
Abstract
Treatment with antibiotics leads to the selection of isolates with increased resistance. We investigated if evolution towards resistance was associated with virulence changes, in the context of P. aeruginosa ventilator-associated pneumonia (VAP). Four patients were selected because they had multiple VAP episodes during short periods (12 days to 5 weeks), with emergence of resistance. We performed whole-genome sequencing of 12 P. aeruginosa from bronchoalveolar lavages or blood culture (3 isolates per patient). Production of quorum sensing-dependent virulence factors, serum resistance, cytotoxicity against A549 cells, biofilm production, and twitching motility were studied. Each patient was infected with a unique strain. For all patients, resistance development was explained by genetic events in ampD, mexR or oprD. Additional variations were detected in virulence- and/or fitness-associated genes (algB, gacA, groEL, lasR, mpl, pilE, pilM, rhlR) depending on the strain. We noticed a convergence towards quorum sensing deficiency, correlated with a decrease of pyocyanin and protease production, survival in serum, twitching motility and cytotoxicity. In one patient, changes in pilM and pilE were related to enhanced twitching. We show that the emergence of resistance in P. aeruginosa is associated with virulence modification, even in acute infections. The consequences of this short-term pathoadaptation need to be explored.
Similar content being viewed by others


Introduction
Pseudomonas aeruginosa is an opportunistic pathogen and one of the main agents of ventilator-associated pneumonia (VAP)1,2. VAP causes significant morbidity with longer intensive care unit stay and additional hospital costs of at least US $10,0193.
P. aeruginosa has a broad arsenal of virulence factors. Their production is finely regulated, in particular by the quorum sensing. This network is composed of four interconnected systems, each consisting of a regulatory protein (transcription factor) and an autoinducing enzyme: LasR / LasI for the las system and RhlR / RhlI for the rhl system. Quorum sensing regulates 10% of P. aeruginosa genome based on bacterial density and probably on environmental stress cues4.
Antibiotic resistance is another key factor in the evolution of P. aeruginosa infections. In recent years, multidrug-resistant P. aeruginosa clones have caused outbreaks of healthcare-associated infections worldwide5. This leads to wonder about the intrinsic virulence of these multidrug-resistant strains6.
The relationship between virulence and resistance in P. aeruginosa is still poorly understood. It is generally thought that resistance implies an energy cost and impairs fitness, negatively impacting virulence. Three studies in rabbit or mouse models showed that multidrug-resistant P. aeruginosa isolates were less virulent, but these studies included a small number of genetically unrelated isolates7,8,9. The in vitro study of Mulet et al. performed on 40 clonally diverse P. aeruginosa showed that multidrug-resistant isolates had reduced motility and fitness5. On the other hand, Skurnik et al. showed that carbapenem-resistant oprD mutants had a greater ability to colonise the mouse gut and to disseminate to the spleen than non-mutated isogenic isolates10. Carbapenem-resistant isolates displayed increased cytotoxicity against murine macrophages and were more resistant to human serum in this study. In addition, P. aeruginosa protein AmpR positively regulates the expression of resistance and virulence genes11. Some specific resistance mechanisms, like loss of OprD or AmpC overexpression, could therefore be associated with modification of virulence.
Pathoadaptation phenomenon has been widely described in hypermutable P. aeruginosa strains, during chronic pulmonary infections in cystic fibrosis patients, with switch to mucoidity, antibiotic resistance and reduced production of virulence factors12, but has been poorly studied in the context of acute infections like VAP. Only recently, Wang et al. investigated the adaptive evolution of a single P. aeruginosa strain responsible for a nosocomial outbreak of VAP infections in China. They found convergent genomic events leading to attenuated virulence13.
Thus, in order to see if acquisition of resistance mechanisms could be associated with changes in strain virulence, we analysed the genetic and phenotypic characteristics of 4 P. aeruginosa strains that developed antibiotic resistance during acute VAP infections in 4 critically ill patients.
Materials and Methods
Bacterial isolates
The 12 P. aeruginosa isolates studied here were collected at Nantes Universitary Hospital in 4 VAP patients hospitalised in intensive care units. For each patient, 3 different isolates were recovered over periods of 12 days to 5 weeks. Eleven isolates were collected from bronchoalveolar lavages (BAL) and one was recovered from blood culture (Table 1). In 2 cases, 2 isolates with different colony morphologies were collected from the same BAL. The clonal relatedness of isolates was investigated by MLST (MultiLocus sequence typing) using the online P. aeruginosa MLST database (www.pubmlst.org). This study was performed on strains isolated from samples sent to the bacteriology lab for diagnosis, without any additional sampling. In Nantes Universitary Hospital, an informed consent is asked for use of data for research purpose on all patients.
Sequencing, assembly and annotation
Bacterial DNA was extracted with iPrep™ PureLink® Virus Kit (Thermo Fisher Scientific) from a single colony of each isolate. DNA concentration was measured by fluorometric method on Quantus™ (Promega®). One μg of each DNA extract was fragmented by sonication on Bioruptor® (Diagenode). The libraries were prepared using TruSeq® DNA PCR-Free Library Prep kit (Illumina®) and sequenced on MiSeq® (Illumina®) with MiSeq Reagent Kit v2 300 cycles (paired-end sequencing 2 × 150 cycles).
