
Abstract
Gliomas are heterogeneous, primary brain tumours that originate from glial cells. The main type of gliomas is astrocytomas. There are four grades (I-IV) of astrocytoma malignancy. Astrocytoma grade IV known as glioblastoma multiforme (GBM) is the most common and aggressive type of astrocytic gliomas. Metallothioneins (MT) are low molecular weight, cysteine rich proteins encoded by a family of metallothionein (MT) genes. MT genes play a crucial role in carcinogenesis of diverse malignancies. We proposed MT genes as prognostic markers for malignant astrocytoma. MT1A, MT1E, MT1X, MT2, MT3 gene expression was elevated in grade IV astrocytomas (glioblastomas) as compared to astrocytomas grade I-III. Statistically significant differences were reached for MT1A and MT2 genes (Mann-Whitney test, p < 0.05). High MT1A, MT1X, MT2, MT3 genes expression was associated with shorter patient survival (Log-rank test, p < 0.05). MT1A gene promoter methylation was decreased in glioblastoma (57.6%) while the gene was highly methylated in grade II-III astrocytoma (from 66.7% to 83.3%) and associated with better patient survival (p < 0.05). MT1A gene methylation showed a trend of being associated with higher mRNA expression level in astrocytomas. Increased MT genes expression in grade IV astrocytomas as compared to I-III grade astrocytomas could be associated with malignant tumour behaviour and progression.
Similar content being viewed by others

Introduction
Gliomas are one of the most common groups of heterogeneous, neuroepithelial primary tumours of the central nervous system (CNS) that arise from glial cells1,2. There are several types of glioma that are named according to the cell type they originate from. One of the most common gliomas is astrocytomas3. Since 2016 the World Health Organization (WHO) has been classifying astrocytic gliomas into four grades of malignancy (grade I-IV astrocytoma) according to histological features such as necrosis, microvascular proliferation, mitoses, abnormal cells and also molecular features such as IDH mutation, 1p/19q deletion or ATRX mutation in addition to histology2,4. The most aggressive type of astrocytoma is grade IV astrocytoma or glioblastoma (GBM). GBM is the most lethal, aggressive type of CNS tumours. GBM is characterized by histological and molecular heterogeneity. Also, GBM is highly invasive and vascularized brain tumour resistant to treatment. The median patient survival time is about 15 months and less than 5% survive for 5 years5. The development of gliomas is associated with genetic and epigenetic alterations that result in the repression of tumour suppressor genes or the activation of oncogenes in the normal brain cells6,7.
Metallothioneins (MT) are the group of low molecular weight, metal binding proteins that are prevalent in many organisms8. There are 16 isoforms of MTs that are divided into 4 classes – MT1 – MT49. MT proteins are encoded by metallothionein (MT) gene family that is located on chromosome 16q138,9. MT2 – MT4 proteins are encoded by a single gene while MT1 is encoded by a set of 13 genes (MT1A, -1B, -1C, -1D, -1E, -1F, -1G, -1I, -1J, -1L, -1M, -1X)8. MTs are involved in tumour biology such as proliferation, differentiation, apoptosis, and angiogenesis10. Also, MT gene expression alterations are associated with carcinogenesis and with aggressive tumour behaviour and even chemotherapy resistance11. However, the expression of MT genes is not specific to any particular cancer type12. For example, MT3 gene has been determined as specific to brain tissue but also it is expressed in kidney, renal carcinoma and bladder cancer12,13,14. Despite this, the importance of MT3 expression changes in tumour is not understood yet. The expression level may depend on the differentiation of tumour, proliferation or gene mutations, for example, p5312. For instance, MT gene expression was associated with tumour grade and cell proliferation rate in germ cell carcinoma and breast cancer12,15. There is not enough information about MT gene expression changes in different grade gliomas. It is known that MT gene expression in glioblastoma tumour samples was related to shorter patients survival16. Thus, it is important to clarify the importance of MTs expression level change during malignant progression. The purpose of this study was to determine MT genes mRNA expression level in different grade glioma tumours and to disclose gene expression associated promoter methylation in gliomas.
