table S2. The results of RT-qPCR matched with the microarray data in all the cases (Fig. 3) with Pearson’s product moment correlation of >0.99 (p-value = 6.54E-17), validating the basic trends of regulation of gene expression found in the microarray analyses.

(A) Heat map and GO analyses of differentially expressed genes. The background-subtracted, p-value filtered and geo-mean cutoff microarray data of the double mutant was subjected to hierarchical clustering using Genespring software ver. 11.5 to generate the heat map. Red, green and yellow represent up-regulated, down–regulated and unregulated genes, respectively. (B) The DEGs were functionally categorized into various biological processes using AgriGO2.0 tool. The p-values of biological processes were log transformed (−log10) and plotted (B). The complete results of AgriGO analyses, which include p-value, FDR and the numbers of DEGs associated with each biological process are listed in the supplementary Table S3.

qPCR validation of differentially expressed genes in gpa1-5gcr1-5 double mutant. A total of 19 DEGs (10 up- and 9 down-regulated) were selected and subjected to RT-qPCR. The experiment was carried out using three biological replicates and the values are presented as log2FC ± SE. qPCR was performed in triplicate and the ratios of statistics were calculated relative to the internal control gene Actin2 (*P < 0.05, **P < 0.01 vs. control).
Gene Ontology and MapMan pathway analyses of double mutant DEGs
To understand the biological effects of loss of both GCR1 and GPA1 function, we performed the GO analyses of the DEGs identified in the double mutant using AgriGO2.0 tool27. The statistically overrepresented GO terms (based on p value and FDR) were considered for further analyses (Fig. 2B and Table S3). All the 656 DEGs were broadly classified into biological processes, molecular functions and cellular components. The over-represented GO terms for biological processes were “response to external stimulus”, “plant-type secondary cell wall biogenesis”, “cell wall organization or biogenesis”, “response to external biotic stimulus”, “response to other organism”, “response to biotic stimulus” and secondary metabolite biosynthetic process” among others. In molecular function category, we observed the significant GO terms were “terpene synthase activity”, “O-methyltransferase activity”, “carbon-oxygen lyase activity”, “acting on phosphates tetrapyrrole binding” and “transcription factor activity” among others whereas “extracellular region” GO term identified for cellular component (Table S3). We also mapped these DEGs into various pathways using MapMan28. Comparative analyses showed a high degree of agreement between GO terms and MapMan pathways. The DEGs were broadly mapped into various pathways (bins) such as metabolic processes (Fig. 4A), different levels of regulation (Fig. 4B) and cellular responses (Fig. 4C). Further insight into these pathways (sub-bins) showed that many DEGs were mapped into biotic and abiotic stress pathways, development, cell wall, lipid and amino acid metabolism, hormone signaling, protein modification and degradation among others. Some DEGs were also classified as receptor like kinases (RLKs), transcriptional regulators and genes regulated by calcium and G-protein signaling (Fig. 4).

Mapping of DEGs found in the gpa1-5gcr1-5 double mutant into various pathways using MapMan. (A) DEGs mapped into metabolic pathways. (B) DEGs associated with regulation. (C) DEGs assigned to cellular responses. Each box represents a DEG while the red and blue colours indicate up- and down-regulated DEGs, respectively.
Sub cellular distribution of DEGs and identification of associated transcription factors
To understand the global cellular context of both GCR1 and GPA1 mutations in terms of the affected subcellular organelles and associated pathways, all the DEGs were subjected to subcellular prediction using YLoc program. We observed that majority fraction of the DEGs were distributed into cytosol (24%), extracellular (22%), nucleus (21%) and plasma membrane (11%) among others (Fig. 5A). This suggests that both GCR1 and GPA1 regulate many processes and pathways that operated within these organelles. Nuclear genome is an important target for myriad signaling pathways that culminate in gene regulation by transcription factors (TFs). A search using the DEGs at the plant transcription factor database (plantTFDB 2.0)29 revealed 64 transcription factors (Table S4) belonging to 22 families. Their regulation was nearly equally distributed in the double mutant, with 34 up-regulated and 30 down-regulated TF genes. (Table S4). Most of them belong to the class of bHLH, C2H2, MYB, WRKY and AP2-EREB families, other than putative and unspecified ones (Fig. 5B). While many of the MYB family members were found to be down-regulated in the double mutant, none of the transcription factors of AP2-EREB and WRKY families were down-regulated. On the other hand, in the bHLH and C2H2 families, the up- and down-regulated transcription factors showed a mixed distribution. Sixteen TFs were commonly regulated in gpa1-5 while none of these TFs were common in the gcr1-5 mutant. The guard cell functions and root differentiation are mediated through G-protein signaling14. The transcriptional regulators such as bHLH, MYB and WD40 are known to regulate these functions30 and many of these regulators were identified as DEGs in our datasets. MYB and WRKY belong to a major TFs class and were reported to be involved in stress responses31,32. Members of AP2/EREB class of TFs have been reported to be involved in storage compound, fatty acid biosynthesis and stress responses33. The up regulation of two TFs, bHLH100 and ERF13 and the down regulation of MYB69 and MYB5 were validated in the double mutant using qPCR (Figs S1 and S2).

Subcellular localization of DEGs and classification of transcription factors among them. (A) Subcellular distributions of the DEGs identified in the double mutant as predicted using YLoc program. (B) Identification and classification of transcription factors among the DEGs in the double mutant using plantTFDB.
Double and single mutants share substantial number of DEGs
In order to gain a comprehensive view of the differential regulation of the affected genes in the single and double mutants of GPA1 and GCR1, we compared the DEGs obtained in gpa1-5gcr1-5 double mutant to those of the single gpa1-517 and gcr1-516 mutants. Out of the 350 GCR1-regulated genes in the single mutant, 115 (or 34%) were common to the 656 DEGs in the double mutant. Similarly, out of the 394 of the GPA1 regulated genes in the single mutant 214 (or 54%) were common to the 656 DEGs in the double mutant. Only 64 DEGs were found to be shared amongst all three mutants (Fig. 6A). The hierarchical clustering of the DEGs from all the mutants revealed that the double mutant (gpa1-5gcr1-5) is closer to gpa1-5 and that gcr1-5 is closer to the wild type (Fig. 6B). This closely parallels the similarity patterns in their phenotypes.

(A) Venn selection of differentially regulated genes between single and double mutants. The DEGs identified in the double mutant in the current study were compared with those identified earlier in the single mutants of gpa1-517 and gcr1-516 and shown as up/down regulated subsets or together. (B) Hierarchical clustering of DEGs obtained from all the 3 mutants shows that gpa1-5gcr1-5 double mutant is closer to the gpa1-5 mutant than the gcr1-5 mutant.