All sequencing data used in this study are available on NCBI BioProject n° PRJNA438112. The Illumina reads were trimmed using Trimmomatic14, quality filtered with the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) and assembled using SPAdes15. SIS software16 and GapFiller version 1.1017 were used to improve the initial set of contigs before sending for annotation using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP)18.
Comparative and phylogenetic analysis
To investigate the epidemiological linkage of our 12 isolates and commonly used strains (e.g. PAO1, PA14, ATCC_27853, NCTC_10332), environmental strains (e.g. B10W, YL84) or clinical strains (e.g. PA_D1 sampled from VAP patients; PA1 from a patient with respiratory tract infection; RP73, DK2 and FRD1 from cystic fibrosis patients), 43 complete genome sequences were retrieved from NCBI (http://www.ncbi.nlm.nih.gov/genome). Core-genome alignment was carried out using PARSNP v. 1.219 and PAO1 genome as reference. Recombinant sites were identified using PhiPack20 and single-nucleotide polymorphisms (SNPs) located in those regions were filtered yielding 172,245 single-nucleotide variants. Phylogenetic analysis was performed considering the 172,245 polymorphic sites of the 55 genomes. A transversion substitution model was selected on the basis of the Akaike’s information criterion with jModelTest221. Maximum likelihood phylogeny was constructed using PhyML22 and visualized using Figtree (http://tree.bio.ed.ac.uk/software/figtree/). The 12 P. aeruginosa genomes of this study were also submitted to CSIphylogeny (https://cge.cbs.dtu.dk/services/CSIPhylogeny/) for pan genomic analysis. Antibiotic resistance genes were predicted from PAO1 genome and the assembled genomes using the ResFinder 3.0 server [https://cge.cbs.dtu.dk/services/ResFinder/]. Comparison of PAO1 genome against our 4 P. aeruginosa strains genomes was done by BLAST search using BLAST Ring Image Generator 0.9523. Genomes were annotated using Prokka v1.12 software24 and proteomes were used for pan-genomic analysis with OrthoMCL software25. Homologous sequences were selected using the all-against-all BlastP algorithm with an E value of <10−5 and 60% nucleotide identity. Functional annotation was performed using COG database (http://weizhongli-lab.org/metagenomic-analysis/server/cog/).
Detection of nucleotide differences
Nucleotide differences were detected and evaluated using CLC GENOMIC WORKBENCH. For each patient, the assembled early isolate genome was annotated and used as reference genome for the other 2 isolates. The paired-end reads in FASTQ format were mapped to the early isolate genome and further compared with each other to generate lists of SNPs and short indels. A frequency cut-off of more than 80% was set to minimise false SNPs due to sequencing error. For confirmation of nucleotide differences, the 3 BAM files from each patient were visualised and compared using the Integrative Genomics Viewer (IGV) version 2.3 (Broad Institute, Cambridge, MA), and questionable regions were analysed by PCR/Sanger sequencing. In order to compare with the reference genome, all paired-end reads were mapped to the annotated genome PAO1.
Antimicrobial susceptibility testing
Antibiotic susceptibility was determined by VITEK® 2 XL (bioMérieux, Marcy l’Etoile, France).
Resistance to human serum
Resistance to pooled human serum was assessed as previously described26 with a final serum concentration of 75%. Responses were graded from 1 to 6. Strains were categorised as sensitive [3-hours count <10% of the inoculum, i.e. grade 1 (1-hour count <10%) and grade 2 (1-hour count between 10% and 100%)], intermediate [1-hour count between 10% and 100%, i.e. grade 3 (3-hours count between 10% and 100%) and grade 4 (3-hours count >100%)] or resistant [1-hour count >100%, i.e. grade 5 (3-hours count between 10% and 100%) and grade 6 (3-hours count >100%)]. Isolates were tested three times.
Proteolytic activity assay
Proteolytic activity of the culture supernatants was assessed as previously described27. Skim milk agar plates containing 10% of skim milk and 1% of agar were prepared. Supernatants of bacterial cultures were added into 9 mm diameter punched holes in skim milk agar and incubated at 37 °C for 24 h. Proteolytic activity was measured by the diameter of the clear zone surrounding the holes. Three independent experiments were performed for each isolate.
Pyocyanin quantification assay
After overnight incubation at 37 °C, 3 mL of chloroform were added to 5 mL culture supernatant and mixed vigorously. One milliliter of 0.2 M hydrochloric acid was added to the organic layer and the absorbance was measured at 520 nm. Pyocyanin concentration (mg/L) was calculated as: P = (OD × 17.072) x (5/3) where OD is optical density at 520 nm, 17.072 is the pyocyanin extinction coefficient, and 5/3 is the dilution factor28. Three independent experiments were performed for each isolate.
