
Abstract
Multidrug-resistant Pseudomonas aeruginosa is one of the worldwide health problems involved in elevated mortality and morbidity. Therefore, it is important to find a therapeutic for this pathogen. In the present study, we have designed a chimeric vaccine against P. aeruginosa with the help of comparative proteomics and reverse vaccinology approaches. Using comparative subtractive proteomic analysis of 1,191 proteomes of P. aeruginosa, a total of twenty unique non-redundant proteomes were selected. In these proteomes, fifteen outer membrane proteins (OMPs) of P. aeruginosa were selected based on the basis of hydrophilicity, non-secretory nature, low transmembrane helix (<1), essentiality, virulence, pathway association, antigenic, and protein-protein network analysis. Reverse vaccinology approach was used to identify antigenic and immunogenic MHC class I, MHC class II and B cell epitopes present in the selected OMPs that can enhance T cell and B cell mediated immunogenicity. The selected epitopes were shortlisted based on their allergenicity, toxicity potentials, solubility, and hydrophilicity analysis. Immunogenic peptides were used to design a multi-epitope vaccine construct. Immune-modulating adjuvants and PADRE (Pan HLA-DR epitopes) sequence were added with epitopes sequence to enhance the immunogenicity. All the epitopes, adjuvants and PADRE sequence were joined by linkers. The designed vaccine constructs (VT1, VT2, VT3, and VT4) were analyzed by their physiochemical properties using different tools. Selected chimeric vaccine constructs (VT1, VT3, and VT4) were further shortlisted by their docking score with different HLA alleles. The final selected VT4 construct was docked with TLR4/MD2 complex and confirmed by molecular dynamics simulation studies. The final vaccine VT-4 construct was in-silico cloned in pET28a. Therefore, the designed construct VT4 may be studied to control the interaction of P. aeruginosa with host and infection caused by P. aeruginosa.
Similar content being viewed by others
Introduction
Hospital-based surveillance studies as well as Infectious Diseases Society of America have begun to refer nosocomial pathogens as ESKAPE pathogens that include Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species. Different molecules such as herbal compounds1,2, secondary metabolites1,3,4 nanomaterial5,6 and in-silico designed drug7,8,9,10 are used to find a suitable alternative to the current antibiotics used against ESKAPE pathogen. In spite of the use of different mechanisms, to cure the ESKAPE pathogen, there is not any permanent treatment available. P. aeruginosa causes opportunistic nosocomial infection such as bacteremia, pneumonia and septicemia11. Mortality rate in bacteremia caused by P. aeruginosa infection is higher as compared to other Gram-negative pathogens12 due to the emergence of resistance in P. aeruginosa against most of the antibiotics13. Damage to the first line of defense, such as the skin or the mucous membranes, enables the colonizing bacteria to enter the bloodstream and cause septicemia14. Recently, there is developing attention in the various bacterial components like virulence factors for the development of vaccines and other therapies15. Extracellular secretory products and membrane of P. aeruginosa have several antigenic components that are involved in the development of immunity in the host against P. aeruginosa hence suitable as a vaccine targets. Major antigenic surface-associated components of P. aeruginosa are lipopolysaccharides (LPS) and outer membrane proteins (OMPs) because of their accessibility on the pathogen surface. In the adhesion and invasion mechanism during the host-pathogen interaction, outer membrane proteins play a major role to invade a host cell and enter in the tissue16. Hence, assessment of epitopes on the surface of these antigenic proteins is crucial for the development of an effective vaccine against P. aeruginosa17. LPS-based vaccines have been successfully tested in animal models as well as in clinical trials. However, the severe side effects observed in vaccinated individuals have made it necessary to develop chimeric vaccines18,19. Numerous vaccine candidates have been tested against this ubiquitous opportunistic pathogen. Immunization with whole cell organism20, killed and live attenuated vaccine21,22, outer membrane vesicles23,24, outer membrane complex11,13,25, flagella26, pilin27 and glycoconjugate vaccine28 have been suggested as efficient vaccination approach against P. aeruginosa due to the abundance of immunogenic components in them. Many such vaccines tested experimentally in pre-clinical trials but only a few have reached the clinical phase, and none of them has been approved for market authorization14.
Current immunization strategies target multiple OMPs as antigens because there is no risks of pathogen reverting back to its virulent form as well as there are few adverse effects as compared to live attenuated or killed whole cell vaccination. This bacterium rapidly acquired the resistance against multiple antibiotics, natural products, herbal products which caused the problems worldwide. Recently, subtractive genomics29,30 and reverse vaccinology31,32 are using as a potent mechanism to filter-out antigenic and immunogenic proteins that can be used as a chimeric vaccine candidate. Subtractive approach subtracts pathogen OMPs that have a role in virulence or contain the essentiality factor which is the required for survival of the P. aeruginosa but not present in the human host33. Once shortlisted, antigenic and immunogenic epitopes of all vaccine targets will be analyzed using different tools. Using these immunogenic epitopes, the chimeric vaccine can be constructed and validated.
Materials and Methods
Proteome of P. aeruginosa
To identify the chimeric vaccine candidates against pathogenic P. aeruginosa, we have downloaded 1191 proteomes of different strains of P. aeruginosa using UNIPORT server. Redundancy and non-redundancy analysis were carried out manually for 1191 P. aeruginosa strains. Reference proteome of P. aeruginosa (strain PAO1) has 5564 proteins were analyzed using different in-silico subtractive proteomics and reverse vaccinology approaches to determine the antigenic and immunogenic proteins/epitopes targets.
Identification of protein location
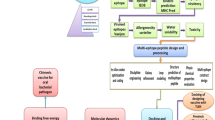
OMPs include a functional role in bacterial pathogenesis, OM permeability barrier, efflux pumps, diffusions of nutrients, membrane integrity and in active transport. For chimeric vaccine construction, bacterial OMPs has selected because of their antigenic and immunogenic behavior34. OMPs that plays an essential and virulent factor role may be useful in therapeutics design. Obtaining a detailed subtractive analysis (Fig. 1) firstly we executed the cellular localization prediction of 5564 proteins using most accurate bacterial localization servers PSORTb 3.0.235, and CELLO36. These softwares identify the localization of proteins as extracellular, outer membrane, cytoplasmic, periplasmic and inner membrane.

Systemic workflow of vaccine OMPs targets identification using comparative subtractive proteomics analysis.
Subtracting non-human homologous proteins
The vaccine candidates that are non-human homologous proteins are considered as a suitable target. This protein targets selection may minimize the probability of cross-reactivity in the host cell. For subtraction of homologous sequence between human host and pathogen, we have carried out BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) analysis of 982 proteins at the NCBI database against the proteome of host Homo sapiens (taxid: 9606). The obtained hits having an E-value (expectation value) of 10−4 or less were considered as homologous proteins and excluded out for further analysis.
Investigation of proteins exported to periplasmic space
Using different servers (PSORTb 3.0.2 and CELLO), we have confirmed the proteins cellular location in the cell. Secretary nature of 918 non-human homologous proteins, were predicted using SignalP 4.037. Proteins being exported from the inner membrane to periplasm following the sec pathway (secretory pathway) were predicted by identifying the presence of N-terminal signal sequences. The positive signal contained proteins were shortlisted to carry out further analysis.