If the genetic interactions are additive, the genes differentially expressed in the double mutant should have been the sum of all the DEGs found in the single mutant. Also, all the DEGs shared by the single mutants should also have been common to the double mutant, but this was not observed using the log2 fold change (log2FC) cut-off of 1.0. The double mutant has almost double the number of DEGs identified in each of the single mutants. 64 DEGs (37 up-regulated; 27 down-regulated) were common to all the three mutants, while the single mutants shared 104 DEGs between them. A closer look at these 104 DEGs revealed that they did not light up in the double mutant due to either the stringent p-value cut-off of 0.05 or log2FC cut-off of 1.0. Out of the 28 such up-regulated genes that did not light up in the double mutant, 10 did not meet the p-value cut-off, 10 had log2FC value of 0.8 and above, while the remaining 8 genes had log2FC value of less than 0.8. Similarly, among the 12 such down-regulated genes, only 4 genes did not meet the log2FC cut-off of −1.0 and the rest did not make it to the list due to p-value cut-off of 0.05 despite having log2FC values beyond −1.0. We validated 10 DEGs from the list shared only by the single mutants and 10 DEGs unique to the double mutant by qPCR (Figs S1 and S2).
Abiotic and biotic stress
Heterotrimeric G-protein-dependent immune regulation34,35,36,37 and abiotic stress-responses4,38,39,40 are well known in plants. Functional analyses of the DEGs revealed that “response to stimulus” constitutes the top most GO category of genes regulated by GPA1 and/or GCR1. Among these DEGs, 32 genes (including ESC, ARR22, and TT7) were reported to be GPA1-regulated17, while 23 (including CAD1, EF1α, and WRKY 53) of them have been reported to be regulated by GCR116, which also includes the 15 genes that are regulated by both GPA1 and GCR1. The genes that are regulated by both GPA1 and GCR1 include Arabidopsis thaliana phloem protein 2 A5 (ATPP2-A5), dark inducible 11 (DIN11), phytoalexin deficient 3 (PAD3), etc. Many DEGs such as DIN11, FMO1, MEE16, and PAD3 have been reported to be differentially regulated in gpa1-517 and gcr1-5 mutants16,41. These include several well-known stress-responsive genes such as low temperature induced 78 (LTI78), plant defensin 2.5 (PDF2.5), ethylene response factor (ERF6), and several peroxidases and transcription factors. Analysis of the DEGs using MapMan revealed them to be involved in abiotic stresses such as cold, heat, drought, salt etc., as well as in biotic stresses. More detailed mapping revealed that 225 out of total 656 DEGs belong to the biotic stress category (Fig. S3), though a few of them are also involved in abiotic stress. A majority of these 225 genes were mapped into signalling, proteolysis, cell wall, PR-proteins and secondary metabolites. The basic trends of their regulation in the mutant have been confirmed by qRT-PCR on two up-regulated genes (peroxidise family protein gene (AT1G49570) and ATPP2A5) and two down-regulated ones (PDR12 and PAD3), as shown in Fig. 3.
Secondary metabolism
The GO class associated with secondary metabolites were found to be an important category, so we checked the involvement of GCR1/GPA1 in regulating the genes of secondary metabolism. We found that 107 DEGs belong to the biosynthesis of flavonoids and isoprenoids based on MapMan as well as pathway analysis using AraCyc database (Fig. S4, Table 2). The genes involved in flavonoid biosynthesis include 2-oxoglutarate, dihydroflavanol-4-reductase (DFR), UDP-glucosyl transferase 73C6 (UGT73C6), etc., while those involved in isoprenoid biosynthesis include dehydrodolichyl diphosphate synthase, myrcene synthase, terpene synthase 21 (TPS21), etc. The basic trends of their regulation in the mutants have been confirmed by qRT-PCR on the up-regulated gene 2-oxoglutarate and two down-regulated ones (FMO1 and DFR), as shown in Fig. 3. Flavonoid biosynthesis was also found to be regulated in our previous studies using single mutants of GPA1 and GCR1, but many more genes belonging to this category are differentially regulated in the double mutant. Thus, out of the 11 DEGs that regulate flavonoid biosynthesis found in the double mutant, only two were differentially regulated in both the single mutants, whereas 5 genes were differentially regulated in gpa1-5 and 3 genes in gcr1-5.
Development
We also detected the association of 80 DEGs in developmental processes (Fig. 4). These genes include senescence-associated gene 12 (SAG12), vegetative storage protein 2 (VSP2), lateral organ boundaries-domain 29 (LBD29), several expansins, etc. Out of these, few genes like expansins are involved in cell wall modification. Genes like transparent testa 8 (TT8), tetratricopeptide repeat 3 (TPR3), cytokinin response factor 4 (CRF4), etc. are involved in development of shoot while transparent testa 16 (TT16), shatterproof 2 (SHP2), flowering locus T (FT), etc. are involved in flower development. GPA1 has been previously reported17 to be involved in developmental processes and hence, shows a larger convergence with 17 genes being common between them. Only four DEGs were found to be common to gcr1-516 in this category. We confirmed the basic trends of regulation in the mutant in this category using qPCR on the up-regulated (AT2G35710 and AT1G78860) as well as down-regulated (VSP2 and AT2G02160) genes (Fig. 3).
Hormone response
G-protein signaling has been implicated in regulation of hormone signaling in plants41,42,43. GO and MapMan analyses showed that 37 of the DEGs were associated with hormone biosynthesis and signaling (Figs 2 and 4). These include genes which are responsive to cytokinin, ethylene, ABA, auxin, salicylic acid, etc. Ethylene is known to down-regulate the expression of AGB144 and the role of GPA1 in ethylene signalling operated in guard cell is known45. Cytokinin oxidase 4 (CKX4) and cytokinin response factor 4 (CRF4) are involved in cytokinin biosynthesis/response; ethylene response factor 6 and 13 (ERF6 and ERF13) and pleiotropic drug resistance 12 (PDR12) are involved in ethylene response. A few others like MYB43, hydroxysteroid dehydrogenase (HSD1), responsive to desiccation 26 (RD26), syntaxin of plants 121 (SYP121) etc., are involved in ABA response. A few auxin-responsive genes like PDR12 and LBD29 were also found among the hormone-responsive genes. Though hormone response was found as a major category in gcr1-516, the overlap to the double mutant in terms of DEGs was limited to only 2 genes. Similarly, only 3 DEGs were found to be common to the gpa117 and double mutant. The basic trends of their regulation in the double mutant have been confirmed by qRT-PCR on the up-regulated genes, CKX4 and ERF13, as well as the down-regulated gene PDR12, as shown in Fig. 3.
Transport
Twenty three genes related to transport were also found to be differentially regulated in the double mutant (Table S3). These include lipid transporters (LPTs), oligopeptide transporters (POT, OPT5), nuclear transport factor (NTF2), as well as nutrient transporters such as methylammonium transporter (TIP 2;3), phosphate transporter (APT1), nitrate excretion transporter (NAXT1) and high affinity K+ transporter (HKT1). A few of these DEGs have been reported earlier in other G-protein mutants41,46. The basic trends of their regulation in the mutant have been confirmed by qRT-PCR on the down-regulated genes PDR12 and AZG2, as shown in Fig. 3. Interestingly, transport was also found to be a major response category in the transcriptomic analyses of the gpa1-5 mutant17, but not in the gcr1-5 mutant16.
