
Abstract
The pandemic 2009 influenza A H1N1 virus is associated with significant mortality. Targeting S1PR1, which is known to modulate the immune response, provides protection against pathogenic influenza virus. The functional role and molecular mechanism of S1PR1 were analysed by generating inducible endothelial cell-specific S1PR1 knockout mice and assessing the therapeutic efficacy of the selective S1PR1 agonist CYM5442 against acute lung injury (ALI) induced by the 2009 influenza A H1N1 virus. Immune-mediated pulmonary injury is aggravated by the absence of endothelial S1PR1 and alleviated by treatment with CYM-5442, suggesting a protective function of S1PR1 signaling during H1N1 infection. S1PR1 signaling does not affect viral clearance in mice infected with influenza. Mechanistically, the MAPK and NF-kB signaling pathways are involved in the ALI mediated by S1PR1 in infected mice. Combined administration of the S1PR1 agonist CYM-5442 and the antiviral drug oseltamivir provides maximum protection from ALI. Our current study provides insight into the molecular mechanism of S1PR1 mediating the ALI induced by H1N1 infection and indicates that the combination of S1PR1 agonist with antiviral drug could potentially be used as a therapeutic remedy for future H1N1 virus pandemics.
Similar content being viewed by others


Introduction
Influenza virus-induced respiratory tract infection often poses considerable medical problems and public health concerns as well as economic hardships to humans and society. The 2009 influenza A H1N1 virus rapidly infected millions worldwide, with estimates of 201,200 respiratory deaths (range 105,700–395,600) and an additional 83,300 (46,000–179,900) cardiovascular deaths1,2. The morbidities and mortalities are often a direct result of a cytokine storm, which defines an early exacerbation and dysregulation of the innate immune response, with robust cytokine/chemokine production3,4,5.
Studying human 2009 influenza A virus infection in animal models3,6,7, we uncovered that cytokine storms were chemically treatable using sphingosine-1-phosphate receptor (S1PR) agonists, which dramatically blunted the production of cytokine/chemokine and inhibited the innate cellular response. Sphingosine-1-phosphate (S1P), the metabolic product of sphingomyelinase activity, is a potent immunomodulatory small molecule8. Platelets, red blood cells and endothelium are believed to be the main sources of S1P9,10,11. S1PR1, a receptor for the bioactive lipid S1P, is expressed on lymphocytes, dendritic cells and endothelial cells3. Endothelial S1PR1 activation is the key regulator of influenza virus-induced cytokine storm3. S1P binding to S1PR1 on the endothelial cell surface can limit the migration of effector lymphocytes to immunologic injury areas, down-modulate the number of virus-specific T cells, blunt cytokine/chemokine expression and reduce the supply of innate inflammatory cells (e.g. natural killer cells, polymorphonuclear leukocytes and macrophages)3,7,12,13,14,15.
Our in vitro study demonstrated the critical role of S1PR1 signaling in regulating the production of cytokine/chemokines by the silencing and over-expressing S1PR1 in cultured human pulmonary microvascular endothelial cells (HPMECs) infected by influenza A virus16. To further validate the functional role of the S1PR1 signaling pathway in immunomodulating effects, animal experiments modelling acute lung injury (ALI) induced by influenza A virus infection are needed. However, S1PR1-deleted mice die at the embryonic stage as a result of oedema, extensive haemorrhage and defective vascular maturation17,18. Thus, we developed inducible genetic loss-of-function models and analysed the therapeutic efficiency of CYM-5442 to elucidate the anti-inflammatory effect of endothelial S1PR1 in the pathogenesis of influenza A H1N1 infection.
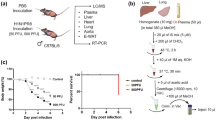
Methods
Virus
The influenza virus in this study was influenza A H1N1 virus strain A/Nanjing/108/2009, (referred to as H1N1 below). H1N1 was isolated and cultured in Madin-Darby canine kidney (MDCK) cells as described before19. Live-virus experiments were performed in Biosafety Level 2 facilities at the Second Affiliated Hospital, Southeast University.
Mice and ethics statement
Wild-type (WT) mice, S1PR1-floxed mice and Tek-CreER T2 mice on the C57BL/6J background were used in these experiments. S1PR1 loxP/loxP mice and Tek-Cre ERT2 mice were provided by the Nanjing Biomedical Research Institute of Nanjing University. Mice were kept in individual cages with free access to water and food under specific-pathogen-free conditions.
S1PR1-floxed mice were crossed to Tek-CreER T2 mice to generate S1PR1 loxP/loxP Tek-CreER T2 mice. Cre-mediated excision was induced in 4-week-old S1PR1 loxP/loxP Tek-CreER T2 male mice by intraperitoneal injection of tamoxifen (TM, 1 mg/day) for five consecutive days20. Males of the same genotype with vehicle (corn oil) injection alone were used as controls.
Mice were challenged intranasally with 105.5 50% tissue culture infective dose (TCID50) of H1N1 virus or an equal volume of virus diluent under 1% (w/v) pentobarbital sodium anaesthesia. Animal experiment proposals were reviewed and approved by the Institutional Animal Care and Ethics Committee of Jinling Hospital, Nanjing University School of Medicine (approval number: JLYY:2013021). We confirm that all experiments were performed in accordance with relevant guidelines and regulations. Experiments were repeated 3 times.