Results
Associations of MT gene level and patient clinical characteristics
For the association analysis between mRNA expression and patient clinical characteristics, the mRNA expression level was assigned to “low expression” and “high expression” groups according to the median value. The mRNA expression below the median was designated as low expression and values above the median as high expression. First, we analyzed the association between MT mRNA expression groups and patients gender. There were no significant associations for MT1E, MT1X genes while MT1A, MT2, MT3 genes showed statistically significant expression differences in males and females (Chi-square test, p < 0.05). Low mRNA expression for MT1A, MT2, and MT3 genes was determined more frequently for man as compared to woman (39.2% vs 11.8%, 41.8% vs. 9.1%, 39.2% vs. 19.6% respectively) (Chi-square test, p < 0.05). Analysis of MT expression in patient age groups have shown that higher MT1A, MT2 and MT3 genes expression level was significantly associated with older patient age (>50 years) (Chi-square test, p < 0.05) (data not shown).
MT1A, MT1E, MT1X, MT2, MT3 expression increases in glioblastoma
The analysis of MT1A, MT1E, MT1X, MT2, MT3 gene mRNA expression was determined in n = 51, n = 53, n = 49, n = 55, n = 51 glioma patients respectively. First, we analysed MT genes mRNA expression between astrocytic gliomas – grade I-III astrocytoma and grade IV astrocytoma, or glioblastoma. Analysis showed the trend for increasing MT1A, MT1E, MT1X, MT2 and MT3 gene expression in higher malignancy tumours, e.g. glioblastomas as compared to I-III grade astrocytomas (Figs 1a,c,e, 2a,c). However, only MT1A and MT2 expression reached a statistically significant level (Mann-Whitney test, p < 0.05), while MT1E and MT3 genes showed the tendency of higher expression in glioblastomas (Mann-Whitney test, p = 0.120, p = 0.058 respectively). It is interesting to notice, that grade I-III astrocytoma group consisted of one gemistocytic astrocytoma sample for which individual data dots plotted for MT1A, MT1E, MT2, and MT3 genes in mRNA expression plots were the largest observations reaching the upper whiskers value which corresponded to the 3-rd quartile of gene expression value in glioblastoma, respectively (Figs 1a,c, 2a,c, sample log2 (2−ddCt) value marked as a red dot). Gemistocytic tumour is a rare variant of astrocytoma grade II with the tendency quickly develop to anaplastic astrocytoma or glioblastoma with poor prognosis17,18.

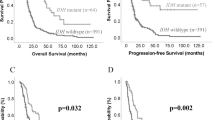
MT1A, MT1E and MT1X gene mRNA expression in different malignancy astrocytomas and expression effect on survival. (a) MT1A gene expression level in grade I-III astrocytoma and glioblastoma (GBM) (Mann-Whitney test, **p < 0.005). (b) Kaplan-Meier overall survival curves according to low and high MT1A gene expression groups (log-rank test: χ2 = 6.400, df = 1, p = 0.011). (c) MT1E gene expression level in grade I-III astrocytoma and glioblastoma (GBM) (Mann-Whitney test, p > 0.05). (d) Kaplan-Meier overall survival curves according to low and high MT1E gene expression groups (log-rank test: χ2 = 3.410, df = 1, p = 0.065). (e) MT1X gene expression level in grade I-III astrocytoma and glioblastoma (GBM) (Mann-Whitney test, p > 0.05). (f) Kaplan-Meier overall survival curves according to low and high MT1X gene expression groups (log-rank test: χ2 = 4.672, df = 1, p = 0.031). The middle line on the scatter plot graph represents the median value, whiskers – the lowest and the highest values. Graph symbols represent different astrocytoma grade: grade I astrocytoma – grey rhombus, grade II astrocytoma – black round, gemistocytic astrocytoma - red dot, grade III astrocytoma – grey triangle, GBM - black rectangular. Gene expression level presented as log2(2−ddCt).

MT2 and MT3 gene mRNA expression in different malignancy astrocytomas and Kaplan-Meier survival curves. (a) MT2 gene expression level in grade I-III astrocytoma and glioblastoma (GBM) (Mann-Whitney test, *p < 0.050). (b) Kaplan-Meier overall survival curves according to low and high MT2 gene expression groups (log-rank test: χ2 = 8.542, df = 1, p = 0.003). (c) MT3 gene expression level in grade I-III astrocytoma and glioblastoma (GBM) (Mann-Whitney test, p > 0.05). (d) Kaplan-Meier overall survival curves according to low and high MT3 gene expression groups (log-rank test: χ2 = 5.694, df = 1, p = 0.017). The middle line on the scatter plot graph represents the median value, whiskers – the lowest and the highest values. Graphs symbols represent different astrocytoma grade: grade I astrocytoma – grey rhombus, grade II astrocytoma – black round, gemistocytic astrocytoma - red dot, grade III astrocytoma – grey triangle, GBM - black rectangular. Gene expression level presented as log2(2−ddCt).