Cellular processes and molecular complexes regulated by both GCR1 and GPA1 function
To understand the function of DEGs detected in the gpa1-5gcr1-5 double mutant, we compared significantly overrepresented GO terms and observed both overlapping as well exclusive biological processes in all three mutant datasets (Table S5). The comparison clearly revealed that processes exclusive to the double mutant predominantly regulate cell wall composition and associated metabolic processes (Fig. 7A). MapMan analyses also revealed the over-representation of cell wall-associated DEGs in the double mutant (Fig. 7B). The results of both AgriGO (Fig. 7A) and MapMan analyses (Fig. 7B) are similar in the sense that the double mutant showed higher number of cell wall-associated exclusive DEGs as compared to either of the single mutants. A combination of both GO and MapMan analyses led to the identification of 36 cell wall-associated exclusive DEGs in the double mutant (Fig. 7C). Majority of these DEGs such as the family members of ANAC, MYC, MYB and pectinesterase were down-regulated, whereas pectinase, expansin-like B3 precursor, proline-rich extensin-like among others up-regulated in the double mutant. To validate the expression level of cell wall associated DEGs identified in the gpa1-5gcr1-5 double mutant, 4 DEGs were selected for qPCR validation. Three DEGs, beta-xylosidase 3 (BXL3), COBRA-like 4 (COBL4), and galacturonosyl transferase 12 (GAUT12), were down-regulated, whereas pectin methylesterase (ATPMEPCRB) was up-regulated in the gpa1-5gcr1-5 double mutant (Fig. S5), confirming their trend on the microarray. The BXL3 is generally localized in the extracellular matrix and is involved in the hydrolysis of arabinan, whereas COBL4, also known as irregular xylem 6 (IRX6), is involved in the secondary cell wall biosynthesis. The loss of function of GAUT12, also known as irregular xylem 8 (IRX8), significantly reduces xylose contents in the cell walls whereas ATPMEPCRB act on cell wall pectin in plant. The modulation of the expression of these genes in the double mutant indicates GCR1 and GPA1 coupling in the regulation of the cell wall.

Heat map of biological processes exclusive to each of the three mutants and cell wall associated DEGs in the double mutant. The GO classes of DEGs exclusive to each of the single and double mutants were used for the analysis. (A) Heat map of the exclusive biological processes generated using heatmapper (http://heatmapper.ca/). The default colour scheme depicts the presence or absence of the exclusive GO classes as yellow or blue respectively. (B) Venn selection of cell wall associated DEGs from all three mutants identified by MapMan. (C) Heat map showing the cell wall associated exclusive DEGs identified in the double mutant using GO and MapMan analyses. Heat map was generated using Multi Experiment Viewer software (http://mev.tm4.org/#/welcome).
To further understand the combinatorial role of GCR1 and GPA1 in cellular response, we used the DEGs from all three mutants to search in the G-protein interactome47, MIND database48, XLGs interactome49 and RGS1 protein networks50. We observed association with known G-protein signaling components in 12, 8 and 16 DEGs in the gcr1-5, gpa1-5 and gpa1-5gcr1-5 mutants, respectively (Fig. 8A). Only two DEGs namely phloem protein 2 A5 and methionine sulfoxide reductase B7 were found to be the common interactor DEGs among all three mutants (Table S6). To further delineate these complex regulations, we developed PPI networks of exclusive DEGs identified in the double mutant and mapped these DEGs into networks. To construct the PPI networks, we retrieved the experimentally validated interactions list from STRING and BioGRID databases and assigned the colour code to the nodes using DEGs expression value. The networks consisting of 2216 nodes and 3499 edges were analysed and viewed in Cytoscape 3.0.051. The PPI network analyses showed many of the DEGs interacting with other components in the networks (Fig. 8B–E). Sub-clustering of the networks using MCODE plugin in Cytoscape revealed 7 highly connected molecular complexes/sub-clusters (Figs 8 and S6). Four molecular complexes having MCODE score >3 with node number >3 (Fig. 8B–E) were selected for further analyses. A total of 5, 18, 4, 7 nodes and 9, 35, 5, 11 edges were detected in sub-cluster 1, 2, 3 and 4, respectively. All seven sub-clusters details are mentioned in Table S7. The sub-cluster 1 includes transcriptional regulators associated with light signaling such as HY5 (Long Hypocotyl 5), COP1 (Constitutive Photomorphogenic1) and HFR1 (Long Hypocotyl in Farred1) (Fig. 8, Table S7). The sub-clusters 4 also include transcription regulators such as ATMYC-2, MYC6.2, ATMYB123, homeodomain-like superfamily protein involve in diverse biological processes. The miscellaneous interactors such as auxin-responsive family protein, glycosyl transferase family 4 protein, nucleotide-sugar transporter family protein, and ubiquitin-conjugating enzyme 34 among others as were identified in sub-cluster 2. AKINBETA1, KIN10, KIN11, and SNF4 genes were identified in sub-cluster 3 and these protein kinases are involved in various cell signaling process. The identification of DEGs in these molecular complexes suggests that associated cellular pathways may be regulated by the combined function of GCR1 and GPA1 in Arabidopsis.

PPI networks of exclusive DEGs identified in the double mutant. (A) Venn diagram showing the overlapping and exclusive DEGs identified as interactors of G-protein signaling components. (B–E) The protein-protein interaction (PPI) networks were constructed with Cytoscape using experimentally validated interactions obtained from BioGRID and STRING databases. Sub-clustering of the PPI networks was performed using the MCODE plugin in Cytoscape and representative networks are shown. The red and dark green nodes represent the up-regulated and down-regulated DEGs, respectively. Interactors that are not among DEGs identified in the double mutant are assigned with light green colour.
Germination of gpa1-5gcr1-5 double mutant is sensitive to low nitrate
The effect of N and N-associated genes on seed germination is well known in plants52,53,54. We analysed the role of G-protein signaling on nitrate-responsive germination in single mutants (gpa1-5, gcr1-5) and their double mutant (gpa1-5gcr1-5) grown on B5 media supplemented with low nitrate (12.5 mM KNO3) optimal nitrate (25 mM KNO3) as per the standard B5 media composition or high nitrate (30 mM KNO3) at 22 °C in a growth chamber. The emergence of radicle was observed every three hours for the next three days (72 h) and total % seeds germinated and time taken for 50% seeds to germinate were used to compare WT and mutants. All of them started germinating around 30 h after soaking and seeds of both the single mutants and wild type were broadly similar at all nitrate doses, both in terms of total germination at 72 h (95–100%) and the time taken for 50% seeds to germinate (Fig. 9). However, the double mutant was sensitive to low nitrate level (12.5 mM) on both counts. It had significantly lower level of total germination (80%) and also significantly slower germination rate, as the time taken for 50% seeds to germinate was delayed by 4 h relative to the WT (Fig. 9B).