Drug treatment
Intratracheal delivery of CYM-5442 (Sigma-Aldrich, MO, USA) (2 mg/kg dissolved in 100 μL of water) or vehicle (100 μL of water) was administered to mice under 1% (w/v) pentobarbital sodium anaesthesia at 1, 13, 25, and 37 hours post-infection. Oseltamivir (Jinling Hospital, Nanjing, China) (2.5 mg/kg dissolved in 100 μL of water) or vehicle (100 μL of water) was administered to mice by oral gavage twice per day for 5 days starting at 1 hour post-infection.
Weight changes and survival rates
After the mice were inoculated intranasally with H1N1 virus, the body weight and survival rates in each group were monitored consecutively for 10 days. The body weight at day 0 served as the baseline.
Lung wet/dry weight ratio measurement
The lung wet/dry weight ratio was used to assess the degree of pulmonary oedema. On day 5 post-infection, the mice were sacrificed. Whole lungs were excised, blotted dry and weighed. Then the lungs were dried in an oven at 55 °C for 24 hours, and the dry weights were weighed and recorded, after which the wet/dry weight ratios were calculated.
Histopathology evaluation
Lungs were harvested on day 5 post-infection and perfused with phosphate-buffered saline (PBS) to remove blood. The lungs were fixed in 10% formalin, paraffin-embedded, sectioned at 4 μm thickness, placed on glass slides, and stained with haematoxylin and eosin. Slides were analysed by three separate pathologists who were blinded to the experiment. The levels of infiltrating inflammatory cells were counted using a normal optical microscope and represented as average cell numbers per 50 randomly chosen regions of lung parenchyma per sample at a magnification of 1000.
Measurements in bronchoalveolar lavage fluid (BALF)
The BALFs were collected at 48 hours post-infection by flushing 0.6 ml of ice-cold PBS back and forth three times through a tracheal cannula with a 20-gauge catheter. The BALFs were centrifuged at 5000 r/min for 10 min at 4 °C and stored at −80 °C until analysis. The total protein concentration in the BALF was examined by a standard BCA assay kit (ThermoFisher, MA, USA). ELISAs were performed on the BALF using mouse CCL2 (MCP-1), CCL3, CCL5 (RANTES), CXCL2, CXCL10 (IP-10), TNF-α and IL-6 kits (R&D Systems, MN, USA). The concentrations of IFN-α, IFN-γ and IL-1α in the BALF were measured with the mouse ELISA kits (SbjBiol Company, Nanjing, China).
Western blot analysis
Lung tissues were lysed in RIPA buffer containing phosphatase inhibitors and protease inhibitors for 30 min on ice. The tissue lysates were then centrifuged at 12 000 rpm for 10 min at 4 °C to obtain the supernatant. Protein concentrations were determined by a standard BCA kit (Thermo_Fisher, MA, USA). Samples were denatured by heating at 95 °C for 10 min after mixing with loading dye. Equal amounts of samples were loaded onto SDS-PAGE gels separated by electrophoresis and then transferred onto polyvinylidene difluoride membranes. The membranes were then blocked with 5% nonfat milk for 1 hour while shaking at room temperature. The membranes were then incubated with primary antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies. Bands were visualized using the enhancing chemiluminescence (ECL) system (Amersham Biosciences, PA, USA). The band intensity was analysed using ImageJ software.
The primary antibodies, including anti-ERK, anti-JNK, anti-phospho-ERK, anti-phospho-JNK, anti-p-p65 subunit and anti-β-actin, were purchased from Cell Signaling Technology (MA, USA). Antibodies against the p65 subunit of NF-kB were purchased from Santa Cruz Biotechnology (TX, USA). HRP-conjugated secondary antibodies were purchased from Abcam (Cambridge, MA, USA).
Immunofluorescence staining and imaging
Lung tissues were removed and immunofluorescence staining was performed via a standard protocol. Nonspecific sites were blocked with 3% bovine serum albumin (BSA) in PBS. Anti-S1PR1 (Proteintech Group, Chicago, IL, USA) and anti-CD31 antibodies (Abcam, Cambridge, MA, USA) were used for immunostaining. After incubation with primary antibodies, the tissues were further incubated with fluorescent secondary antibodies (Alexa Fluor dyes). Nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma, St. Louis, MO, USA). Finally, confocal immunofluorescence images were captured by Olympus Fluo View confocal microscopy.
Quantitative real-time RT-PCR
The studies measured the levels of influenza H1N1 virus RNA from mice lung samples on day 3 post-infection. The levels of viral RNA were quantified by a standard real-time RT-PCR assay for detection of the influenza A virus M1 gene. For the real-time PCR, cDNA was made from RNA isolated from mouse lung tissue. A total amount of 1 μg of RNA was used for reverse-transcription to obtain cDNA with a RT-PCR kit (Thermo Fisher Scientific, MA, USA), following the manufacturer’s instructions. Real-time analysis for the H1N1 M1 gene and GAPDH was performed by using the SYBR Green Master Mix (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. Relative mRNA expression values of H1N1 M1 genes were normalized by GAPDH. The fold change in mRNA expression was calculated by the equation: 2 (ΔCt of M1 gene-ΔCt of GAPDH). The melting curve for the H1N1 M1 gene was analysed to ensure the specificity of each amplification.