Next, gene expression data were grouped into “low” and “high” gene expression groups according to median values, and further MT gene mRNA expression associated with tumour malignancy grade was analysed. It was shown that higher percentage of glioblastomas fall into MT high expression group (from 37.7% for MT1E to 43.6% for MT2) as compared to grade I-III astrocytomas (from 5.5% for MT2 to 13.2% for MT1E). However, only the MT2 gene reached statistically significant difference (Chi-square test, p < 0.05).
MT1A gene methylation decreases in glioblastoma
The analysis of MT1A gene promoter methylation using MS-PCR was determined in 50 out of 55 of the glioma patient samples used for gene expression analysis. The methylation status was estimated according to the signal appearing on a gel. In the case of methylated or both signals, the gene was designated as methylated (Fig. 3a). Our analysis showed that the MT1A gene was methylated in 62% (31/50) of gliomas. The highest methylation frequency was determined in grade II-III astrocytoma (from 66.7% to 83.3%) and the lowest – in glioblastoma (57.6%). The association analysis between MT1A gene methylation status and patients gender showed no statistically significant differences (Chi-square test, p > 0.05) (data not shown). We looked for MT1A gene promoter methylation associated with gene expression. The association analysis between MT1A gene mRNA expression and gene promoter methylation status was not statistically significant (Chi-square test, p > 0.05) (Fig. 3c).

Methylation analysis of the MT1A gene. (a) Representative electrophoregram of MT1A gene MS-PCR. L-molecular weight marker, M- primer for methylated DNA, U-primer for unmethylated DNA, 1- methylated DNA control, 2- unmethylated DNA control, 3 - water control, TI-TIV- tumour samples. TI-TII samples estimated as methylated gene promoter while TIII, TIV as unmethylated gene promoter. (b) Kaplan-Meier overall survival curves according to the methylation status of MT1A gene. M – methylated, U – unmethylated. Unmethylated gene promoter is associated with shorter patients survival (log-rank test: χ2 = 4.948, df = 1, p = 0.026). (c) mRNA expression of MT1A gene in methylated (M) and unmethylated (U) promoter status groups (Mann-Whitney test, p > 0.05). The middle line on the graph represents a median value, whiskers – the lowest and the highest values.
MTs affects on patient survival
For Kaplan-Meier analysis, MT gene’s “low” and “high” expression groups were utilised to compare survival curves. Significant relation was shown between MT1A, MT1X, MT2 and MT3 genes and survival time when high gene expression was associated with poor patients survival (log-rank test for MT1A: χ2 = 6.400, df = 1, p = 0.011; MT1X: χ2 = 4.672, df = 1, p = 0.031; MT2: χ2 = 8.542, df = 1, p = 0.003; MT3: χ2 = 5.694, df = 1, p = 0.017). MT1E higher gene expression showed the tendency to be associated with worse patient outcome (log-rank test: χ2 = 3.410, df = 1, p = 0.065) (Figs 1b,d,f, 2b,d). Median survival time within high gene expression group was 11.3 months for MT1A, MT1X genes, 11.5 months for MT1E, MT2 and MT3 genes, while in low expression group 21.6 months for MT1A, MT1E genes, 25.9 months for MT1X, 28.98 months for MT2 and MT3 genes. Although, Kaplan-Meier analysis for survival curves in patient groups having tumours with methylated and unmethylated MT1A gene was performed. The statistical analysis showed significant survival difference when unmethylated gene promoter was associated with poor patients survival (log-rank test: χ2 = 4.948, df = 1, p = 0.026) (Fig. 3b). Median survival time within methylated gene promoter patient group was 22.47 months while within unmethylated 11.27 months.