Lipoprotein signal peptide detection
In bacteria, lipoproteins are determined as antigenic and immunogenic targets. To predict potential lipoproteins among the obtained exported proteins, we were used highly sensitive and specific PRED-LIPO software38. Proteins that contained the positive PRED-LIPO signal were considered as good antigenic targets. Shortlisted 482 secretary proteins were analyzed by PRED-LIPO program. 82 proteins were found to be lipoproteins and were separated from the list and the remaining 400 proteins were considered for further experiment. Out of these 400 proteins, 101 proteins are located in OMPs and extra-cellular proteins (ECPs) hence considered for further analysis.
Identification of trans-membrane alpha-helices
The trans-membrane integral inner membrane proteins (IMPs) are the plasma membrane-spanning proteins and contain α-helices. To subtract those proteins that are being exported and getting embedded in the plasma membrane, the 101 obtained proteins were tested in TMHMM (Trans-membrane helices hidden Markov model for topology prediction) 2.039 server. The P. aeruginosa exported proteins that contain more than one TM α-helix with the standard settings were considered as integral IMPs. Non-IMPs proteins (<1 TM α-helix) were selected for further experiments.
Proteins follow signal peptide-independent secretion pathway
Classical pathway transports protein from cytosol to periplasm while the non-classical pathway transports protein from periplasm to extracellular region. Non-classical pathway secreted proteins do not contain the signal peptides but still found in the bacterial extracellular region. With the help of SecretomeP 2.0 program40, 83 proteins were excluded due to its non-classical secreted extracellular proteins. The proteins with a SecP score of 0.5 and above were considered to be non-classically secreted. Finalized OMPs were BLAST against the 10 randomly selected non-redundant proteome using Ensembl database.
Identification of OMPs with an essential role
The database of essential genes (DEG) includes experimentally identified essential proteins which determined the pathogen essential proteins41. In DEG, BLASTp search was performed with a cut-off of 1e−04 E-value, and a bit score of 100, as well as the BLOSUM62 matrix that shortlisted fifteen OMPs of P. aeruginosa.
Identification of virulence-associated OMPs
Virulence factors associated with OMPs have a major role in host-pathogen interaction and modulate host-defense mechanism. VFDB database is used for the identification of virulence-associated OMPs42. P. aeruginosa identified 15OMPs were subjected to BLASTp against VFDB protein core dataset (R1) with E-value was 1e−04 and default cut-off bit score >100. All 15 OMPs were predicted to be virulent.
Identification of proteins involved in pathogen-specific pathways
Kyoto encyclopedia of gene and genome (KEGG) pathway database43 explains the role of proteins in different metabolic pathways. To identify the unique and common pathways between the human host and P. aeruginosa, we have compared all enlisted metabolic pathways manually. The 15 OMPs were segregated based on their role in pathogen-specific (unique), and common (pathogen & host both) pathways. We have also identified the functional role of these 15 OMPs via pathways dependent or pathway independent analysis.
Antigenicity prediction of OMPs
As discussed above, comparative subtractive proteomics approach filtered-out 15 virulent OMPs out of 5564 proteins of P. aeruginosa. These 15 OMPs were considered as potential targets for chimeric vaccine design. Antigenicity of the virulent OMPs can enhance the immune response in the host cell; hence, we performed reverse vaccinology analysis outlined in Fig. 2. Firstly, we carried out antigenicity analysis using VaxiJen server44. The proteins that contained the score value > 0.5 were considered for afterward analysis as an antigenic OMPs.

Illustration of chimeric multi-epitope vaccine design using reverse vaccine workflow.
Protein-protein interaction network analysis
A chimeric vaccine against OMPs will affect the different protein expressions in the bacterial cell. These vaccines may affect the intra-species OMPs interacting partners that resulted reduce the pathogenesis of P. aeruginosa infection. All OMPs protein-protein intra-species interaction analysis was executed to find out the proteins network for identified OMPs using protein interaction database STRING45.
T cell MHC Class I epitope prediction
Antigenic 15 virulent OMPs were selected for the reverse vaccinology analysis. We have performed different T-cell epitope analysis for potent epitopes identification, using four independent servers like IEDB MHC-I prediction server46, MHC-NP47, NetCTLpan1.148, and NetMHCpan3.049. In the host cell proteasomal complex, pathogenic proteins naturally proceed into epitopes. The immune response against the pathogen is initiated by T-cell that interacts with MHC class I represented epitopes. The IEDB (http://tools.immuneepitope.org/processing/) MHC-I processing predictions server identify epitopes that can interact with MHC I molecules. We have selected a MHC allele reference file that contains 27 different alleles that cover more than 97% of the world population. Other parameters of prediction tools were set to default. The MHC-NP server (http://tools.immuneepitope.org/mhcnp/) used the MHC elution experiment to predict the probability that a given peptide is naturally processed. The NetCTLpan1.1 (http://www.cbs.dtu.dk/services/NetCTLpan/) server is predicted proteins cytotoxic lymphocyte (CTL) epitopes. This server uses weight matrix, artificial neural networks (ANN), and TAP transport efficiency to predict peptides that have the potential for MHC-I binding, and proteasomal C terminal cleavage site. NetMHCpan3.0 (http://www.cbs.dtu.dk/services/NetMHCpan/) server using ANN predicts the ability of peptide-MHC class I binding. All 15 OMPs were analyzed by all four prediction methods individually. The top predicted epitopes from all the proteins were selected. Only those epitopes were selected for the further analysis presented in all four servers.
Class I immunogenicity prediction
IEDB server (http://tools.immuneepitope.org/immunogenicity/), a MHC I immunogenicity prediction tool was applied to identify MHC-Peptide complexes with potential immunogenicity in the infected host cell50. The shortlisted epitopes were analyzed using this server with default parameters. The epitopes that gained a positive immunogenicity score were selected for further analysis.
T-cell MHC Class II epitope prediction
IEDB, MHC Class II prediction server is used to identify T-cell epitopes that can bind to MHC class II molecules51. The consensus (stabilization matrix alignment, and average relative binding matrix) prediction approach used to identify the potent MHC II binding epitopes.
Analysis of antigenic and toxicity behavior of epitopes
Obtained immunogenic epitopes (MHC I and MHC II) were analyzed for their antigenic and toxicity behavior. Both analyses were carried out using VaxiJen version 2.044 and ToxinPred tool52 respectively. VaxiJen server (>0.5) predicts the antigenic behavior of epitopes by their physicochemical behavior. In the host cell, the specific immune response induced that will target only bacterial cell or also host cell was analyzed by the ToxinPred tool.
B-cell epitope prediction
To initiate humoral immunity, epitopes should be recognized by B lymphocytes. B-cell epitopes were predicted by the different server like BCPREDS53, the FBCPred server54 and IEDB server55. BCPRED and FBCPred cut-off score used in the present study is >0.8. Epitopes that predicted by all three servers were chosen for further study.
Comparison of predicted epitopes to select final epitopes for vaccine design
Final chimeric vaccine sequence was designed using B-cell epitopes, MHC-I epitopes, and MHC-II epitopes after manual comparative analysis. Overlapping epitopes sequences were merged and used to constructs the final vaccine.
Hydropathy analysis of epitopes
For chimeric OMPs vaccine, all epitopes should have the hydrophilic nature (present on the surface) otherwise the epitopes will not be able to initiate the immune response in the host cell. Hydrophilic epitopes of OMPs are present on the surface of bacterium while hydrophobic epitopes are spanning in the bacterial membrane. GRAVY score analysis of epitopes was done using the ProtParam tool. The GRAVY (grand average of hydropathy) value of protein is calculated as the total of all the amino acids hydropathy values, divided by the number of amino acid residues in a given protein. A positive GRAVY value indicates that the protein is hydrophobic whereas a negative value indicates that protein contains hydrophilic properties.