Statistical analysis
Data were presented as the mean ± SEM. The differences between groups were analysed by Student’s t tests for comparing two groups or ANOVA with Tukey’s post hoc test for multiple group comparisons. Survival data were analysed using Kaplan-Meier survival analysis. Statistical analysis was conducted using GraphPad Prism 5 software. P < 0.05 indicated statistical significance.
Results
Deletion of endothelial S1PR1 promotes immune-mediated pulmonary injury induced by H1N1 infection
To study the role of endothelial S1PR1 in virus-induced pulmonary injury, we used the endothelial cell-specific inducible S1PR1 knockout mouse line (referred to as S1PR1 ECKO below) and compared it with control mice. We combined the floxed S1PR1 allele with a Tek-CreER T2 transgene that expresses a TM-inducible Cre recombinase under the control of the Tek promoter20. At 4 weeks of age, S1PR1 loxP/loxP Tek-CreER T2 mice were injected with TM, thus ablating S1PR1 in a wide variety of tissues. After completion of TM injections, the S1PR1 protein was markedly reduced in most of the organs examined (lung, heart, liver, kidney and spleen) (Fig. 1A). The efficiency of deletion was verified by immunofluorescence staining with an almost complete loss of S1PR1 immunofluorescence in the pulmonary endothelium (Fig. 1B).

Endothelial cell-specific inducible S1PR1 knockout mouse. (A) Immunoblotting verified the levels of S1PR1. The S1PR1 protein level was markedly reduced in lung, heart, liver, kidney and spleen tissues after tamoxifen (TM) injection for 5 days. VEH: samples from vehicle-administered S1PR1 loxP/loxP Tek-Cre ERT2 mice; TM: samples after completion of TM administration. Each immunoblot is from a single experiment and is representative of three separate experiments. (B) Immunofluorescent staining of S1PR1 (green) and CD31 (red) in lung tissue. S1PR1 immunofluorescence was undetectable in pulmonary vascular endothelial cells of S1PR1 ECKO mice. Magnification: 1000× ; scale bars, 20 μm. Images are representative of 10 mice per group.
Compared to similarly infected control mice, mice with a deletion of endothelial S1PR1 displayed significantly shortened survival (median survival: 7 days versus 5 days, respectively; P = 0.008) (Fig. 2A). The lung wet/dry weight ratios in S1PR1 ECKO mice were significantly higher than those in control mice (P = 0.028) (Fig. 2B). We next examined whether S1PR1 gene ablation results in an increased degree of inflammatory response induced by virus infection. At 5 days post-infection, histological evaluation of S1PR1 ECKO mouse lungs showed an aggravation of lung injury evidenced by massive pulmonary tissue consolidation, excessive exudation in the alveolar air sacs, vascular haemorrhaging, increased alveolar wall thickness, hyaline membrane formation and considerable inflammatory cell infiltration (Fig. 2C). A significant increase in the number of infiltrating inflammatory cells was evident in the lungs of S1PR1 ECKO mice (P = 0.030) (Fig. 2D). At 48 hours post-infection, the concentrations of total protein in S1PR1 ECKO mouse BALF were significantly increased compared with those in control mouse BALF (P = 0.024) (Fig. 2E), while the abundance of cytokine/chemokine was markedly upregulated in S1PR1 ECKO mouse BALF (Fig. 2F). Analysis of IFN-α, IFN-γ, CCL2, CCL3, CCL5, CXCL2, CXCL10, TNF-α and IL-6 in BALF revealed significantly higher levels in S1PR1 ECKO mice than in control mice.

Endothelial S1PR1 deletion aggravates ALI induced by human pathogenic H1N1 2009 influenza virus infection in mice. Control and S1PR1 ECKO mice were infected with 105.5 TCID50 of influenza A virus. (A) Kaplan-Meier survival curves (n = 15). Control mice survived longer than S1PR1 ECKO mice (P = 0.008). (B) Wet/dry ratios of lungs on day 5 post-infection (n = 6). (C) Representative pathological images of the lungs before and after infection. Histopathologic analysis on day 5 post-infection showed progressive lung lesions with increased infiltration of inflammatory cells, alveolar haemorrhage, hyaline membrane formation and oedema in S1PR1 ECKO mice. Magnification: 200× ; scale bars, 100 μm. (D) Relative number of infiltrating inflammatory cells in lung tissues on day 5 post-infection (n = 5). (E) Total protein in BALF at 48 hours post-infection (n = 5). (F) Concentrations of cytokines/chemokines were measured at 48 hours post-infection by ELISA in BALF (n = 5). *P < 0.05, **P < 0.01 and ***P < 0.001 compared with control mice. These data are representative of three experiments and are shown as the means ± SEM.