Discussion
In this study, we performed a thorough analysis of metallothionein gene activity in I to IV grade astrocytomas. Here we checked MT1A, MT1E, MT1X, MT2, MT3 gene expression level and epigenetic regulation for one of the highly expressed metallothioneins, MT1A, in different grade gliomas and looked for clinical significance of MT gene activity in glioma tissue in terms of patient survival. We distributed our study into three steps – determination of MT genes expression in I-IV grade glioma, thorough analysis of MT1A gene determining expression regulation through promoter methylation and clinical importance of MT gene activity.
We analysed mRNA expression level and methylation status association with patient gender and age. Analysis for MT1A, MT2, MT3 showed associations with female gender and patient age older than 50 years. We assume, that those gender associations could be due to unequal gender distribution between different malignancy groups while patients within glioblastoma group gathered in a study period comprised mainly women. Likewise, we determined that the methylated MT1A gene promoter was associated with younger patient age (37.1% vs. 27.4%, respectively). To our knowledge, there are no reports about MT promoter methylation associations with age. As glioblastoma develop more frequently in older patients, we simulate that unmethylated promoter was more frequently observed in glioblastomas as compared to I-III astrocytomas due to the patient age distribution differences.
Next, MT gene mRNA analysis showed an association between higher gene expression value and more aggressive tumour type, in particular, GBM. Furthermore, survival analysis revealed poor prognosis for patients having high gene expression. Our results are in accordance with Shai-Mehrian et al. which analysed 67 glioblastoma samples and determined MT gene expression, including MT1A, MT1E, MT3 increase in glioblastoma and association with poor patients survival16. Up-regulation of MT genes are also reported for other types of cancer, for example, breast, renal or bladder15,19,20. According to obtained results, increased MT expression is closely associated with higher malignancy grade of astrocytic gliomas. Expression of metallothionein genes is inducible by a number of cell harmful agents such as heavy metals, as well as oxidative stress8,21. Our results suggest that MT1A and MT2 genes could be used as markers for malignancy of astrocytic gliomas. While, MT1A, MT1X, MT2, MT3 genes could be used to prognosticate a patient’s survival.
Epigenetic dysregulation is associated with various tumours, including gliomas22. One of the proposed MT genes expression regulation mechanisms is epigenetic9. MT genes play a crucial role in many solid tumours and the importance of epigenetic modifications of MT genes should be more analysed. Moreover, it is known that hypermethylation of specific CpG regions in MT genes could be used as a prognostic and diagnostic marker for some tumours, for example, MT1M and MT1G in hepatocellular carcinoma (HCC)23. Also, it is suggested that methylation of MT1 gene cluster (MT1F, MT1M) is associated with the gene silencing and oncogenic process in breast cancer24. In our study, methylation analysis of MT1A gene revealed gene promoter methylation in 57.6% of glioblastoma while methylation in grade III astrocytoma reached 83.3%. These findings suggest that MT1A gene could act as an oncogene because of MT1A gene up-regulation and progressively apparent unmethylation in glioblastoma. This is the opposite to Yu et al. who performed CpG island methylation analysis for MT1A gene using conventional PCR in astrocytic gliomas. The Yu group determined no statistically significant MT1A gene methylation differences between I-IV grade astrocytoma, while a slight increase in methylation abundancy was detected in glioblastoma (21.4%, 33%, 25.1% and 41.6% for I-IV grades respectively)21. These discrepancies suggest that the implication of MT gene hypermethylation in astrocytic glioma pathology deserves further analysis.
In conclusion, this study is one of the most detailed analyses of MT genes expression in different grade glioma. Our study suggests that MT genes expression is associated with tumour malignancy grade and higher gene expression level is more expected in grade IV astrocytoma (glioblastoma). Furthermore, MT1A gene expression and methylation analysis showed that shorter patient survival is strongly associated with unmethylated and highly expressed gene.
Methods
The subject
There were investigated 55 post-operative tissues of patients with different grade gliomas: pilocytic astrocytoma, grade I (n = 2), diffuse astrocytoma, grade II (n = 8), gemistocytic astrocytoma, grade II (n = 1), anaplastic astrocytoma, grade III (n = 6), glioblastoma (GBM), grade IV (n = 38). All the tissues were surgically resected and gathered at the Department of Neurosurgery of the Lithuanian University of Health Sciences, Kaunas Clinics, Lithuania, from 2014 to 2016. The patient samples were diagnosed according to the histopathology test results and WHO classification at the Department of the Pathological Anatomy of the Lithuanian University of Health Sciences. All patients underwent standard treatment, comprised of resection followed by radiotherapy plus concurrent temozolomide (TMZ) chemotherapy. All study methodologies were performed in accordance with the regulations and guidelines and were approved by the Regional Bioethics Committee of the Lithuanian University of Health Sciences, Kaunas, Lithuania (No. P2-9/2003). All patients gave their written informed consent before surgery.