MHC restricted alleles using cluster analysis
Using IEDB analysis, we have shortlisted the MHC class I and class II-restricted epitopes. MHCcluster v2.0 server was the additional crosscheck of identified epitopes, to confirm the prediction56. The server describes peptides and HLA functional relationship in the form of a static heat map and graphical tree.
Construction of design model vaccine
To design chimeric vaccine targeted to OMPs, we were joined all the selected OMPs epitopes (HTL, CTL and B epitopes) using amino acid linkers (HEYGAEALERAG and GGGS linkers). Immunogenicity of constructs was enhanced by the addition of different adjuvants using ‘EAAAK’ linkers at both terminus (N and C). These adjuvant were 50 s ribosomal L7/L12 protein57, beta-defensin58, HBHA protein (M. tuberculosis, accession no. AGV15514.1), and HBHA conserved sequence59 respectively. To improve the vaccine efficacy and potency non-natural pan DR (PADRE) 13 amino acid epitope (AKVAAWTLKAAAC) that induce CD4+ T-cells were also combined along with the adjuvants. Alexander et al. also explain that if PADRE glycoconjugates (i.e KXVAAWTLKAAZC, where X is l-cyclohexylalanine & Z is aminocaproic acid) is used then it will further enhance the IgG production60. Polymorphism of HLA-DR molecules in the worldwide population is overcome by PADRE sequence that has 100-fold more potency than the universal T helper epitopes. It was found that vaccine construct with PADRE sequence exhibited better CTL responses than a vaccine without it61. All adjuvant proteins are agonist to different TLR complexes that polarizes CTL responses which have a robust immuno-stimulatory effect62.
Allergenicity, antigenicity and solubility prediction of design vaccine constructs
For a selection of the suitable vaccine constructs, all four vaccine constructs (VT1, VT2, VT3, and VT4) were analyzed using on the basis of their antigenicity, allergenicity and soluble prediction methods. For the allergenic behavior of vaccine, the constructs were analyzed by the AlgPred server63. Antigenicity of vaccine constructs was predicted using two servers: ANTIGENpro64 and VaxiJen 2.044 server. Vaccine constructs solubility and corresponding probability (≥0.5) were predicted using SOLpro server64.
Physicochemical behavior analysis of vaccine constructs
Vaccine constructs physiochemical properties were characterized using Expasy ProtParam server65. This server is widely used for determining the number of amino acids, PI values, molecular weight, aliphatic index, instability index, and hydropathicity GRAVY values. Instability index of protein predicts proteins stability (<40). Aliphatic index value explains about vaccines thermostability. GRAVY explains the proteins hydrophilic or hydrophobic nature.
Prediction of the secondary structure
Various vaccine constructs were used for predicting its secondary structure components using PSIPRED v3.3 program66. PSIPRED 3.2 server accuracy is 81.6% which predicts the proteins alpha and beta helix and coil structure.
Molecular Docking study and Molecular Dynamics Simulation
Finalized three vaccine constructs (VT1, VT3, and VT4) were intensively modeled using the Phre2 online tool. Different HLA alleles PDB ID i.e. DRB1*03:01(1A6A), DRB1*15:01(1BX2), DRB3*02:02(3C5J), DRB5*01:01(1H1S), HLA-B*44:03(1SYS), HLA-B*53:01(1A1M), HLA-B*15:01(1XR8), HLA-B*39:01(4O2E), HLA-B*58:01(5IM7) and HLA-B*35:01(1ZSD) were downloaded from protein data bank (RCSB). With the help of PatchDock server, docking of three vaccine constructs was performed with 10 different HLA alleles. Similarly, we also docked the vaccine construct (VT4) with TLR 4/MD2 complex (PDB ID 3FXI) with the help of PatchDock67. The 10 best solutions of PatchDock result were further refinement using FireDock. Docking of the vaccine construct (VT-4) with TLR 4/MD2 complex was also performed using ClusPro68. VT4-TLR4 complex molecular dynamics simulation was performed using Gromacs v5.1.2 as the published method69.
Codon optimization of design vaccine construct VT-4 and In-silico cloning
Java Codon Adaptation Tool (JCAT) was developed to improve heterologous protein production in E. coli host strain70. Vaccine VT4 construct was reverse translated, and subsequently adapted for codon usage to E. coli. The prokaryotic ribosome binding sites, rho-independent transcription terminators, and few restriction sites were avoided. Gene sequence of final design vaccine construct VT-4 was cloned in E. coli pET28a vector, by Snapgene tool71 that make sure vaccine construct expression.
Results
Comparative subtractive proteomic approach shortlisted P. aeruginosa strains
With the help of UNIPORT server, proteomes of different strains of Pseudomonas genospecies were downloaded that contains the list of 1224 strains. From 1224 different Pseudomonas strains, we have filtered out the 1191 strains of P. aeruginosa for further analysis. Redundancy and non-redundancy analysis were carried out manually for the proteome of 1191 P. aeruginosa strains. During comparison, 11 different groups of non-redundant strains were made. Nine strains of P. aeruginosa did not show any redundancy with the other Pseudomonas strains. Therefore, a total number of 20 (11 + 9) (Supplementary Table ST-1) strains of Pseudomonas were selected for further analysis. Proteome of P. aeruginosa PAO1 strain is considered as a reference proteome in the present study.
Identification of subcellular localization of the proteins
Vaccine design using subtractive proteome analysis against the pathogen could minimize time, and resources for developing the therapeutic agent that have large targets. To achieve the potent proteins for vaccine construct, firstly, we have filtered all 5564 proteins (of reference proteome) on the basis of their sub-cellular localization. With the help of CELLO and PSORTb tool, we have filtered 982 different proteins out of 5564 proteins of P. aeruginosa PAO1. These 982 proteins localization is in the periplasm, the extracellular and outer membrane of the bacterial cell.
Identification of bacterial proteins that are non-human homologous
To exclude out the cross-reactivity of vaccine with the human host cell, the potent vaccine should be designed against non-human homologous proteins. We found 918 non-human homologous proteins out of 982 proteins using BLASTp. These non-human homologous bacterial proteins were lead to vaccine construct that only interacts with the proteins of P. aeruginosa.
Signal peptide detection
The exported proteins are secreted out across the inner membrane and enter the periplasmic space. With the help of a positive signal in SignalP server, 482 P. aeuginosa proteins (out of 918 proteins) were selected as exported proteins. The exported proteins have more chance to be transported and located in the bacterial surface that might be a better target for vaccine design. Therefore, proteins with exported capacity were selected for further experiments.
Lipoproteins of P. aeruginosa
With the help of acyl chain, bacterial lipoproteins are located in the outer membrane, inner membrane, and periplasm. With using PRED_LIPO program, we successfully identified 82 P. aeruginosa proteins as a lipoprotein. These lipoproteins can also be considered as potent antigenic targets, but in the present study, we are targeting the OMPs. Therefore, for designing chimeric vaccine constructs, we have excluded out these 82 lipoproteins and rest 400 proteins were considered in further experiments.
TM α-helices containing proteins
In bacteria, secretory exported proteins have different locations. Some of the proteins located in the periplasm, some of them embedded into the outer membrane and some of them embedded in the periplasmic side of the inner membrane. These IMPs contain TM hydrophobic α-helices that act as an internal signal and helps in getting them embedded in the membrane. Out of 400 proteins, we only considered 101 proteins which are located in the OMPs. Using TMHMM server, we have found 18 P. aeruginosa proteins that contain more than one TM α-helices. These 18 proteins are considered to be inner membrane proteins and exclude out. Finally, filtered 83 were considered for further analysis.