N-responsive germination in single and double mutants. Thirty seeds each of the wild-type (Ws2) and all three mutants, gpa1-5, gcr1-5 and gpa1-5gcr1-5, were surface sterilized and stratified at 4 °C in dark for 48 h. These seeds were placed on 1X B5 agar plates supplemented with different concentrations of KNO3 as shown for optimal (A), low (B) and high (C) dose of nitrate. The plates were transferred to growth chambers maintained at 22 ± 1 °C and after 12 h germination was monitored at every 3 h until 72 h. The data are plotted as a percentage of germinated seeds along with standard error bars. The data was statistically analysed using ANOVA in the GraphPad Prism 6.0 (*P < 0.05, **P < 0.01, ***P < 0.001).
In order to investigate whether these mutants are affected in the genes encoding nitrate uptake and metabolism, we grew them along with WT in low (12.5 mM) and high (30 mM) nitrate conditions for 14 days, harvested their root tissues and analyzed the expression of known nitrate-regulated genes, nitrate transporter (NRT1) and nitrate reductase (NR2) by real time PCR. At 12.5 mM nitrate level, the expression of NRT1 was higher in all three mutants as compared to wild type, whereas NRT1 expression was reduced at 30.0 mM nitrate (Fig. S7). Considering that NRT1 is known to be a low affinity nitrate transporter and sensor or transceptor, 12.5 mM nitrate may have been perceived as inadequate due to GCR1 and/or GPA1 mutation, triggering higher expression of NRT1, which was not the case at 30.0 mM nitrate. This is consistent with our previously reported role for Gα signaling in N-response and nitrate reductase expression/activity5,17. Accordingly, the perceived nitrate-limited condition in both the single and double mutants also explains the observed down-regulation of nitrate reductase (NR2) transcript level at 12.5 mM nitrate but not at 30 mM nitrate, except in the gpa1-5 mutant (Fig. S7). We also demonstrate for the first time that gcr1-5 mutant shows altered dose-dependent differential N-response for both NRT1 and NR2 gene expression, implying GCR1-GPA1 coupling in N-signaling.
Discussion
It is well recognized that heterotrimeric G-proteins play important roles in several plant processes, despite the limited diversity of their components24. For example, all the functions of the Gα subunit were previously attributed to GPA1 in Arabidopsis, till it was shown that a few of these functions are attributed to XLGs3,55. The existence of multiple γ subunits necessitated the classification of downstream signalling partners/pathways in Arabidopsis56. Normally, this would also be expected for molecules upstream of G-proteins, as their diversity facilitates the perception, discrimination and transduction of diverse signals. Instead, they were viewed from a predominantly all-or-none approach that initially relied only on GPCRs14 and subsequently relied only on RGS20, arguing explicitly that only one of the two possibilities can exist24 till recently. We have provided the first evidence against such exclusive approach using parallel functional genomic analyses of mutants of Arabidopsis GCR116/GPA117 from a gene discovery perspective. We showed there by Venn selection that 30% of all GCR1-responsive genes and 57% of all GCR1-regulated processes were similar to those of GPA1, though there were also many that did not overlap with those of GPA1. This was by far the most compelling indication, not only in favour of the GCR1-GPA1 partnership, but also in favour of its possible coexistence with other alternative partnerships (GCRx-GPA1, GCR1-GPAx, non-GCR-partnership with GPA1 or GCR1 partnership with a non-G-protein).
In this study, we extended this approach further by microarray analysis of a double-mutant generated from the confirmed single mutants of GCR116 and GPA117 in Arabidopsis to further confirm the genes/processes co-regulated by GCR1-GPA1 partnerships, as well as to predict other possible partnerships based on the observed responses. This double mutant (gpa1-5gcr1-5) is different from the only other double mutant reported so far11, not only because it is in a different ecotype, but also with respect to the specific loci of mutations in their single mutant parents we generated and used for crossing, as described earlier for gpa1-517 and gcr1-516. The double mutant was found to be phenotypically similar to the previously published double mutant11 as well as closer to the gpa1-5 parent (Fig. 1B–E)17, further confirmed by hierarchical clustering (Fig. 2). We found 656 DEGs in the double mutant spanning all 5 chromosomes, with nearly equal proportion of up/down-regulated genes. Nineteen of these genes (10 up-regulated and 9 down-regulated) have been verified by qRT-PCR (Fig. 3) and a larger list of the top 10 DEGs is given in Table 1. Functional annotation and MapMan pathway enrichment analysis showed that these DEGs were involved in many pathways such as response to external stimulus, primary and secondary cell wall modulation/biosynthetic processes, plant immunity, secondary metabolism, nitrogen signaling and light signaling among others.
The genes/processes identically regulated in all 3 mutants can be best explained by GCR1 and GPA1 working together in the same G-protein signalling pathway, though co-regulation by convergence of independent pathways cannot be ruled out, till the clinching biochemical evidence for the functional coupling of GCR1 and GPA1 is obtained. On the other hand, independent signalling pathways of GCR1 and GPA1 provide the most plausible explanation for the regulation of the 51 additional genes in the double mutant shared only with the GCR1 mutant, as well as for the 150 additional genes shared only between the double mutant and the GPA1 mutant. At least some of these DEGs in the double mutant common to either of the two single mutants (but not both) belong to the same process categories including stress, response to stimulus, transcription, etc. that are shared by all three mutants. This means that even when GCR1 and GPA1 follow independent pathways involving other partners to regulate different genes, some of them seem to achieve similar regulatory outcomes at the process level. This is indeed the best explanation for 195 unshared genes from the GCR1 single mutant and 140 unshared genes from the GPA1 single mutant belonging to 41 shared processes in the double mutant. These processes include response to stress, cell wall modification, development, hormone response, etc.
To understand the functional association of DEGs and associated processes regulated by GCR1 but independent of GPA1, we compared the list of DEGs and found that 51 DEGs in the double mutant shared only with the gcr1-5 mutant and not with the gpa1-5 mutant constitute about 44% of the 115 total DEGs shared between them (as the remaining 64 are common to all 3 mutants). Their identical pattern of regulation in the gcr1-5 mutant and the double mutant clearly indicates that the effects of GCR1 mutation are carried over to the double mutant but GPA1 mutation has no effect on these genes, either in the gpa1-5 mutant or in the double mutant. The best explanation for this is that GCR1 regulates these genes through some other partner, which may be another GPA-like isoform that is yet to be identified, or the Gβ and/or Gγ, RGS, XLG components of heterotrimeric G-protein complex, or through a totally different, non-G-protein signalling mechanism. While testing these possibilities is beyond the scope of the current study, it does offer a list of genes regulated through such a partnership as a starting point to test these hypotheses.