For the MT gene expression and methylation analysis in total 55 samples were analysed as follows: I-III malignancy grade astrocytic gliomas (n = 17) and GBM (n = 38). The grade I-III astrocytic gliomas group comprised of 88.2% (n = 15) men and 11.8% (n = 2) women. The patient age ranged from 21 to 63 yr and the median age was 32 yr. The median survival time was 27 months and ranged from 10.3 to 64.2 months. The GBM group comprised of 47.4% (n = 18) men and 52.6% (n = 20) women. The age of GBM patients ranged from 36 to 82 yr and the median age was 61 yr. The median survival time was 11.8 months and ranged from 1.2 to 34.6 months.
Quantitative real-time polymerase chain reaction (qRT-PCR) with SYBR Green Dye
Total RNA was extracted from 50–100 mg of tumourous tissue with “mirVana miRNA Isolation Kit” (ThermoFisher Scientific, USA). cDNA synthesis was made of 2 μg of total RNA with “High-Capacity cDNA Reverse Transcription Kit“ (Applied Biosystems, USA). The mRNA expression level of MT genes (MT1A, MT1E, MT1X, MT2, MT3) and β-actin as reference gene were detected with qRT-PCR with SYBR Green dye using real-time PCR system “AB 7500 Fast” (Applied Biosystems, USA). The reaction was performed in 12 µl of the total volume of mixture which included 6 µl Maxima SYBR Green/ROX qPCR Master Mix (2x) (ThermoFisher Scientific, USA), 15 ng cDNA, water, nuclease-free and gene primers, with the concentration of 0.2 µM for MT1A, MT1E, MT1X and 0.1 µM for MT2, MT3, and β-actin. MT gene-specific primers were used as follows: 5′-CTTGGGATCTCCAACCTCAC-3′ (forward), 5′-AGGAGCAGCAGCTCTTCTTG-3′ (reverse) for MT1A gene; 5′-GGGCTCCATTCTGCTTTCCA-3′ (forward), 5′-TTGGGGTCCATTTCGAGCAA-3′ (reverse) for MT1E gene; 5′-CTGCTTCTCCTTGCCTCGAA-3′ (forward), 5′-TGTCTGACGTCCCTTTGCAG-3′ (reverse) for MT1X gene; 5′-ATCCCAACTGCTCCTGCGCCG-3′ (forward), 5′-CAGCAGCTGCACTTGTCCGACG-3′ (reverse) for MT2 gene; 5′-CTGAGACCTGCCCCTGCCCTT-3′ (forward), 5′-TGCTTCTGCCTCAGCTGCCTCT-3′ (reverse) for MT325. MT1A gene expression primers were designed using Primer 326. As an endogenous control β-actin gene was used with the primer sequences published27: 5′-AGAGCTACGAGCTGCCTGAC-3′ (forward), 5′-AGCACTGTGTTGGCGTACAG-3′ (reverse). The amplification cycling conditions included: the initial denaturating at 95 °C for 10 min, 40 cycles at 95 °C for 15 s, annealing at 60–68 °C (for MT1A, MT1E, β-actin 60 °C, for MT1X 66 °C, for MT2, MT3 68 °C) for 30 s, 72 °C for 30 s and the final stage for melting curve at 95 °C for 15 s, 60 °C for 1 min, 95 °C for 15 s, 60 °C for 15 s. Each tumour sample was analysed in triplicates. MT gene expression was normalized to healthy human brain RNA samples (RHB) “Total RNA-Human Adult Normal Tissue 5 donor Pool: Brain“ (BioChain) using 2−ΔΔCt method where ΔΔCt = ΔCttumour tissue (CtMT − Ctβ-actin)− ΔCtRHB (CtMT − Ctβ-actin). For the graphical representation, the log2 scale was used.