S1PR1 activation by CYM-5442 suppresses the excessive immune response induced by H1N1 infection
Control mice treated with CYM-5442 were much heavier than the vehicle-treated controls (P < 0.001), whereas there was no significant difference in the weight of S1PR1 ECKO mice between those treated with vehicle or with CYM-5442 (Fig. 3A). The survival rates of the four groups were recorded (Fig. 3B). Treatment with CYM-5442 significantly improved the survival rate (45%) after influenza challenge compared with that of mice receiving vehicle (10%) in control mice (P = 0.033), but there was no effect in S1PR1 ECKO mice. The pulmonary oedema was measured by wet/dry ratios (Fig. 3C). On day 5 post-infection, the CYM-5442 treatment significantly reduced wet/dry ratios (P = 0.012) in control mouse lungs but not in S1PR1 ECKO mouse lungs. Histopathology of lungs showed that CYM-5442 administration greatly ameliorated immune-mediated lung damage with markedly decreased tissue consolidation and haemorrhage, thinner alveolar walls, and reduced inflammatory cell accumulation versus lungs from control mice receiving the vehicle (Fig. 3D). Compared to those of vehicle-treated control mice, there was a significant reduction in the number of inflammatory cells in the lungs (Fig. 3E) and in the cytokines/chemokines in the BALFs (Fig. 3F) of CYM-5442-treated mice. In S1PR1 ECKO mice, compared with vehicle recipients, the levels of cytokines/chemokines in CYM-5442 recipients showed a declining trend, although there was no statistical significance.

Administration of CYM-5442 ameliorates ALI induced by H1N1 virus infection in control mice, but no effect was observed on S1PR1 ECKO mice. Control and S1PR1 ECKO mice were infected with 105.5 TCID50 of influenza A virus and treated with either vehicle or CYM-5442. (A) Body weight change curves (n = 20). The differences between vehicle and CYM-5442-treated control mice were significant (P < 0.001). (B) Kaplan-Meier survival curves (n = 20). Control mice treated with CYM-5442 survived longer than untreated control mice (P = 0.033). (C) Wet/dry ratios of lungs from infected mice (n = 6). (D) Representative pathological images of the lungs on day 5 post-infection. Lung injury was significantly less in control mice receiving CYM-5442 with less pulmonary consolidation, exudation in alveolar air space, vascular haemorrhage and inflammatory infiltrates than lung injury in vehicle recipients. Magnification: 200× ; scale bars, 100 μm. (E) Relative average quantification of inflammatory cells in lung tissues on day 5 post-infection (n = 5). (F) Concentrations of cytokines/chemokines were measured at 48 hours post-infection by ELISA in BALF (n = 5). (G) Viral loads of 2009 H1N1 in lung tissues on day 3 post-infection (n = 5). Using uninfected mice lung RNA as a baseline, equivalent levels of influenza virus RNA occurred in all four groups. *P < 0.05, **P < 0.01 and ***P < 0.001. These data are representative of three experiments and are shown as the means ± SEM.
The S1PR1 signaling pathway does not affect viral infection
The levels of influenza virus RNA from mouse lung samples at 3 days post-infection were determined (Fig. 3G). Data are presented as the fold increase over that of uninfected mouse lung tissue. The RNA levels of the H1N1 M1 gene did not show a significant difference between S1PR1 ECKO mice receiving vehicle and control mice receiving vehicle. Moreover, the levels of viral RNA in CYM-5442 recipients were similar to those in vehicle recipients in control mice, which is inconsistent with the mortality and histopathology data.
CYM-5442 decreases activation of the MAPK and NF-kB pathways in infected mice
WT C57BL/6J mice were infected with 105.5 TCID50 of H1N1 or virus diluent and either vehicle or CYM-5442 was administered to the mice. Lungs were harvested at 48 hours post-infection, and the effect of CYM-5442 on MAPK and NF-kB pathways was examined by western blot analysis.
To confirm activation of the MAPK pathways, the activated forms of ERK1/2 and JNK1/2 were detected in lung tissue extracts by using antibodies that selectively recognize the phosphorylated (activated) forms. CYM-5442 had no effect on the expression levels of phospho-ERK1/2 and phospho-JNK1/2 in uninfected mice (Fig. 4A). Both ERK1/2 and JNK1/2 were phosphorylated at significantly higher levels in H1N1-infected mice than in uninfected mice (Fig. 4A). The phosphorylation levels of ERK1/2 and JNK1/2 were significantly diminished in infected mice treated with CYM-5442 (Fig. 4B). To verify activation of the NF-kB pathway, lung tissue extracts were analysed for changes in levels of phosphorylated p65. The results demonstrated that CYM-5442 did not affect the expression level of phospho-p65 in uninfected mice (Fig. 4C). The phosphorylation of p65 appeared to be enhanced significantly in infected mice. Administration of CYM-5442 in infected mice resulted in significantly decreased levels of phospho-p65 (Fig. 4D). Our data demonstrated that CYM-5442 treatment attenuated the activation of the MAPK and NF-kB signaling pathways, which were induced by virus infection.