The significant overlap of DEGs (150 using stringent cut-offs) between gpa1-517 and the double mutant suggests their regulation via GPA1 but independent of GCR1 function. Even though they form a minority of the 656 DEGs identified in the double mutant, they constitute 70% of all the 214 DEGs shared between gpa1-5 and the double mutant (as the remaining 64 GPA1-regulated genes are shared between all 3 mutants). Their huge overlap and identical differential regulation explains the sheer predominance of the effects of GPA1 mutation in the double mutant, in terms of the 92% similarity in the 79 processes to which their shared DEGs belong as well as their phenotypic traits.
We detected a higher number of DEGs (including exclusive DEGs) in the double mutant than in either of the single mutants. The exclusive biological processes in all three datasets revealed overrepresentation of cell wall modification/biogenesis/organization, response to light intensity, flavonoid biosynthetic and metabolic processes among others in the double mutant; hydrogen peroxide metabolic and catabolic processes in the gpa1-5 mutant and response to starvation, phosphate starvation and nutrient levels in the gcr1-5 mutant (Fig. 7A). This clearly suggests that modulation of cell wall composition requires both GCR1 and GPA1 function. MapMan pathway analyses also showed significant enrichment of cell wall associated DEGs in the double mutant as compare to either of the single mutants (Fig. 7B).
PPI network analysis yielded 7 molecular complexes/sub-clustered genes, of which sub-cluster 1 revealed light regulated transcription factors HY5, HFR1, COP1, MYB18 and HFY1. Out of them, HY5 and HFR1 acts downstream of phytochrome A (phyA) mediated signaling and regulate phyA-responsive gene expressions in Arabidopsis. HY5 and HFR1 both are positive regulators of phyA signaling and interact with COP1 E3 ligase, which is negative regulator of photomorphogenesis57. HFR1 was up-regulated in the double mutant, which suggests that GPA1 and GCR1 may regulate these molecular complexes through HFR1 function and accordingly their associated phenotypic traits and biological responses. We identified another important hub (Fig. 8C) involved in nitrate (N) response regulation in Arabidopsis. KIN10 and KIN11 show significant homology with human adenosine monophosphate-activated protein kinase (AMPKα1). It has been shown that loss of KIN10 and KIN11 function reduces mutant sensitivity to N level58. Further, the circadian clock-dependent activities of these kinases are regulated by N level and control the flowering time in Arabidopsis58. Though KIN10 and KIN11 were not identified as DEGs in our mutants but we detected AKINBETA1 (5′-AMP-activated protein kinase beta-2 subunit) as an up-regulated DEG, which is interacting with both KIN10 and KIN11 to constitute molecular complexes (Fig. 8C). This leads to a testable hypothesis that both GCR1 and GPA1 control the N-regulated flowering time via modulating KIN10, KIN11 and associated molecular complexes in plants. The role of HY5 has been established as phloem mobile signal that enhances the nitrate uptake from roots59. The NIN-like protein 8 (NLP8), a transcription factor and positive regulator of nitrate signaling, is essential for nitrate-regulated seed germination in Arabidopsis53. Our physiological data on the sensitivity of seed germination to low nitrate in the double mutant (Fig. 9) further support the involvement of G-protein signaling17 as a regulator of nitrate response. Our molecular evidence on the differential transcript accumulation of the low affinity nitrate transporter/transceptor (NRT1) and nitrate reductase (NR2) in the root tissues of single and double mutants at low N (Fig. S7) confirms the role of GCR1 and GPA1 coupling in nitrate signaling. Further examination of G-protein signaling in N response and NUE is in order, in view of these and earlier studies17,60 in this regard. Hormones control developmental and defense responses by orchestrating cellular pathways. GO and MapMan analyses showed many DEGs associated with hormone biosynthesis as well as signaling (Fig. 4 Table S3). The DEGs involved in auxin and ethylene biosynthesis were overrepresented among other hormonal pathways (Fig. 4). We also detected the auxin-related molecular complexes comprised of indole-3-acetic acid inducible 31 (IAA31), auxin response factor 16 (ARF16) and indole-3-acetic acid inducible 5 (AA5). IAA3 was down-regulated in the double mutant, but how GPA1 and GCR1 coordinate these hormonal responses involving IAA3 is yet to be discovered. The GO terms for response to stimulus and biotic stresses belong to highly enriched biological processes (Fig. 2B). MapMan analyses also highlighted the biotic stress as a major pathways/bin (Fig. 4). Further sub-clustering of PPI networks showed that regulatory protein (NPR1), NPR1-like protein 3 (NPR3), and AHBP-1B (bZIP transcription factor family protein) are involved in the formation of molecular complexes (Fig. S6). NPR1 and NPR3 are salicylic acid receptors and AHBP-1B interacts with these receptors to modulate the expression of PR genes in Arabidopsis42. AHBP-1B was up-regulated in the double mutant, which suggests that combined function of GPA1 and GCR1 modulate plant immunity. Further investigation is needed to understand the mechanism of immune regulation by co-functionality of GPA1 and GCR1 in Arabidopsis.
Conclusions
This is the first comprehensive transcriptome analysis of gpa1-5 gcr1-5 double mutant that goes beyond abiotic stress6, and provides compelling genetic evidence to our earlier findings based on the single mutants16,17 on: a) the role of GCR1 in G-protein signalling and b) the combinatorial involvement of GCR1 and/or GPA1 in regulating different gene sets and c) specific evidence of GCR1-GPA1 coupling in mediating nitrate response. Our analysis reveals the genes/processes identically regulated in both single and double mutants, providing the strongest genetic evidence thus far for GCR1-GPA1 coupling, at least in Arabidopsis. They include cell wall composition/processes, plant immunity, nitrogen signaling and biosynthesis of isoprenoids, stress, development and nutrient transport, among others. PPI network analyses and MCODE sub-clustering led to the identification of seven hub key genes, which are regulated by coupling of GPA1 and GCR1. Our comparative analysis of the mutants also reveal the genes/processes that are affected only by either GPA1 or GCR1 in the single mutants but not in the double mutant, providing a starting point to find their other signaling partners, including, but not limited to other isoforms of GCR/GPA. Most importantly, we identified genes uniquely regulated in the double mutant but not in any of the single mutants, though the processes to which they belong may not be so exclusive.
Methods
Isolation of double mutant
The gpa1-5 gcr1-5 double mutants were obtained by crossing the gcr1-5 mutant16 to gpa1-5 mutant17. First, a number of homozygous gpa1-5 gcr1-5 individuals were identified among the F2 progeny due to their characteristic phenotype i.e. enlarged roundish rosette leaves under the short-day growth conditions. Second, these individuals were subjected to the PCR analyses to test for the absence of the GPA1 and GCR1 gene copies. Predicted gpa1-5 gcr1-5 double mutant individuals were allowed to self-pollinate, and homozygosity for both gene mutations were verified using S2 segregation analyses on drugs (BASTA and Kanamycin).