Selection of OMPs after elimination of the non-classical secretory proteins
Classical secretion pathway involved signal peptide while non-classical secretion system does not involve signal peptide sequence. To eliminate the non-classical secretory proteins, we have used SecretomeP 2.0 program. In SecretomeP 2.0 server, we have analyzed the 83 OMPs proteins of P. aeruginosa. In secP server, proteins that scored > 0.5 were considered as non-classical secretory proteins. We found 68 out of 83 P. aeruginosa proteins as non-classical secretory proteins. These proteins may be located in the extracellular region of the bacterial cell hence excluded from further analysis. This analysis leads to the selection of the final 15 unique OMPs proteins (Table 1). BLAST using Ensembl genome database showed that all the 15 OMPs have sequence similarity with the 10 randomly selected non-redundant proteomes.
OMPs have an essential protein of P. aeruginosa
Essential proteins as vaccine targets will cause the major effect on the bacterium, Hence, we have analyzed 15 OMPs proteins using DEG server (Table 1). Out of 15 proteins, one protein Q9HU51; uncharacterized protein is considered essential for the survival of P. aeruginosa strain.
All selected OMPs have a role in the virulence of P. aeruginosa
A chimeric vaccine against the virulent factor associated OMPs will enhance the potency and efficiency of the protein. The VFDB database result (Table 1) showed that all 15 proteins are involved in the virulence of P. aeruginosa. Hence, these proteins are also considered as an essential target to inhibit the pathogenesis of P. aeruginosa.
Pathogen-specific OMPs have been selected
P. aeruginosa and human host metabolic pathways (present in KEGG Database) manual comparison have done to find the shortlisted outer membrane pathogenic proteins role in the different pathways. This result identifies 42 unique pathogen pathways (Supplementary Table ST-2) with respect to P. aeruginosa and the remaining 79 pathways were common and found in both pathogen and host. Eight out of fifteen OMPs found to posse’s metabolic pathways dependent and 7 remaining proteins were metabolic pathway independent proteins (Table 1).
Outer membrane proteins are antigenic
From the subtractive proteome analysis, we have filtered out 15 OMPs that may use as promiscuous vaccine targets. We analyzed the proteins antigenicity using VaxiJen web server that found all are the potent antigenic OMPs (Table 1). All 15 proteins were identified with the prediction score ranging from 0.5043 to 0.7906 .
Selected proteins showed intra-species protein-protein interaction
STRING predictions of fifteen OMPs of P. aeruginosa showed the different intra-species interaction between the proteins. With the help of all interaction study, we might understand the protein networking of 15 OMPs. A chimeric vaccine against these 15 OMPs may affect all interacting network proteins that further enhance the efficacy of the chimeric vaccine. All proteins networking is shown in Supplementary Figure SF 1.
Selected OMPs have MHC I epitopes
T-cell epitopes of 15 OMPs were used for MHC-I binding prediction by IEDB server. The epitopes with higher affinity (IC < 50 nM) and good percentile rank (≤0.2) were selected for analysis. IEDB MHC1, MHC-NP, netCTLpan1.1, and netMHCpan3.0 B cell epitope analysis identified the potent epitopes. From all servers, we have identified 221 epitopes (Supplementary Table ST-3) in the 15 antigenic OMPs. To improve accuracy, we have filtered epitopes that were predicted by all four tools. In total, 80 common epitopes out of 221 epitopes were selected from these four servers.
Selected MHC I epitopes are Immunogenic
Selected 80 MHC class I epitopes from different servers were analyzed for a class I immunogenicity and toxicity. A high immunogenicity score showed high potency to stimulate naive T cells and also induce cell-mediated immunity. In IEDB immunogenicity prediction, 80 epitopes gained a score range from 2.9388 to −0.0193 (Supplementary Table ST-3). One protein i.e. uncharacterized protein (Q9HU51) identified potent epitopes did not show the positive values score hence excluded from further analysis. In this analysis, 50 epitopes of 14 different OMPs contained the positive value score were selected for further analysis (Table 2).
Selection of MHC-II epitopes in OMPs for a chimeric vaccine design
MHC-II binding predictions of all 15 OMPs were subjected using IEDB server. On the basis of percentile rank as well as IC50 value (<50 nM), 41 epitopes (Supplementary Table ST-5) were selected for afterwards analysis. MHC II prediction showed that thirteen out of 15 OMPs showed low percentile rank and higher affinity epitopes. Two OMPs (Q9I0W0; Outer membrane protein CzcC, and Q9I0E2; OpdB proline porin) did not show a good percentile for MHC II class epitopes.
Toxicity and antigenicity prediction of MHC class I and MHC class II epitopes
Epitopes induced cross-reactivity in the host tissue was identified using the ToxinPred server. The 50 MHC I epitopes and 41 MHC II epitopes were found to be non-toxic (Supplementary Tables ST-4 and ST-5). Antigenicity of potent MHC I epitope and MHC II epitopes were analyzed using VaxiJen web server. MHC I epitopes of protein (Q9I319, type III secretion outer membrane protein PscC) and MHC II epitopes of protein (Q9I319, Type III secretion outer membrane protein PscC and Q9HU51, uncharacterized protein) antigenicity value was less than 0.5, so these proteins epitopes were excluded as non-antigenic proteins. The result showed a total of 52 (30 + 22) epitopes contained the potent antigenicity. These 52 (30 + 22) (Supplementary Table ST-4 and Table 3) epitopes of MHC class I and MHC class II were selected.
Selection of B-cell epitopes in the OMPs for development of a chimeric vaccine
These 15 OMPs were analyzed to predict the B-cell epitope that enhances the humoral immune response. B-cell epitopes were identified by three different IEDB BepiPred linear epitope prediction servers, BCPREDS, and FBCPred. All 15 OMPs BepiPred linear diagrams have shown in Fig. 3. To improve the accuracy of epitope prediction, we selected epitopes which were predicted by all three tools. In total, 31 different epitopes (Table 4) of fifteen antigenic OMPs were selected from these three servers. These three servers gave the different length epitopes, and we have selected the epitopes on their antigenicity score by the vaxiJen server. In this, we selected 15 antigenic epitopes from all 15 proteins.

Identification of B cell epitopes of antigenic Proteins (UNIPORT ID) using Bepipred linear epitope server.
Physiochemical analysis of epitopes
To explore the epitopes physiochemical properties, we have used GRAVY (grand average of hydropathy) analysis using ProtParam tool. Firstly, we have selected epitopes with the high antigenic score from each 15 OMPs. After shortlisting of high scored 40 (13 + 12 + 15) epitopes for MHC I, II and B cell (Table 5), we have proceeded for GRAVY analysis. In GRAVY result, we have gained the negative value score of total 28 (5 + 8 + 15) epitopes of MHC I, II and B cell respectively. These shortlisted hydrophilic epitopes may be present in the outer surface and have more chance to elicit the high immunogenicity in the host cell. These hydrophilic epitopes were selected to design a chimeric vaccine construct.
Selected epitopes were analyzed for MHC restriction and cluster analysis
After physiochemical analysis, the selected epitopes were validated for the MHC interaction analysis using the MHCcluster. Interacted alleles were further assessed by cluster analysis and the result is shown as a heat map (Fig. 4) and dynamic tree (Supplementary Figure SF 2) concerning to MHC class I and MHC class II. The epitopes are clustered by interaction with HLA. Red color suggesting strong interaction while yellow color indicates weak interaction.