The MAPK and NF-kB pathways are involved in ALI induced by virus infection mediated by S1PR1 signaling. WT mice were infected with 105.5 TCID50 of influenza A virus or virus diluent, and either vehicle or CYM-5442 were administered to mice. Lungs were harvested at 48 hours post-infection for western blot analysis. Densitometry was performed and fold changes in protein expression were quantified. (A) Activation of ERK1/2 and JNK1/2 in infected mice. (B) Inhibition of ERK1/2 and JNK1/2 phosphorylation by CYM-5442. (C) Virus infection increased the phosphorylation of NF-kB p65. (D) Decreased phosphorylation of NF-kB p65 in infected mice after CYM-5442 treatment. *P < 0.05, **P < 0.01 and ***P < 0.001. Data are shown as the means ± SEM from five mice/group. Each immunoblot is from a single experiment and is representative of three separate experiments.
CYM-5442 combined with oseltamivir provides maximum protection against H1N1 viral challenge
Of the WT mice infected with 105.5 TCID50 of H1N1, one group received vehicle alone, a second group was treated with CYM-5442 alone, a third group received oseltamivir administration alone, and a combination of CYM-5442 and oseltamivir was administered to a fourth group.
Combined CYM-5442 and oseltamivir treatment resulted in 75% survival at 10 days post-infection and provided greater survival than either CYM-5442 (45%; P = 0.03) or oseltamivir (30%; P = 0.001) alone (Fig. 5A). The pulmonary oedema measured by wet/dry ratios was substantially ameliorated (Fig. 5B) and accompanied with an improved survival rate, when mice were administered by CYM-5442 and oseltamivir in combination. Histological examination of the lungs revealed that combined treatment dramatically reduced tissue injury (Fig. 5C). Mice treated with CYM-5442 alone showed smaller areas of tissue consolidation, less numerous vascular haemorrhages, and less inflammatory cell infiltration than those received oseltamivir alone at 5 days post-infection. Notably, minimal injury was recorded in mice receiving combined CYM-5442 and oseltamivir, which was consistent with the number of infiltrating inflammatory cells (Fig. 5D). Compared with vehicle- and oseltamivir-treated mice, CYM-5442 as well as combined CYM-5442 and oseltamivir recipients had significantly diminished concentrations of total protein in BALF on day 2 post-infection (Fig. 5E). As shown in Fig. 5F, CYM-5442-treated mice produced significantly reduced levels of IFN-α, CCL2, CCL5, CXCL10 and IL-6 in their BALF tested at 48 hours post-infection. Oseltamivir treatment alone significantly inhibited the production of CCL2. Importantly, combined CYM-5442 and oseltamivir administration resulted in significantly reduced levels of IFN-α, IFN-γ, CCL2, CCL5, CXCL10, TNF-α, IL-6 and IL-1α compared with those of vehicle treated mice.

Combination therapy of CYM-5442 and oseltamivir provides the greatest therapeutic benefit. Wild-type (WT) mice were infected with 105.5 TCID50 of H1N1 virus and either vehicle, CYM-5442, oseltamivir or combined CYM-5442 and oseltamivir were administered to mice. (A) Kaplan–Meier survival curves (n = 20). For mice treated with vehicle alone, 15% survived at 10 days after infection. Compared with vehicle administration, CYM-5442 administration alone enhanced survival (45% vs. 15%; P = 0.02). Combined treatment further enhanced survival (75% vs. 15%; P < 0.001) and was more efficient than CYM-5442 (75% vs. 45%; P = 0.03) and oseltamivir (75% vs. 30%; P = 0.001) treatments alone. (B) Lung wet/dry weight ratios on day 5 post-infection (n = 6). (C) Histopathologic analysis of pulmonary tissues on day 5 post-infection. Pathological images showed less inflammatory cell infiltration, haemorrhage and pulmonary oedema in the three treatment groups than in the vehicle group. Tissue injury was minimal in the combined treatment group. Magnification: 200× ; scale bars, 100 μm. (D) Assessment of inflammatory cell accumulation on day 5 post-infection (n = 5). (E) Analysis of total protein in BALF at 48 hours post-infection (n = 5). (F) Levels of cytokines/chemokines in BALF at 48 hours post-infection (n = 5). *P < 0.05, **P < 0.01 and ***P < 0.001. These data are representative of three experiments and are shown as the means ± SEM.
Discussion
Most patients infected with the 2009 H1N1 virus suffered respiratory illness and some severe cases developed to acute respiratory distress syndrome (ARDS)21. The fatal ARDS patients had higher plasma levels of proinflammatory cytokines/chemokines than the non-fatal ARDS or the mild-disease patients22. In 2011, Teijaro, et al. used the S1PR1 selective agonist CYM-5442 to suppress innate immune responses, resulting in inhibition of cytokine/chemokine secretion and reducing mortality significantly in H1N1-infected mice3,4.
To further validate the involvement of endothelial S1PR1 signaling in the control of immunopathology induced by H1N1 infection, endothelial cell-specific S1PR1 genetic loss in mice was analysed. The lungs of virally infected S1PR1 ECKO mice were highly oedematous and extensively exudative, as reflected by significant increases in wet/dry weight ratios and BALF total protein concentration. H1N1 infection induced a progressive disease pathogenesis in S1PR1 ECKO mice, which was demonstrated by diffuse alveolar damage, vascular haemorrhage and excessive inflammatory infiltration. In agreement with the mortality and histopathology results, the concentrations of responsive cytokines/chemokines in the BALF were significantly upregulated in S1PR1 ECKO mice, indicating an aberrant pulmonary immune response.