Growth conditions and phenotypic characterization
Both the mutant and wild type seeds were surface-sterilized using 70% ethanol and washed thrice with autoclaved ultrapure water and stratified at 4 °C for two days on half-strength B5 agar plates. The plates were incubated in a growth chamber at 22 ± 1 °C with a light intensity of 150 μM sec−1 m−2 and a photoperiod of 16:8 (light:dark). Ten days old plants were transferred to 3.5 cm pots containing a mixture of soilrite and vermiculite (1:1), supplemented with full-strength B5 media and regularly watered using sub-irrigation. The plants were used for the measurement of phenotypic characters throughout their life cycle.
RNA isolation and microarray analysis
Total RNA was isolated from 23 days old whole plants as described previously16. RNA samples were analyzed for quality, quantity and suitability for microarray using Nanodrop spectrophotometer and Bioanalyzer (Agilent Technologies, Santa Clara, USA). The same RNA preparations were also used for confirming the knockout mutants using RT-qPCR with gene-specific primers. The Cy3 labelled cRNAs from independent biological duplicates of the wild type (Ws2) and gpa1-5gcr1-5 double mutant were subjected to microarray analysis using Agilent 8 × 60k Arabidopsis arrays (AMADID 037661) as described6. Overall the microarray images were clean, with uniform intensity and very low background noise. The data were extracted using Feature Extraction 10.7 software (Agilent Technologies) and normalized using the recommended ‘Per Chip and Per Gene Normalization’ feature of GeneSpring GX Version 11.5. The correlation coefficients of replicates were obtained by principal component analysis. Log2fold change value of 1.0 and p-value of 0.05 was used as a cut-off for differential-regulation. The Benjamini Hochberg FDR procedure at a cut-off value of p ≤ 0.05 was used for multiple testing corrections. Area-proportional Venn selections were done using the DEG lists in the gpa1-5, gcr1-5 and the double mutants using the online software (http://bioinforx.com/free/bxarrays/venndiagram.php).
Functional classification/meta-analysis of DEGs
The DEGs were assigned gene ontology terms according to the TAIR 10 database61. The DEG lists were subjected to enriched GO categorization using AgriGO2.0 with default settings. The DEGs were mapped into various pathways (bins) using MapMan tool. The coloured boxes in each bin represent the DEGs log2FC values. Further, pathway analysis of the DEGs was done to obtain the list of changed pathways using plant MetGenMAP, which takes AraCyc as the background. Differentially expressed transcription factors were compared with the Plant Transcription Factor Database (plantTFDB ver 2.0).
Data validation using qPCR
A few DEGs were selected from microarray data for its validation based on their roles in different biological processes. The genes were selected in a manner such that at least two up-regulated and two down-regulated genes figured in each of the described biological category. The RT-qPCR was carried out using 1.0 μl of 1:50 diluted cDNA, reverse transcribed form 5 μg of DNase treated RNA. PCR amplifications were performed in 20 μl reactions using the KAPA SYBR® FAST Master Mix (2X) Universal (Kapa Biosystems, USA) with 100 nmoles of each gene-specific primer in Stratagene Mx3000P (Agilent technologies). The amplifications were carried out using biological triplicates, two of which were the same as those used for microarray. Serial dilutions were used to check for primer efficiency and only those primers that worked at 100 ± 10% efficiency were used for all qPCR analyses. The specificity of primer pairs was confirmed by melting curve analysis of the amplicons. Actin2 (ACT2) was used as an internal control for normalization. Quantification of the relative changes in gene expression was performed by the standard curve method.
Construction of PPI networks and sub-clustering analyses
The exclusive DEGs identified in the double mutants were used to retrieve the interactors from STRING (https://string-db.org/) and BioGRID (https://thebiogrid.org/) databases. The experimentally validated interactions were considered to create the PPI networks and DEGs were mapped using Cytoscape version 3.0. Molecular complex detection (MCODE) plugin was used to perform the sub-clustering of the networks and identification of the molecular complexes associated with various pathways.
N-responsive seed germination assay
Seeds of Arabidopsis wild-type (Ws2) and all three mutants (gpa1-5, gcr1-5, gpa1-5gcr1-5) were surface-sterilized using 70% ethanol for 5 minutes and subsequently washed 5 times with ultrapure water. The stratification of seeds was carried out at 4 °C in total darkness for 48 h to facilitate uniform germination. These stratified seeds were placed on 1X B5 agar plates supplemented with different concentrations of KNO3 [optimal nitrate as per standard B5 media composition (25 mM), low nitrate (12.5 mM) and high nitrate (30 mM)]. Plates were transferred to the growth chamber maintained at 22 ± 1 °C with photoperiod (12 h of light/dark period). After 12 h, we examined the seed germination at every 3 h till 72 h.
For qPCR analyses, surface sterilized and stratified seeds of the wild type and all three mutants were grown in B5 medium containing 12.5 and 30 mM KNO3 at 22 °C ± 1 in a growth chamber. Root tissues (~100 mg) were used to extract their total RNA using Trizol (Invitrogen, USA) as described by the manufacturer. DNase I treated total RNAs were transcribed into cDNAs using RevertAid first strand cDNA synthesis kit (Thermo Fisher Scientific). The qPCR reaction was performed using KAPA SYBR FAST Master Mix (2x) Universal (Kapa Biosystems, USA) or Brilliant III Ultra-Fast SYBR Green QPCR Master Mix on Agilent MxPro3000P machine. The comparative C(T) method was used for relative quantitation of the transcript and the expression of the genes was normalized using actin as a reference gene.
Data Availability
GEO accession number GSE 40217 (GSM 988511 and GSM 988512).
References
Lee, Y.-R. J. & Assmann, S. M. Arabidopsis thaliana ‘extra-large GTP-binding protein’ (AtXLG1): a new class of G-protein. Plant Molecular Biology 40, 55–64 (1999).
Ding, L., Pandey, S. & Assmann, S. M. Arabidopsis extra-large G proteins (XLGs) regulate root morphogenesis. Plant J 53, 248–263, https://doi.org/10.1111/j.1365-313X.2007.03335.x (2008).
Urano, D. et al. Saltational evolution of the heterotrimeric G protein signaling mechanisms in the plant kingdom. Sci. Signal. 9, ra93–ra93 (2016).
Jangam, A. P., Pathak, R. R. & Raghuram, N. Microarray analysis of rice d1 (RGA1) mutant reveals the potential role of G-protein alpha subunit in regulating multiple abiotic stresses such as drought, salinity, heat, and cold. Frontiers in Plant Science 7, 11 (2016).
Ali, A., Sivakami, S. & Raghuram, N. Regulation of activity and transcript levels of NR in rice (Oryza sativa): Roles of protein kinase and G-proteins. Plant Science 172, 406–413 (2007).