Methylation-specific polymerase chain reaction (MS-PCR)
For the MT1A gene methylation analysis DNA was extracted from 50–100 mg of tumourous tissue samples by salting-out method using 10% SDS (Invitrogen, USA), proteinase K (ThermoFisher Scientific, Germany), followed by Trichlormetan/Chloroform (Carl Roth® GmbH, Germany) and 6 M sodium chloride (Carl Roth® GmbH, Germany) extraction and DNA precipitation with 96% ethanol. A total of 400 ng of extracted DNA was treated with sodium bisulfite using “EpiJET Bisulfite Conversion Kit“ (ThermoFisher Scientific, USA) according to the manufacturer’s protocol. The methylation status of the MT1A gene was determined by MS-PCR. The pair of primers were designed using “MethPrimer“ tool28. The primers for methylated MT1A allele 5′-TTTACGTTTTGTATTACGTCGATTC-3′ (forward) and 5′-CAACCTTAACGTTTCGATCG-3′ (reverse), PCR amplicon size 127 bp and for unmethylated allele 5′-TATGTTTTGTATTATGTTGATTTGG-3′ (forward) and 5′-CCAACCTTAACATTTCAATCACC-3′ (reverse), PCR amplicon size 126 bp (Metabion International AG, Germany). The reaction was performed in 15 µl of the total volume which comprised of 7.5 µl Maxima Hot Start Green PCR Master Mix (2x) (ThermoFisher Scientific, USA), 1 µM of each primer, 1 µl bisulfite treated DNA and nuclease-free water. The cycling conditions included: the initial denaturation at 95 °C for 5 min, 38 cycles at 95 °C for 15 s, 60 °C for 30 s, 72 °C for 30 s and 72 °C for 5 min. The reaction was performed with “Veriti 96 Well Thermal Cycler” (Applied Biosystems, Singapore). After PCR, the amplicons were separated on 2% agarose gel and visualized under UV gel imaging system (GelDoc XR System, BioRad, USA).
Statistical analysis
The statistical analyses were performed by SPSS 22 and GraphPad Software “Prism 5” programmes. Nonparametric Mann-Witney test was used to compare continuous variables in two different variable groups (tumour grade groups, promoter methylation groups). The Chi-square test was used to analyse the association between mRNA expression, methylation status, and patients clinical characteristics. Kaplan-Meier analysis (Log-rank test) was used to determine the differences between patient survival curves in different gene expression or gene methylation groups. The p < 0.05 value was considered statistically significant.
Data Availability
The datasets generated and analysed in the current study are available under request from the corresponding author.
References
Delgado-Lopez, P. D., Corrales-Garcia, E. M., Martino, J., Lastra-Aras, E. & Duenas-Polo, M. T. Diffuse low-grade glioma: a review on the new molecular classification, natural history and current management strategies. Clin. Transl. Oncol. 19, 931–944 (2017).
Alcantara Llaguno, S. R. & Parada, L. F. Cell of origin of glioma: biological and clinical implications. British Journal of Cancer 115, 1445–1450 (2016).
Adamson, D. C., Rasheed, B. A. K., McLendon, R. E. & Bigner, D. D. Central nervous system. Cancer Biomark. 9, 193–210 (2010).
Toro-Moreno, A. C. et al. Tumores de sistema nervioso central en pediatría: Presente y futuro del abordaje diagnóstico. Rev. Ecuatoriana Neurol. 26, 283–288 (2017).
Lee, D. H., Ryu, H.-W., Won, H.-R. & Kwon, S. H. Advances in epigenetic glioblastoma therapy. Oncotarget 8, 18577–18589 (2017).
Nagarajan, R. P. & Costello, J. F. Molecular epigenetics and genetics in neuro-oncology. Neurotherapeutics 6, 436–446 (2009).
Boudreau, C. R., Yang, I. & Liau, L. M. Gliomas: advances in molecular analysis and characterization. Surg. Neurol. 64(discussion 294), 286–94 (2005).
Ruttkay-Nedecky, B. et al. The Role of Metallothionein in Oxidative Stress. International Journal of Molecular Sciences 14, 6044–6066 (2013).
Kimura, T. & Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. International Journal of Molecular Sciences 17 (2016).
Arriaga, J. M., Bravo, A. I., Mordoh, J. & Bianchini, M. Metallothionein 1G promotes the differentiation of HT-29 human colorectal cancer cells. Oncology Reports 37, 2633–2651 (2017).