In contrast, S1PR1 activation by CYM-5442 effectively dampens immune-mediated lung injury after influenza challenge. The changes in body weight and improved survival rates suggested that CYM-5442 treatment had a protective effect against virus infection. The therapeutic efficiency of CYM-5442 was confirmed by assessing pulmonary oedema and histoathology, which were substantially ameliorated, accompanied by reduced levels of total protein and cytokines/chemokines in the BALF. In our study, the immunopathology induced by influenza virus infection was not improved by CYM-5442 in S1PR1 ECKO mice, indicating that S1PR1 expression on endothelial cells is critical in CYM-5442-induced lung protection.
We systematically demonstrate that the MAPK and NF-kB signaling pathways, which are upstream signaling molecules in inflammatory reactions, are mediated by S1PR1 signaling in 2009 H1N1-infected mice. The MAPK family has been shown to play important roles in influenza virus-induced proinflammatory cytokine expression19,23,24. Therefore, we investigated the effect of CYM-5442 on the activation (phosphorylation) of MAPK components induced by virus infection. We noted that influenza virus infection increased the activation of MAPK pathway components, including ERK1/2 and JNK1/2, whereas CYM-5442 inhibited the activation of the MAPK pathway, which was accompanied by alterations in the levels of cytokines/chemokines. As the NF-κB pathway is a key signaling pathway that mediates the production and release of antimicrobial molecules to activate the immune response25,26, we assessed the phosphorylation of the p65 subunit, which is essential for the activation and translocation of NF-κB heterodimers. We found that activation of p65 was upregulated by H1N1 infection and downregulated by CYM-5442 administration in infected mice, confirming a previous in vitro report16. Mechanistically, our study provided in vivo validation of the critical role of MAPK and NF-kB signaling pathways through which S1PR1 signaling may ameliorate ALI induced by influenza virus infection.
The combination therapy of CYM-5442 and oseltamivir gave the greatest therapeutic benefit in virulent influenza virus infections. The survival rate, lung histopathology and pulmonary oedema alterations in the combination-treated mice were significantly improved. During inflammation, increased levels of cytokines/chemokines result in the recruitment of immune cells into inflamed sites27,28, while an aberrant immune response with robust cytokines/chemokines secretion and excessive inflammatory cell recruitment to the lung is the key contributor to the morbidity and mortality of 2009 H1N1 infections3,22. After treatment with a combination of CYM-5442 and oseltamivir, the levels of responsive cytokines/chemokines were significantly reduced in the infected mice.
Lung injury resulting from influenza virus infection depends on virus infection and an excessive inflammatory immune response with elevated expression of cytokines/chemokines6,7,29. K. B. Walsh7 demonstrated that S1PR1 agonist treatment can blunt dendritic cell activation and hamper the proliferation of T cells, resulting in a smaller number of virus-specific T cells. Thus, activation of S1PR1 generates a sufficient anti-influenza virus T-cell response that terminates the infection. We found that CYM-5442 did not disrupt the kinetics of viral clearance in vivo but successfully limited immune-mediated pulmonary injury. Oseltamivir inhibits influenza virus neuraminidase activity, preventing the release of newly replicated virions from the surface of lysed cells, ultimately reducing viral load. The joint regimen of oseltamivir and CYM-5442 produced the best therapeutic effects in treatment of H1N1 infection in mice model. Therefore, utilizing a combination of the S1PR1 agonist CYM-5442 which controls immunopathologic injury and the antiviral neuraminidase inhibitor oseltamivir which inhibits viral replication, could achieve optimal results.
Multiple studies over the past decade have demonstrated that S1PR1 signaling is critical in the suppression of enhanced and unneeded inflammation, maintaining endothelial barrier function and protecting the host from immunopathology3,4,30,31,32. The sphingosine analogue FTY720 (fingolimod) was approved by the US Food and Drug Administration as a first-line treatment for relapsing forms of multiple sclerosis, highlighting the potential use of S1P receptor agonists33. Signaling by nonessential but injurious S1P receptors that are associated with several side effects, e.g., airway hyperresponsiveness, bradycardia, lymphopenia and vascular barrier disruption, is likely to limit their utility in ALI syndromes34,35,36,37, which indicates that selectivity of the S1P receptor is important. Our data provide strong evidence in vivo that activated S1PR1 in endothelial cells is an essential component for dampening the destructive immune response against influenza virus infection. Hence, a therapeutic approach using selective S1PR1 agonists is of particular interest.
We acknowledge some limitations in our study. The absence of analysis of delayed administration at the time when the immune response is most destructive is a shortcoming in the study. In actual real world situations, people will not receive treatment until 1–2 days after infection. A delayed therapy, e.g, starting at 6 or 24 hours or later after infection, needs further investigation. Additionally, we did not perform experiments to show which cell types and how these cells contribute to cytokine induced pathology during infection in S1PR1 ECKO mice. However, previous literature has demonstrated that endothelial cells are a source of cytokines/chemokines in the lung during influenza infection, and that S1PR1 signaling in the endothelium provides a mechanism for inhibiting cytokine storms and blunting the accumulation of innate inflammatory infiltrates characterized as macrophages/monocytes, neutrophils, and NK cells3,7. Another limitation is that the role of the CYM-5442 ligand in virus-specific adaptive immune cell recruitment was not checked. It will be an important future direction of our research.