Chakraborty, N., Singh, N., Kaur, K. & Raghuram, N. G-protein signaling components GCR1 and GPA1 mediate responses to multiple abiotic stresses in Arabidopsis. Frontiers in Plant Science 6, 1000 (2015).
Raghuram, N., Chandok, M. R. & Sopory, S. K. Light regulation of nitrate reductase gene expression in maize involves a G-protein. Molecular Cell Biology Research Communications 2, 86–90 (1999).
Liu, J. et al. Heterotrimeric G proteins serve as a converging point in plant defense signaling activated by multiple receptor-like kinases. Plant Physiology 161, 2146–2158 (2013).
Aranda-Sicilia, M. N. et al. Heterotrimeric G proteins interact with defense-related receptor-like kinases in Arabidopsis. Journal of Plant Physiology 188, 44–48 (2015).
Tunc-Ozdemir, M., Urano, D., Jaiswal, D. K., Clouse, S. D. & Jones, A. M. Direct modulation of a heterotrimeric G protein-coupled signaling by a receptor kinase complex. Journal of Biological Chemistry, jbc C116, 736702 (2016).
Apone, F. et al. The G-protein-coupled receptor GCR1 regulates DNA synthesis through activation of phosphatidylinositol-specific phospholipase C. Plant Physiology 133, 571–579 (2003).
Colucci, G., Apone, F., Alyeshmerni, N., Chalmers, D. & Chrispeels, M. J. GCR1, the putative Arabidopsis G protein-coupled receptor gene is cell cycle-regulated, and its overexpression abolishes seed dormancy and shortens time to flowering. Proceedings of the National Academy of Sciences 99, 4736–4741 (2002).
Chen, J.-G. et al. GCR1 can act independently of heterotrimeric G-protein in response to brassinosteroids and gibberellins in Arabidopsis seed germination. Plant Physiology 135, 907–915 (2004).
Pandey, S. & Assmann, S. M. The Arabidopsis putative G protein–coupled receptor GCR1 interacts with the G protein α subunit GPA1 and regulates abscisic acid signaling. The Plant Cell 16, 1616–1632 (2004).
Warpeha, K. M. et al. G-protein-coupled receptor 1, G-protein Gα-subunit 1, and prephenate dehydratase 1 are required for blue light-induced production of phenylalanine in etiolated Arabidopsis. Plant Physiology 140, 844–855 (2006).
Chakraborty, N. et al. Transcriptome analysis of Arabidopsis GCR1 mutant reveals its roles in stress, hormones, secondary metabolism and phosphate starvation. Plos One 10, e0117819, https://doi.org/10.1371/journal.pone.0117819 (2015).
Chakraborty, N. et al. G-protein α-subunit (GPA1) regulates stress, nitrate and phosphate response, flavonoid biosynthesis, fruit/seed development and substantially shares GCR1 regulation in A. thaliana. Plant Molecular Biology 89, 559–576 (2015).
Johnston, C. A. et al. GTPase acceleration as the rate-limiting step in Arabidopsis G protein-coupled sugar signaling. Proceedings of the National Academy of Sciences 104, 17317–17322 (2007).
Urano, D. et al. G protein activation without a GEF in the plant kingdom. PLoS Genetics 8, e1002756 (2012).
Urano, D. & Jones, A. M. “Round up the usual suspects”: a comment on nonexistent plant G protein-coupled receptors. Plant Physiology 161, 1097–1102 (2013).
Gookin, T. E., Kim, J. & Assmann, S. M. Whole proteome identification of plant candidate G-protein coupled receptors in Arabidopsis, rice, and poplar: computational prediction and in-vivo protein coupling. Genome Biology 9, R120 (2008).
Jones, J. C., Jones, A. M., Temple, B. R. & Dohlman, H. G. Differences in intradomain and interdomain motion confer distinct activation properties to structurally similar Gα proteins. Proceedings of the National Academy of Sciences, 201202943 (2012).
Urano, D. et al. Endocytosis of the seven-transmembrane RGS1 protein activates G-protein-coupled signaling in Arabidopsis. Nature Cell Biology 14, 1079–1088 (2012).
Urano, D., Chen, J.-G., Botella, J. R. & Jones, A. M. Heterotrimeric G protein signalling in the plant kingdom. Open Biology 3, 120186 (2013).
Hackenberg, D. et al. Gα and regulator of G‐protein signaling (RGS) protein pairs maintain functional compatibility and conserved interaction interfaces throughout evolution despite frequent loss of RGS proteins in plants. New Phytologist 216, 562–575 (2017).
Hackenberg, D., Sakayama, H., Nishiyama, T. & Pandey, S. Characterization of the heterotrimeric G-protein complex and its regulator from the green alga Chara braunii expands the evolutionary breadth of plant G-protein signaling. Plant Physiology 163, 1510–1517 (2013).
Tian, T. et al. agriGO v2. 0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Research 45, W122–W129 (2017).
Thimm, O. et al. MAPMAN: a user‐driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant Journal 37, 914–939 (2004).
Zhang, H. et al. PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Research 39, D1114–D1117 (2010).
Ramsay, N. A. & Glover, B. J. MYB–bHLH–WD40 protein complex and the evolution of cellular diversity. Trends in Plant Science 10, 63–70 (2005).
Wu, K.-L., Guo, Z.-J., Wang, H.-H. & Li, J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Research 12, 9–26 (2005).
Dubos, C. et al. MYB transcription factors in Arabidopsis. Trends in Plant Science 15, 573–581 (2010).
Cernac, A. & Benning, C. WRINKLED1 encodes an AP2/EREB domain protein involved in the control of storage compound biosynthesis in Arabidopsis. The Plant Journal 40, 575–585 (2004).
Bacete, L., Mélida, H., Miedes, E. & Molina, A. Plant cell wall‐mediated immunity: cell wall changes trigger disease resistance responses. The Plant Journal 93, 614–636 (2018).
Cheng, Z. et al. Pathogen-secreted proteases activate a novel plant immune pathway. Nature 521, 213 (2015).
Torres, M. A., Morales, J., Sánchez-Rodríguez, C., Molina, A. & Dangl, J. L. Functional interplay between Arabidopsis NADPH oxidases and heterotrimeric G protein. Molecular Plant-Microbe Interactions 26, 686–694 (2013).
Llorente, F., Alonso‐Blanco, C., Sánchez‐Rodriguez, C., Jorda, L. & Molina, A. ERECTA receptor‐like kinase and heterotrimeric G protein from Arabidopsis are required for resistance to the necrotrophic fungus plectosphaerella Cucumerina. The Plant Journal 43, 165–180 (2005).
Colaneri, A. C., Tunc-Ozdemir, M., Huang, J. P. & Jones, A. M. Growth attenuation under saline stress is mediated by the heterotrimeric G protein complex. BMC Plant Biology 14, 129 (2014).