Cluster analysis of the HLA alleles for both MHC molecules through heat map representation. (A) Representing the cluster of the MHC-I (QEAHWTINL, RSWQLRYDY, YESGYTEGL, GEYGTRFSL, and FGADHETAY) epitopes. (B) Representing the cluster of MHC-II molecules (GETFVVDPRVKGQVS, ALELAGVLQQRLDDQ, GGPVLLDQRLSGDTS, QGFLLRYESGYTEGL, GNAIELDARGRYFAA, DAKYRAGAAALSDRL, AEARYAYLNAWLRLR, and DNGLELDGTLLRAKS) epitopes.
Construction of chimeric vaccine
All shortlisted epitopes were used to design the chimeric vaccine. Using different analysis, we were shortlisted 5 different (QEAHWTINL, RSWQLRYDY, YESGYTEGL, GEYGTRFSL, and FGADHETAY) MHC I epitopes from 15 OMPs. Eight different MHC II epitopes (GETFVVDPRVKGQVS, ALELAGVLQQRLDDQ, GGPVLLDQRLSGDTS, QGFLLRYESGYTEGL, GNAIELDARGRYFAA, DAKYRAGAAALSDRL, AEARYAYLNAWLRLR, and DNGLELDGTLLRAKS) were selected using different analysis. From 15 OMPs, we have shortlisted 15 epitopes for B-cell (IVPNAEAKTEAGGG, PGESPGT, NPGLNAEYPAGTGCCSDGES, DSGSGSGGTGLLPA, RSTDEGGSRVNNRA, WRKEELEDRSVNTAGDAS, QGRGHPQAGDPNAR, RDRYSEGGGDNSRS, FEEGEGTRTDLLET, ALSDAPPRKDVPFRRKQDDR, EGSAGVSGGAGNGW, DLESAYTPGRVGFGLDLHGF, SKYDQTARDGRGER, GSQRGDDPRAKWDG, and DSEEGGGAGNGAVG). For vaccines construct, we have joined all epitopes with the help of linkers. To increase the vaccine immunogenicity, we have also added different adjuvants and PADRE sequence with the epitope sequences. We were made 4 different vaccines construct (VT1, VT2, VT3, and VT4). VT1 vaccine was merged with HBHA adjuvant, VT2 vaccine with HABA conserved sequence, VT3 vaccine with beta-defensin adjuvant and VT4 vaccine with 50 ribosomal L7/L12 protein. All detailed vaccine construct sequence was shown in Table 6.
Antigenicity, allergenicity, and solubility prediction of design vaccine constructs
Four vaccine constructs (VT1, VT2, VT3, and VT4) were further analyzed using the AlgPred, ANTIGENpro, VaxiJen 2.0 and SOLpro server (Supplementary Table ST-6). AlgPred server found all vaccine constructs have non-allergenic behavior. Antigenicity analysis of constructs was contained antigenicity score of more than 0.569 in ANTIGENpro and 1.5596 in VaxiJen 2.0 that suggests a good antigenic property of these four vaccines constructs. Using SOLpro, all four vaccine constructs were showed the good solubility (>0.9820) during its heterologous expression in the E. coli.
Physicochemical analysis of design vaccine constructs
All four vaccine physicochemical properties were analyzed using ProtParam server. In this, all vaccine constructs molecular weight range have found between 59–72 kDa. Different physicochemical values of all vaccine construct demonstrate that the protein is stable in respective pH (Table 6). GRAVY (a hydropathic index) was found to be a negative value (−0.544), that suggest the hydrophilic nature of the design constructs and presents strong interactions with water molecules. The high aliphatic index range (49.85 to 59.07) of all vaccines construct indicates the protein stability in several temperatures. Three (VT1, VT3, and VT4) out of four vaccine constructs showed Instability score (<40) which explain that protein has good stability to initiate an immunogenic reaction. The total negative amino acid residues (Asp & Glu) and the total positive amino acid residues (Arg & Lys) were shown in the respective tables.
Structure prediction of selected vaccine constructs
Secondary structures of the final three vaccine constructs were predicted using PSIPRED. The secondary structure of VT1, VT3 and VT4 vaccine constructs are shown in Fig. 5. The structure of all vaccine constructs has alpha helix, beta-sheet, and beta turn. The model of VT1, VT3 & VT4 construct were generated and validated by the Ramachandran plot. The modeled structure and Ramachandran plot of VT4 is shown in Fig. 6.

Secondary structure of vaccine constructs (VT1, VT3, and VT4) using PESIPRED server.

Tertiary structure prediction and validation of vaccine construct VT4. (A) The tertiary structure of model vaccine VT4. (B) Ramachandran plot of the modeled VT4 showing 92% residues in the allowed region.
Interaction of vaccine constructs with HLA allele’s protein
A vaccine can interact with different HLA alleles of human populations. We have docked final 3 vaccine constructs (VT1, VT3, and VT4) with 10 different HLA allele’s proteins. VT4 have the least global binding energy value with different HLA alleles i.e. 1A6A (HLA-DR B1*03:01); −17.92, 1BX2 (HLA-DRB1*15:01); −22.69, 3C5J (HLA-DR B3*02:02); −14.16, 1H1S (HLA-DR B5*01:01); −5.26, 1SYS (HLA- B*44:03); −32.07, 1A1M (HLA-B*53:01); −20.03, 1XR8 (HLA-B*15:01); −11.61, 4O2E (HLA-B*39:01); −10.10, 5IM7 (HLA-B*58:01); −0.37, and 1ZSD (HLA-B*35:01); −10.10 as shown in (Supplementary Table ST-7). We have analyzed all three different constructs and finalized the one suitably, and best vaccine constructs i.e. VT4 that can be developed to control P. aeruginosa infection.
Interaction and Molecular Dynamics Simulation of VT4 with TLR 4
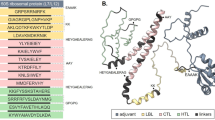
The interaction between the TLR4 and adjuvants enhance the immune response. Hence, we have performed interaction study between VT4 with TLR 4/MD2 complex (PDB 3FXI). VT4 contains the adjuvant 50 s ribosomal L7/L12 protein. TLR 4 is agonist to an L7/L12 ribosomal protein that can increase the several immune responses in the host cell. The Patchdock docking result showed that negative (−11.11) binding energy that suggests a good interaction between VT4 and TLR-4/MD2 complex. This VT4-TLR4 complex further analyzed for docking using ClusPro. Best modeled ClusPro complex have minimum energy scored of −1009 which explained the interaction of VT4 with TLR-4 (Fig. 7). To further validate the interaction of the best-docked complex (from CluSPro), molecular dynamics simulation (MDS) was performed. MDS result also confirms that VT4-TLR-4/MD2 complex get stabilizes at 6 ns (Fig. 8) that further confirms the interaction between VT4 with TLR-4/MD2 complex.

Docked complex of vaccine constructs VT4 with the human TLR4-MD2 complex. The vaccine construct VT-4 docked within the TLR-4 receptor.

Molecular dynamics simulation of VT4- TLR4/MD2 complex. The result shows the RMSD obtained for the complex which showed that complex is stable at 6 ns.