Cumulatively, our data assist in elucidating the functional role of endothelial S1PR1 in the lung tissue pathogenesis of H1N1 infection. We systematically illustrate that MAPK and NF-kB signaling pathways are involved in ALI mediated by activated S1PR1 in 2009 H1N1-infected mice. Moreover, our results show that a therapeutic treatment consisting of the specific S1PR1 agonist CYM-5442 and the antiviral drug oseltamivir may be useful for a future influenza A (H1N1) virus pandemic.
Data Availability
All relevant data are within the paper.
Change history
10 May 2023
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41598-023-34694-8
28 July 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41598-020-69835-w
References
Novel Swine-Origin Influenza, A. V. I. T. et al. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. The New England journal of medicine 360, 2605–2615, https://doi.org/10.1056/NEJMoa0903810 (2009).
Dawood, F. S. et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis 12, 687–695, https://doi.org/10.1016/S1473-3099(12)70121-4 (2012).
Teijaro, J. R. et al. Endothelial Cells Are Central Orchestrators of Cytokine Amplification during Influenza Virus Infection. Cell 146, 980–991, https://doi.org/10.1016/j.cell.2011.08.015 (2011).
Walsh, K. B., Teijaro, J. R., Rosen, H. & Oldstone, M. B. Quelling the storm: utilization of sphingosine-1-phosphate receptor signaling to ameliorate influenza virus-induced cytokine storm. Immunologic research 51, 15–25, https://doi.org/10.1007/s12026-011-8240-z (2011).
Kobasa, D. et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445, 319–323, https://doi.org/10.1038/nature05495 (2007).
Teijaro, J. R. et al. Protection of ferrets from pulmonary injury due to H1N1 2009 influenza virus infection: immunopathology tractable by sphingosine-1-phosphate 1 receptor agonist therapy. Virology 452–453, 152–157, https://doi.org/10.1016/j.virol.2014.01.003 (2014).
Walsh, K. B. et al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proceedings of the National Academy of Sciences of the United States of America 108, 12018–12023, https://doi.org/10.1073/pnas.1107024108 (2011).
Huwiler, A., Kolter, T., Pfeilschifter, J. & Sandhoff, K. Physiology and pathophysiology of sphingolipid metabolism and signaling. Bba-Mol Cell Biol L 1485, 63–99, https://doi.org/10.1016/S1388-1981(00)00042-1 (2000).
Hanel, P., Andreani, P. & Graler, M. H. Erythrocytes store and release sphingosine 1-phosphate in blood. Faseb J 21, 1202–1209, https://doi.org/10.1096/fj.06-7433com (2007).
Schaphorst, K. L. et al. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. American journal of physiology. Lung cellular and molecular physiology 285, L258–267, https://doi.org/10.1152/ajplung.00311.2002 (2003).
Venkataraman, K. et al. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circulation research 102, 669–676, https://doi.org/10.1161/Circresaha.107.165845 (2008).
Marsolais, D. et al. A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proceedings of the National Academy of Sciences of the United States of America 106, 1560–1565, https://doi.org/10.1073/pnas.0812689106 (2009).
Rosen, H., Gonzalez-Cabrera, P. J., Sanna, M. G. & Brown, S. Sphingosine 1-phosphate receptor signaling. Annual review of biochemistry 78, 743–768, https://doi.org/10.1146/annurev.biochem.78.072407.103733 (2009).
Rosen, H., Sanna, M. G., Cahalan, S. M. & Gonzalez-Cabrera, P. J. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends in immunology 28, 102–107, https://doi.org/10.1016/j.it.2007.01.007 (2007).
Rosen, H., Stevens, R. C., Hanson, M., Roberts, E. & Oldstone, M. B. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annual review of biochemistry 82, 637–662, https://doi.org/10.1146/annurev-biochem-062411-130916 (2013).
Jiang, H., Shen, S. M., Yin, J., Zhang, P. P. & Shi, Y. Sphingosine 1-phosphate receptor 1 (S1PR1) agonist CYM5442 inhibits expression of intracellular adhesion molecule 1 (ICAM1) in endothelial cells infected with influenza A viruses. PloS one 12, e0175188, https://doi.org/10.1371/journal.pone.0175188 (2017).
Liu, Y. J. et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 106, 951–961, https://doi.org/10.1172/Jci10905 (2000).
Allende, M. L., Yamashita, T. & Proia, R. L. G-protein-coupled receptor S1P(1) acts within endothelial cells to regulate vascular maturation. Blood 102, 3665–3667 (2003).
Gao, W. et al. Distinct regulation of host responses by ERK and JNK MAP kinases in swine macrophages infected with pandemic (H1N1) 2009 influenza virus. PloS one 7, e30328, https://doi.org/10.1371/journal.pone.0030328 (2012).