Krizkova, S. et al. An insight into the complex roles of metallothioneins in malignant diseases with emphasis on (sub)isoforms/isoforms and epigenetics phenomena. Pharmacol. Ther. 183, 90–117 (2018).
Cherian, M. G., Jayasurya, A. & Bay, B.-H. Metallothioneins in human tumors and potential roles in carcinogenesis. Mutat. Res. 533, 201–209 (2003).
Ghazi, I. E., Martin, B. L. & Armitage, I. M. Metallothionein-3 Is a Component of a Multiprotein Complex in the Mouse Brain. Exp. Biol. Med. 231, 1500–1506 (2006).
Sens, M. A. et al. Metallothionein isoform 3 as a potential biomarker for human bladder cancer. Environmental Health Perspectives 108, 413–418 (2000).
Jin, R. et al. Metallothionein 2A expression is associated with cell proliferation in breast cancer. Carcinogenesis 23, 81–86 (2002).
Mehrian-Shai, R. et al. High metallothionein predicts poor survival in glioblastoma multiforme. BMC Medical Genomics 8 (2015).
Avninder, S. et al. Gemistocytic astrocytomas: histomorphology, proliferative potential and genetic alterations–a study of 32 cases. J. Neurooncol. 78, 123–127 (2006).
Yang, H. J. et al. The significance of gemistocytes in astrocytoma. Acta Neurochir. (Wien). 145(discussion 1103), 1097–103 (2003).
Somji, S., Sens, M. A., Lamm, D. L., Garrett, S. H. & Sens, D. A. Metallothionein isoform 1 and 2 gene expression in the human bladder: evidence for upregulation of MT-1X mRNA in bladder cancer. Cancer Detect. Prev. 25, 62–75 (2001).
Pal, D., Sharma, U., Singh, S. K., Mandal, A. K. & Prasad, R. Metallothionein gene expression in renal cell carcinoma. Indian Journal of Urology: IJU: Journal of the Urological Society of India 30, 241–244 (2014).
Yu, J. et al. Methylation profiles of thirty four promoter-CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer 4, 65 (2004).
Kondo, Y., Katsushima, K., Ohka, F., Natsume, A. & Shinjo, K. Epigenetic dysregulation in glioma. Cancer Science 105, 363–369 (2014).
Ji, X.-F. et al. MT1M and MT1G promoter methylation as biomarkers for hepatocellular carcinoma. World Journal of Gastroenterology: WJG 20, 4723–4729 (2014).
Jadhav, R. R. et al. Genome-wide DNA methylation analysis reveals estrogen-mediated epigenetic repression of metallothionein-1 gene cluster in breast cancer. Clinical Epigenetics 7 (2015).
Han, Y.-C. et al. Metallothionein 1 h tumour suppressor activity in prostate cancer is mediated by euchromatin methyltransferase 1. The Journal of pathology 230, 184–193 (2013).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Research 40, e115 (2012).
Skiriutė, D. et al. Glioma Malignancy-Dependent NDRG2 Gene Methylation and Downregulation Correlates with Poor Patient Outcome. Journal of Cancer 5, 446–456 (2014).
Li, L.-C. & Dahiya, R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18, 1427–1431 (2002).
Acknowledgements
This work was supported partially by a grant (No. MIP-052/2015) from the Research Council of Lithuania and the Medical Faculty Science Fund of Lithuanian University of Health Sciences. We kindly thank Ms. Jūratė Žeglienė for tumour sample and patient data collection.
Author information
Authors and Affiliations
Contributions
D.S. generated an idea, conception, and design of the study, B.M. designed qPCR primers, B.M. & I.V. performed MTs gene expression analysis. B.M. performed MSP assay, data analysis and wrote a manuscript draft. D.S. & A.T. contributed to data interpretation and final approval of the written manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Masiulionytė, B., Valiulytė, I., Tamašauskas, A. et al. Metallothionein Genes are Highly Expressed in Malignant Astrocytomas and Associated with Patient Survival. Sci Rep 9, 5406 (2019). https://doi.org/10.1038/s41598-019-41974-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41974-9
This article is cited by
-
Coexpression network analysis identified MT3 as a hub gene that promotes the chemoresistance of oral cancer by regulating the expression of YAP1
BMC Oral Health (2023)
-
Differences in stromal component of chordoma are associated with contrast enhancement in MRI and differential gene expression in RNA sequencing
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