In-silico cloning of VT4 construct for its heterologous expression in E. coli
Chimeric vaccine cloning and expression within the expression vector were analyzed by Java Codon Adaptation Tool. Reverse translation generates cDNA sequence that will be used for in-silico cloning. Codon optimization analysis of VT4 construct showed 77.50% GC content of constructs. The CAI value indicates the heterologous expression of the selected gene. VT-4 have CAI value of 1.0 that suggests that it will be highly expressed in E. coli cell. The DNA sequence of restriction sites for EcoRI and NdeI restriction enzyme was added at 5′ and 3′ ends respectably. VT4 was in-silico cloned into pET28a vector for its heterologous expression in E. coli using EcoRI and NdeI restriction enzyme (Fig. 9).

In-silico restriction cloning of gene sequence of final vaccine construct VT4 into pET28a expression vector showing VT4 sequence red colour surrounded between EcoRI (192) and NdeI (2257).
Discussion
The burden of disease caused by P. aeruginosa is the most common pathogen causing acute healthcare-associated pneumonia in the critically ill, elderly and immune-compromised person is substantial. Treatment is complicated by the organism’s resistance to multiple antibiotics and its capacity to form aggregates and biofilms on mucosal membranes and medical device surfaces. The establishment of persistent P. aeruginosa infection in those with chronic pulmonary disorders is also of considerable importance. Both acute and chronic P. aeruginosa infections are associated with significant morbidity, increased mortality and considerable cost to the health system and the community. There is no permanent cure and prevention for the multidrug-resistant P. aeruginosa. Studies have been done to screen herbal compounds1,2, nanomaterial5,6 and in-silico design therapeutics7,8 to find some alternative to the current therapeutics currently used against P. aeruginosa. Recently, proteome-based approaches (Subtractive genomics and reverse vaccinology) are used to design multi-epitope vaccine in A. baumannii by our group.
In the present work, we have shortlisted the non-redundant 20 strains with the reference proteome of P. aeruginosa. Using reference proteome i.e P. aeruginosa PAO1 strain, 5564 proteins were analyzed on the basis of different subtractive approaches. In the subtractive analysis, we have shortlisted the proteome on the basis of cellular localization, lipoproteins, a transmembrane helix, and classical and nonclassical secretary analysis. After the shortlisting, we have identified 15 antigenic outer membrane proteins. These 15 OMPs were classified on the basis of essentiality using DEG server, virulence factor using VFDB, and pathway-dependent and independent analysis using KEGG database. These 15 OMPs were selected for chimeric vaccine design using the reverse vaccinology. Antigenic, non-allergenic, nontoxic MHC I, MHC II and B cell epitopes were identified using different servers. All selected epitopes were merged with different adjuvants and linkers to enhance the immune responses. After joining with the different adjuvants, four vaccine constructs (VT1, VT2, VT3, and VT4) have been made. These vaccine constructs were further analyzed for their antigenic, allergenic and toxicity behavior. The non-toxic and non-allergenic vaccine may be the good immunotherapy against the pathogenic P. aeruginosa. On the basis of physiochemical behavior, we have shortlisted the three vaccine constructs (VT1, VT3, and VT4) for further analysis. Furthermore, docking based interaction analysis was performed to elucidate the binding affinity of designed vaccine with the different HLA molecules i.e. DRB1*03:01, DRB1*15:01, DRB3*02:02, DRB5*01:01, HLA-B*44:03, HLA-B*53:01, HLA-B*15:01, HLA-B*39:01, HLA-B*58:01 and HLA-B*35:01. With the help of docking analysis, we have shortlisted the VT4 as a potential vaccine that may enhance the immune response in the host cell. We have added different adjuvants to enhance immune response. We have also validated the interaction of VT4 with the TLR4/MD2 complex. Chimeric vaccine construct VT-4 against the P. aeruginosa may be used to generate the immune response and have the potential to interact with various HLA alleles. In the design vaccine constructs VT4, we have also added adjuvant L7/L12 ribosomal protein, Pan-DR epitopes, and linker along with multi-epitope sequences that may also enhance the significant P. aeruginosa specific immune responses. Therefore, in brief, we have taken possible significant factors that may induce the immunogenicity and feasibility of the vaccine construct VT-4.
Conclusion
The availability of complete proteome of bacteria is facilitating many computational approaches. The data presented here demonstrate that stepwise prioritization of proteome using different comparative proteomics and reverse vaccinology approaches which are an effective way of rapidly reducing the unwanted number of P. aeruginosa proteins. This process is an efficient way of enriching the potential target, and for identifying those that are critical for normal cell function and have a virulent role in the host cell. Such a strategy will enable us to locate antigenic and immunogenic OMPs vaccine targets that have a role in pathogenesis. The antigenic, non-allergenic, nontoxic MHC I, MHC II and B cell epitopes were merged with different adjuvants and linkers to enhance the immune responses that further improves the effectiveness of the final chimeric multi-epitope vaccine VT-4. In addition to this, the designed vaccine VT-4 needs to be experimentally validated to ensure its efficacy against P. aeruginosa infections. The recommended vaccine (VT-4) need to be also validated in animal models before use against P. aeruginosa.
Data Availability
All the data are available in the manuscript.
References
Tiwari, V., Roy, R. & Tiwari, M. Antimicrobial active herbal compounds against Acinetobacter baumannii and other pathogens. Frontiers in Microbiology 6, https://doi.org/10.3389/fmicb.2015.00618 (2015).
Ulloa-Urizar, G., Aguilar-Luis, M. A., De Lama-Odría, M. D. C., Camarena-Lizarzaburu, J. & del Valle Mendoza, J. Antibacterial activity of five Peruvian medicinal plants against Pseudomonas aeruginosa. Asian Pacific. Journal of Tropical Biomedicine 5, 928–931, https://doi.org/10.1016/j.apjtb.2015.07.016 (2015).
Tiwari, M., Raghav, R. & Tiwari, V. Comparative Anti-Bacterial Activity of Differently Capped Silver Nanomaterial on the Carbapenem Sensitive and Resistant Strains of Acinetobacter baumannii. Journal of Nanomedicine and Nanotechnology 6, 314 (2015).
Tiwari, M., Roy, R. & Tiwari, V. Screening of Herbal-Based Bioactive Extract Against Carbapenem-Resistant Strain of Acinetobacter baumannii. Microb Drug Resist 22, 364–371, https://doi.org/10.1089/mdr.2015.0270 (2016).
Ramasamy, M. & Lee, J. Recent Nanotechnology Approaches for Prevention and Treatment of Biofilm-Associated Infections on Medical Devices. BioMed Research International 2016, 1851242, https://doi.org/10.1155/2016/1851242 (2016).
Tiwari, V., Tiwari, M. & Solanki, V. Polyvinylpyrrolidone-Capped Silver Nanoparticle Inhibits Infection of Carbapenem-Resistant Strain of Acinetobacter baumannii in the Human Pulmonary Epithelial Cell. Front Immunol 8, 973, https://doi.org/10.3389/fimmu.2017.00973 (2017).
Sahner, J. H. et al. Combining in Silico and Biophysical Methods for the Development of Pseudomonas aeruginosa Quorum Sensing Inhibitors: An Alternative Approach for Structure-Based Drug Design. Journal of Medicinal Chemistry 56, 8656–8664, https://doi.org/10.1021/jm401102e (2013).
Verma, P. & Tiwari, V. Targeting Outer Membrane Protein Component AdeC for the Discovery of Efflux Pump Inhibitor against AdeABC Efflux Pump of Multidrug Resistant Acinetobacter baumannii. Cell Biochem Biophys, https://doi.org/10.1007/s12013-018-0846-5 (2018).