Xu, P., Zang, A., Chen, H. & Cai, W. The Small G Protein AtRAN1 Regulates Vegetative Growth and Stress Tolerance in Arabidopsis thaliana. Plos One 11, e0154787 (2016).
Lee, C., Ahn, J. & Choi, Y. The G-protein alpha-subunit gene CGA1 is involved in regulation of resistance to heat and osmotic stress in Chlamydomonas reinhardtii. Cell & Molecular Biology (Noisy le Grand) 63 (2017).
Okamoto, H. et al. The α-subunit of the heterotrimeric G-protein affects jasmonate responses in Arabidopsis thaliana. Journal of Experimental Botany, erp060 (2009).
Zhao, Z., Stanley, B. A., Zhang, W. & Assmann, S. M. ABA-regulated G protein signaling in Arabidopsis guard cells: a proteomic perspective. Journal of Proteome Research 9, 1637–1647 (2010).
Wang, R.-S. et al. Common and unique elements of the ABA-regulated transcriptome of Arabidopsis guard cells. BMC Genomics 12, 1 (2011).
Steffens, B. & Sauter, M. G proteins as regulators in ethylene-mediated hypoxia signaling. Plant Signaling & Behavior 5, 375–378 (2010).
Ge, X. M. et al. Heterotrimeric G protein mediates ethylene‐induced stomatal closure via hydrogen peroxide synthesis in Arabidopsis. The Plant Journal 82, 138–150 (2015).
Pandey, S. et al. Boolean modeling of transcriptome data reveals novel modes of heterotrimeric G‐protein action. Molecular Systems Biology 6 (2010).
Klopffleisch, K. et al. Arabidopsis G‐protein interactome reveals connections to cell wall carbohydrates and morphogenesis. Molecular Systems Biology 7, 532 (2011).
Jones, A. M. et al. Border control—a membrane-linked interactome of Arabidopsis. Science 344, 711–716 (2014).
Liang, Y., Gao, Y. & Jones, A. M. Extra large G-protein interactome reveals multiple stress response function and partner-dependent XLG subcellular localization. Frontiers in Plant Science 8, 1015 (2017).
Jaiswal, D. K., Werth, E. G., McConnell, E. W., Hicks, L. M. & Jones, A. M. Time-dependent, glucose-regulated Arabidopsis Regulator of G-protein Signaling 1 network. Current Plant Biology 5, 25–35 (2016).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research 13, 2498–2504 (2003).
Sharma, N. et al. Phenotyping for nitrogen use efficiency (NUE) I: Rice genotypes differ in N-responsive germination, oxygen consumption, seed urease activities, root growth, crop duration and yield at low N. Frontiers in Plant Science 9, 1452 (2018).
Yan, D. et al. NIN-like protein 8 is a master regulator of nitrate-promoted seed germination in Arabidopsis. Nature Communications 7, 13179 (2016).
Osuna, D., Prieto, P. & Aguilar, M. Control of Seed Germination and Plant Development by Carbon and Nitrogen Availability. Frontiers in Plant Science 6, 1023 (2015).
Chakravorty, D., Gookin, T. E., Milner, M., Yu, Y. & Assmann, S. M. Extra-large G proteins (XLGs) expand the repertoire of subunits in Arabidopsis heterotrimeric G protein signaling. Plant Physiology 169(1), 512–529 (2015).
Chakravorty, D. et al. An atypical heterotrimeric G‐protein γ‐subunit is involved in guard cell K+-channel regulation and morphological development in Arabidopsis thaliana. The Plant Journal 67, 840–851 (2011).
Jang, I.-C., Henriques, R. & Chua, N.-H. Three transcription factors, HFR1, LAF1 and HY5, regulate largely independent signaling pathways downstream of phytochrome A. Plant and Cell Physiology 54, 907–916 (2013).
Yuan, S. et al. Arabidopsis cryptochrome 1 functions in nitrogen regulation of flowering. Proceedings of the National Academy of Sciences 113, 7661–7666 (2016).
Chen, X. et al. Shoot-to-Root Mobile Transcription Factor HY5 Coordinates Plant Carbon and Nitrogen Acquisition. Current Biology 26, 640–646 (2016).
Sun, H. et al. Heterotrimeric G proteins regulate nitrogen-use efficiency in rice. Nature Genetics 46, 652–656 (2014).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Research 40, D1202–1210, https://doi.org/10.1093/nar/gkr1090 (2012).
Acknowledgements
This work was supported by research grant to NR[38(1246)/10/EMRII] and research fellowships to NC(09/806(015)/2008-EMRI) from the Council of Scientific and Industrial Research (CSIR), Government of India, whereas DKJ and NG were supported by fellowship from the Indo-UK Virtual Nitrogen Centre on Nitrogen Efficiency of Whole-cropping Systems (NEWS) BT/IN/UK-VNC/44/NR/2015-16. AM thanks the GGS Indraprastha University for her STRF fellowship and the help of Ashu Tyagi in growing the plants for RT-qPCR analyses for the revised manuscript. KK would like to acknowledge funding by the Biotechnology and Biological Sciences Research Council (BBSRC) of the UK through the Designing Future Wheat (DFW) Institute Strategic Programme (BB/P016855/1).
Author information
Authors and Affiliations
Contributions
N.C. performed the mutant phenotypic analysis, RNA isolation, microarray and RT-qPCR experiments, analyzed the data and wrote the initial draft of the manuscript; K.K. generated and back-crossed the mutants; R.H. conceived, planned and led the mutant isolation and multiplication; D.K.J. added to the GO and network analysis, helped in manuscript editing and revision, N.G. contributed to further data analysis, A.K. and V.A. performed the germination experiments and RT-qPCR of NRT1 and NR2, while A.M. validated the expression of cell wall associated DEGs by RT-qPCR. N.R. conceived, planned and supervised the transcriptome analysis and data interpretation, as well as edited, revised and finalized the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakraborty, N., Kanyuka, K., Jaiswal, D.K. et al. GCR1 and GPA1 coupling regulates nitrate, cell wall, immunity and light responses in Arabidopsis. Sci Rep 9, 5838 (2019). https://doi.org/10.1038/s41598-019-42084-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42084-2
This article is cited by
-
Life, death and resurrection of plant GPCRs
Plant Molecular Biology (2023)
-
Nitrate-responsive transcriptome analysis of rice RGA1 mutant reveals the role of G-protein alpha subunit in negative regulation of nitrogen-sensitivity and use efficiency
Plant Cell Reports (2023)
-
An emerging role of heterotrimeric G-proteins in nodulation and nitrogen sensing
Planta (2023)
-
Heterotrimeric G-protein α subunit (RGA1) regulates tiller development, yield, cell wall, nitrogen response and biotic stress in rice
Scientific Reports (2021)
-
Transcriptomic and network analyses reveal distinct nitrate responses in light and dark in rice leaves (Oryza sativa Indica var. Panvel1)
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