Forde, A., Constien, R., Grone, H. J., Hammerling, G. & Arnold, B. Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis 33, 191–197, https://doi.org/10.1002/gene.10117 (2002).
Belongia, E. A. et al. Clinical characteristics and 30-day outcomes for influenza A 2009 (H1N1), 2008–2009 (H1N1), and 2007–2008 (H3N2) infections. Jama 304, 1091–1098, https://doi.org/10.1001/jama.2010.1277 (2010).
To, K. K. et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 50, 850–859, https://doi.org/10.1086/650581 (2010).
Marchant, D. et al. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. Journal of virology 84, 11359–11373, https://doi.org/10.1128/JVI.00804-10 (2010).
Hui, K. P. et al. Induction of proinflammatory cytokines in primary human macrophages by influenza A virus (H5N1) is selectively regulated by IFN regulatory factor 3 and p38 MAPK. Journal of immunology 182, 1088–1098 (2009).
Baker, R. G., Hayden, M. S. & Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell metabolism 13, 11–22, https://doi.org/10.1016/j.cmet.2010.12.008 (2011).
Hayden, M. S. & Ghosh, S. Shared principles in NF-kappaB signaling. Cell 132, 344–362, https://doi.org/10.1016/j.cell.2008.01.020 (2008).
Ley, K., Laudanna, C., Cybulsky, M. I. & Nourshargh, S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature reviews. Immunology 7, 678–689, https://doi.org/10.1038/nri2156 (2007).
Nourshargh, S. & Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 41, 694–707, https://doi.org/10.1016/j.immuni.2014.10.008 (2014).
La Gruta, N. L., Kedzierska, K., Stambas, J. & Doherty, P. C. A question of self-preservation: immunopathology in influenza virus infection. Immunology and cell biology 85, 85–92, https://doi.org/10.1038/sj.icb.7100026 (2007).
Wang, L. & Dudek, S. M. Regulation of vascular permeability by sphingosine 1-phosphate. Microvascular research 77, 39–45, https://doi.org/10.1016/j.mvr.2008.09.005 (2009).
Sammani, S. et al. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. American journal of respiratory cell and molecular biology 43, 394–402, https://doi.org/10.1165/rcmb.2009-0223OC (2010).
Krump-Konvalinkova, V. et al. Stable knock-down of the sphingosine 1-phosphate receptor S1P1 influences multiple functions of human endothelial cells. Arteriosclerosis, thrombosis, and vascular biology 25, 546–552, https://doi.org/10.1161/01.ATV.0000154360.36106.d9 (2005).
Brinkmann, V. et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nature reviews. Drug discovery 9, 883–897, https://doi.org/10.1038/nrd3248 (2010).
Pelletier, D. & Hafler, D. A. Fingolimod for multiple sclerosis. The New England journal of medicine 366, 339–347, https://doi.org/10.1056/NEJMct1101691 (2012).
Hale, J. J. et al. Selecting against S1P3 enhances the acute cardiovascular tolerability of 3-(N-benzyl)aminopropylphosphonic acid S1P receptor agonists. Bioorg Med Chem Lett 14, 3501–3505, https://doi.org/10.1016/j.bmcl.2004.04.070 (2004).
Takuwa, N. et al. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovascular research 85, 484–493, https://doi.org/10.1093/cvr/cvp312 (2010).
Niessen, F. et al. Dendritic cell PAR1-S1P3 signaling couples coagulation and inflammation. Nature 452, 654–658, https://doi.org/10.1038/nature06663 (2008).
Acknowledgements
This project was supported by the National Natural Science Foundation of China (81470206), and the Natural Science Foundation of Jiangsu Province (BK20151372).
Author information
Authors and Affiliations
Contributions
Conception and design: Jiangnan Zhao and Yi Shi; Supply of reagents and knockout mice: Meiying Zhu and Simen Shen; Animal experiments: Jiangnan Zhao and Hao Jiang; Sample analyses and statistical analyses: Jiangnan Zhao and Xin Su; Drafting the manuscript for important intellectual content: Jiangnan Zhao and Yi Shi. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41598-023-34694-8
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Zhu, M., Jiang, H. et al. RETRACTED ARTICLE: Combination of sphingosine-1-phosphate receptor 1 (S1PR1) agonist and antiviral drug: a potential therapy against pathogenic influenza virus. Sci Rep 9, 5272 (2019). https://doi.org/10.1038/s41598-019-41760-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41760-7
This article is cited by
-
Emerging Targets for Modulation of Immune Response and Inflammation in Stroke
Neurochemical Research (2023)
-
Receptor-dependent effects of sphingosine-1-phosphate (S1P) in COVID-19: the black side of the moon
Molecular and Cellular Biochemistry (2023)
-
Sphingosine 1-phosphate receptor-targeted therapeutics in rheumatic diseases
Nature Reviews Rheumatology (2022)
-
Present and future treatment strategies for coronavirus disease 2019
Future Journal of Pharmaceutical Sciences (2021)
-
Alterations of lipid metabolism provide serologic biomarkers for the detection of asymptomatic versus symptomatic COVID-19 patients
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