Verma, P., Maurya, P., Tiwari, M. & Tiwari, V. In-silico interaction studies suggest RND efflux pump mediates polymyxin resistance in Acinetobacter baumannii. J Biomol Struct Dyn, 1–9, https://doi.org/10.1080/07391102.2017.1418680 (2017).
Tiwari, V., Tiwari, M. & Biswas, D. Rationale and design of an inhibitor of RecA protein as an inhibitor of Acinetobacter baumannii. J Antibiot (Tokyo) 71, 522–534, https://doi.org/10.1038/s41429-018-0026-2 (2018).
Chirani, A. S. et al. The effect of in silico targeting Pseudomonas aeruginosa patatin-like protein D, for immunogenic administration. Computational biology and chemistry 74, 12–19, https://doi.org/10.1016/j.compbiolchem.2018.02.001 (2018).
Burrows, L. L. The Therapeutic Pipeline for Pseudomonas aeruginosa Infections. ACS infectious diseases, https://doi.org/10.1021/acsinfecdis.8b00112 (2018).
Wessel, A. K., Liew, J., Kwon, T., Marcotte, E. M. & Whiteley, M. Role of Pseudomonas aeruginosa Peptidoglycan-Associated Outer Membrane Proteins in Vesicle Formation. Journal of Bacteriology 195, 213–219, https://doi.org/10.1128/JB.01253-12 (2013).
Doring, G. & Pier, G. B. Vaccines and immunotherapy against Pseudomonas aeruginosa. Vaccine 26, 1011–1024, https://doi.org/10.1016/j.vaccine.2007.12.007 (2008).
Yang, F. et al. PA0833 Is an OmpA C-Like Protein That Confers Protection Against Pseudomonas aeruginosa Infection. Frontiers in Microbiology 9, https://doi.org/10.3389/fmicb.2018.01062 (2018).
Solanki, V., Tiwari, M. & Tiwari, V. Host-bacteria interaction and adhesin study for development of therapeutics. International journal of biological macromolecules 112, 54–64, https://doi.org/10.1016/j.ijbiomac.2018.01.151 (2018).
Chirani, A. S. et al. Immunological study on integrated PilQ and disulphide loop region of PilA against acute Pseudomonas aeruginosa infection: In silico analysis and in vitro production. Journal of Acute Disease 5, 131–142, https://doi.org/10.1016/j.joad.2015.11.006 (2016).
Priebe, G. P. & Goldberg, J. B. Vaccines for Pseudomonas aeruginosa: a long and winding road. Expert review of vaccines 13, 507–519, https://doi.org/10.1586/14760584.2014.890053 (2014).
Qian, F. et al. Conjugating recombinant proteins to Pseudomonas aeruginosa ExoProtein A: a strategy for enhancing immunogenicity of malaria vaccine candidates. Vaccine 25, 3923–3933, https://doi.org/10.1016/j.vaccine.2007.02.073 (2007).
Cripps, A. W. et al. Safety and immunogenicity of an oral inactivated whole-cell pseudomonas aeruginosa vaccine administered to healthy human subjects. Infection and immunity 74, 968–974, https://doi.org/10.1128/iai.74.2.968-974.2006 (2006).
Kamei, A., Coutinho-Sledge, Y. S., Goldberg, J. B., Priebe, G. P. & Pier, G. B. Mucosal vaccination with a multivalent, live-attenuated vaccine induces multifactorial immunity against Pseudomonas aeruginosa acute lung infection. Infection and immunity 79, 1289–1299, https://doi.org/10.1128/iai.01139-10 (2011).
Meynet, E. et al. Killed but metabolically active Pseudomonas aeruginosa-based vaccine induces protective humoral- and cell-mediated immunity against Pseudomonas aeruginosa pulmonary infections. Vaccine 36, 1893–1900, https://doi.org/10.1016/j.vaccine.2018.02.040 (2018).
Zhang, X. et al. Immunization with Pseudomonas aeruginosa outer membrane vesicles stimulates protective immunity in mice. Vaccine 36, 1047–1054, https://doi.org/10.1016/j.vaccine.2018.01.034 (2018).
Zhao, K., Deng, X., He, C., Yue, B. & Wu, M. Pseudomonas aeruginosa outer membrane vesicles modulate host immune responses by targeting the Toll-like receptor 4 signaling pathway. Infection and immunity 81, 4509–4518, https://doi.org/10.1128/iai.01008-13 (2013).
Liu, X., Li, W. G. & Luo, G. X. [Study on Construction of Recombinant Bb-pGEX-OprI Vaccine of Pseudomonas aeruginosa and Its Protection Effect]. Sichuan da xue xue bao. Yi xue ban = Journal of Sichuan University. Medical science edition 49, 13–17 (2018).
Saha, S. et al. Blocking of the TLR5 activation domain hampers protective potential of flagellin DNA vaccine. Journal of immunology (Baltimore, Md.: 1950) 179, 1147–1154 (2007).
Horzempa, J. et al. Immunization with a Pseudomonas aeruginosa 1244 pilin provides O-antigen-specific protection. Clinical and vaccine immunology: CVI 15, 590–597, https://doi.org/10.1128/cvi.00476-07 (2008).
Micoli, F., Costantino, P. & Adamo, R. Potential targets for next generation antimicrobial glycoconjugate vaccines. FEMS Microbiology Reviews 42, 388–423, https://doi.org/10.1093/femsre/fuy011 (2018).
Rashid, M. I., Naz, A., Ali, A. & Andleeb, S. Prediction of vaccine candidates against Pseudomonas aeruginosa: An integrated genomics and proteomics approach. Genomics 109, 274–283, https://doi.org/10.1016/j.ygeno.2017.05.001 (2017).
Uddin, R. & Jamil, F. Prioritization of potential drug targets against P. aeruginosa by core proteomic analysis using computational subtractive genomics and Protein-Protein interaction network. Computational biology and chemistry 74, 115–122, https://doi.org/10.1016/j.compbiolchem.2018.02.017 (2018).
Rappuoli, R., Bottomley, M. J., D’Oro, U., Finco, O. & De Gregorio, E. Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. The Journal of Experimental Medicine 213, 469–481, https://doi.org/10.1084/jem.20151960 (2016).
Solanki, V. & Tiwari, V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Scientific Reports 8, 9044, https://doi.org/10.1038/s41598-018-26689-7 (2018).
Dutta, A. et al. In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori. In silico biology 6, 43–47 (2006).
Vij, R. et al. A targeted boost-and-sort immunization strategy using Escherichia coli BamA identifies rare growth inhibitory antibodies. Scientific Reports 8, 7136, https://doi.org/10.1038/s41598-018-25609-z (2018).
Yu, N. Y. et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics (Oxford, England) 26, 1608–1615, https://doi.org/10.1093/bioinformatics/btq249 (2010).
Yu, C. S., Chen, Y. C., Lu, C. H. & Hwang, J. K. Prediction of protein subcellular localization. Proteins 64, 643–651, https://doi.org/10.1002/prot.21018 (2006).
Petersen, T. N., Brunak, S., von Heijne, G. & Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature methods 8, 785–786, https://doi.org/10.1038/nmeth.1701 (2011).
Bagos, P. G., Tsirigos, K. D., Liakopoulos, T. D. & Hamodrakas, S. J. Prediction of lipoprotein signal peptides in Gram-positive bacteria with a Hidden Markov Model. Journal of proteome research 7, 5082–5093, https://doi.org/10.1021/pr800162c (2008).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of molecular biology 305, 567–580, https://doi.org/10.1006/jmbi.2000.4315 (2001).
Bendtsen, J. D., Kiemer, L., Fausboll, A. & Brunak, S. Non-classical protein secretion in bacteria. BMC microbiology 5, 58, https://doi.org/10.1186/1471-2180-5-58 (2005).
Gao, F., Luo, H., Zhang, C. T. & Zhang, R. Gene essentiality analysis based on DEG 10, an updated database of essential genes. Methods in molecular biology (Clifton, N.J.) 1279, 219–233, https://doi.org/10.1007/978-1-4939-2398-4_14 (2015).
Chen, L. et al. VFDB: a reference database for bacterial virulence factors. Nucleic acids research 33, D325–328, https://doi.org/10.1093/nar/gki008 (2005).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic acids research 44, D457–D462, https://doi.org/10.1093/nar/gkv1070 (2016).
Doytchinova, I. A. & Flower, D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC bioinformatics 8, 4, https://doi.org/10.1186/1471-2105-8-4 (2007).
Szklarczyk, D. et al. STRINGv10: protein–protein interaction networks, integrated over the tree of life. Nucleic acids research 43, D447–D452, https://doi.org/10.1093/nar/gku1003 (2015).
Kim, Y. et al. Immune epitope database analysis resource. Nucleic acids research 40, W525–530, https://doi.org/10.1093/nar/gks438 (2012).
Giguere, S. et al. MHC-NP: predicting peptides naturally processed by the MHC. Journal of immunological methods 400−401, 30–36, https://doi.org/10.1016/j.jim.2013.10.003 (2013).
Stranzl, T., Larsen, M. V., Lundegaard, C. & Nielsen, M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62, 357–368, https://doi.org/10.1007/s00251-010-0441-4 (2010).
Nielsen, M. & Andreatta, M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome. Medicine 8, 33, https://doi.org/10.1186/s13073-016-0288-x (2016).
Calis, J. J. et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS computational biology 9, e1003266, https://doi.org/10.1371/journal.pcbi.1003266 (2013).
Wang, P. et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC bioinformatics 11, 568, https://doi.org/10.1186/1471-2105-11-568 (2010).
Gupta, S. et al. In silico approach for predicting toxicity of peptides and proteins. PloS one 8, e73957, https://doi.org/10.1371/journal.pone.0073957 (2013).
El-Manzalawy, Y., Dobbs, D. & Honavar, V. Predicting linear B-cell epitopes using string kernels. Journal of molecular recognition: JMR 21, 243–255, https://doi.org/10.1002/jmr.893 (2008).
El-Manzalawy, Y., Dobbs, D. & Honavar, V. Predicting flexible length linear B-cell epitopes. Computational systems bioinformatics. Computational Systems Bioinformatics Conference 7, 121–132 (2008).
Larsen, J. E., Lund, O. & Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome research 2, 2, https://doi.org/10.1186/1745-7580-2-2 (2006).
Thomsen, M., Lundegaard, C., Buus, S., Lund, O. & Nielsen, M. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics 65, 655–665, https://doi.org/10.1007/s00251-013-0714-9 (2013).
Lee, S. J. et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PloS one 9, e104351, https://doi.org/10.1371/journal.pone.0104351 (2014).
Mei, H. F. et al. beta-defensin 2 as an adjuvant promotes anti-melanoma immune responses and inhibits the growth of implanted murine melanoma in vivo. PloS one 7, e31328, https://doi.org/10.1371/journal.pone.0031328 (2012).
Rana, A. & Akhter, Y. A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure based approach. Immunobiology 221, 544–557, https://doi.org/10.1016/j.imbio.2015.12.004 (2016).
Alexander, J. et al. Linear PADRE T Helper Epitope and Carbohydrate B Cell Epitope Conjugates Induce Specific High Titer IgG Antibody Responses. The Journal of Immunology 164, 1625–1633, https://doi.org/10.4049/jimmunol.164.3.1625 (2000).
Wu, C. Y., Monie, A., Pang, X., Hung, C. F. & Wu, T. C. Improving therapeutic HPV peptide-based vaccine potency by enhancing CD4+ T help and dendritic cell activation. Journal of biomedical science 17, 88, https://doi.org/10.1186/1423-0127-17-88 (2010).
Yang, Y. et al. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Human vaccines & immunotherapeutics 11, 795–805, https://doi.org/10.1080/21645515.2015.1012017 (2015).
Saha, S. & Raghava, G. P. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic acids research 34, W202–209, https://doi.org/10.1093/nar/gkl343 (2006).
Cheng, J., Randall, A. Z., Sweredoski, M. J. & Baldi, P. SCRATCH: a protein structure and structural feature prediction server. Nucleic acids research 33, W72–76, https://doi.org/10.1093/nar/gki396 (2005).
Gasteiger, E. et al. In The Proteomics Protocols Handbook (ed. John M. Walker) 571–607 (Humana Press, 2005).
Buchan, D. W. A., Minneci, F., Nugent, T. C. O., Bryson, K. & Jones, D. T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic acids research 41, W349–W357, https://doi.org/10.1093/nar/gkt381 (2013).
Dina, S.-D. et al. Taking geometry to its edge: Fast unbound rigid (and hinge-bent) docking. Proteins: Structure, Function, and Bioinformatics 52, 107–112, https://doi.org/10.1002/prot.10397 (2003).
Kozakov, D. et al. The ClusPro web server for protein-protein docking. Nature protocols 12, 255–278, https://doi.org/10.1038/nprot.2016.169 (2017).
Verma, P., Tiwari, M. & Tiwari, V. In silico high-throughput virtual screening and molecular dynamics simulation study to identify inhibitor for AdeABC efflux pump of Acinetobacter baumannii. J Biomol Struct Dyn 36, 1182–1194, https://doi.org/10.1080/07391102.2017.1317025 (2018).
Grote, A. et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic acids research 33, W526–531, https://doi.org/10.1093/nar/gki376 (2005).
Adames, N. R. et al. GenoLIB: a database of biological parts derived from a library of common plasmid features. Nucleic acids research 43, 4823–4832, https://doi.org/10.1093/nar/gkv272 (2015).
Acknowledgements
Vishvanath Tiwari thanks Central University of Rajasthan, Bandarsindri, Ajmer for a provided research facility to conduct the experiment. VS thanks UGC for fellowship (2061530812/21/06/2015(i)-EU-V). The present manuscript is performed in the absence of funding.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: V.T., Performed the experiments: V.S. and V.T. Analysed the data: V.T., Wrote the manuscript: V.S., M.T. and V.T., Proofread and correction in final version: M.T. and V.T.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Solanki, V., Tiwari, M. & Tiwari, V. Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Sci Rep 9, 5240 (2019). https://doi.org/10.1038/s41598-019-41496-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41496-4
This article is cited by
-
Protein profiling and immunoinformatic analysis of the secretome of a metal-resistant environmental isolate Pseudomonas aeruginosa S-8
Folia Microbiologica (2024)
-
Rational design of multi-epitope-based vaccine by exploring all dengue virus serotypes proteome: an immunoinformatic approach
Immunologic Research (2024)
-
Immunoinformatics design of multi-epitope vaccine using OmpA, OmpD and enterotoxin against non-typhoidal salmonellosis
BMC Bioinformatics (2023)
-
Designing of multi-epitope chimeric vaccine using immunoinformatic platform by targeting oncogenic strain HPV 16 and 18 against cervical cancer
Scientific Reports (2022)
-
In-Silico Vaccine Design Based on a Novel Vaccine Candidate Against Infections Caused by Acinetobacter baumannii
International Journal of Peptide Research and Therapeutics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