Cytotoxicity assay
A549 human lung cells monolayers seeded in 24-well tissue culture plates were infected by bacterial suspensions (multiplicity of infection of 100). After 6 hours of incubation, cytotoxicity towards A549 cells was quantified by measuring the amount of lactate dehydrogenase (LDH) released into the culture supernatant using the CytoTox 96® Non-Radioactive Cytotoxicity Assay kit (Promega®). Cytotoxicity was calculated relative to mortality of uninfected cells (set at 0%) and mortality of cells lysed with Triton X-100 1% (positive control, 100%). Assays were carried out in triplicate in three independent experiments for each isolate.
Twitching motility assay
Twitching motility was studied as previously described29. Single colonies were inoculated to the bottom of a 1% LB agar plate. The plates were incubated for 24 hours at 37 °C. The agar was carefully removed after incubation and the adherent bacteria stained with crystal violet dye, followed by washing with tap water to remove unbound dye. Twitching zone areas were measured. Three independent experiments were performed for each isolate.
Biofilm assay
For each isolate, a 0,5 Mac Farland suspension was prepared in BHI, diluted 1/100, and used to inoculate four wells of a 96-well polystyrene microtiter plate. After 24 H at 30 °C without shaking, the bacterial suspensions were removed, the wells were washed with physiological water, air-dried for 20 mn and stained with Gram’s safranin solution for 5 mn. The plates were washed four times to wash off unbound safranin and air-dried for one hour. The adherent bacteria (biofilm) were then dissolved in 200 microliters of ethanol, and the absorbance at 560 nm was measured. The biofilm assay was performed three times independently.
Statistical analysis
When appropriate, results were expressed as mean ± standard deviation. Comparative analyses were performed with the Kruskal–Wallis test. Data were analysed using GraphPad Prism v6.0 (Graphpad Software, San Diego, CA, USA). All analyses were two-tailed. A p-value < 0.05 was considered statistically significant.
Results
Genomic characterisation of Pseudomonas aeruginosa isolates
Three isolates per patient for a total of 4 patients were sequenced and genomes characteristics investigated. Phylogenetic analysis and MLST typing showed that in each patient individually, the 3 P. aeruginosa isolates were very closely related. However, the isolates belonged to different clades from one patient to another (Supplementary Fig. S1).
To specify the relationship between those isolates, we performed pan genomic analysis of the 12 genomes. Isolates from the same patient shared more than 99% of their gene content. However, strains from different patients shared between 91.7% (PA-VAP-3 versus PA-VAP-4) and 97.1% (PA-VAP-2 versus PA-VAP-3) (Fig. 1). We predicted a total of 3,854 (79.45%) orthologous genes representing the core genome (genes shared by all isolates) and 1034 orthologous genes and singletons representing the accessory genome (genes encoded in one or more isolates but not in all). P. aeruginosa accessory genome encoded mostly proteins involved in an unknown function (38.72%), products involved in DNA and RNA processing (12.90%), cell wall biogenesis (12.51%) and metabolism and transport (34.94%). Comparative analysis of the 12 P. aeruginosa genomes revealed that strains from each patient harboured several strain-specific genomic regions. Isolates from patient 1, 2, 3 and 4 encoded respectively 169, 118, 337 and 187 unique genes (only present in these genomes). Most of these genes had unknown functions (93.4%, 94.1%, 93.1% and 92.5%, respectively).

Pan genomic analyses of the 12 isolates: heatmap representation of the gene content comparison between all isolates. The gene content is calculated as the number of orthologous genes between two isolates/the total number of genes.
We further analysed SNPs and short indels between the earliest isolate and late isolates from each patient by mapping the reads of late isolates to the genome of earliest isolate. All the detected variations were non-synonymous SNPs, deletions or insertions (Table 1). Some of these variations affected genes involved in antibiotic resistance (ampD, oprD, mexR). Others concerned genes involved in bacterial virulence and/or fitness (lasR, gacA, groEL, pilM, pilE, algB, rhlR, rhlB, rhlA, mpl).
Antimicrobial susceptibility
Prediction of resistance genes using ResFinder 3.0 showed that all 12 isolates harboured genes involved in resistance to beta-lactams (blaOXA-50 and blaPAO), aminoglycosides (aph(3′)-IIb), phenicols (catB7) and fosfomycin (fosA). Besides, for each patient, one of the sequential isolates showed a different antibiotic susceptibility pattern. For most patients, the emergence of resistance was correlated with the antibiotics dispensed (Table 1). Thus, patient 1 was treated with ceftazidime after isolate 1A was recovered. Isolate 1C exhibited increased resistance to beta-lactams, explained by a nonsense mutation in ampD. Moreover, strains collected from patients 2 and 3 acquired resistance to imipenem, explained by insertion (isolate 2B) or deletion (isolate 3C) in oprD. Whereas a carbapenem treatment was administered to patient 3, surprisingly, patient 2 did not receive imipenem. Finally, patient 4 was treated with cefepime and levofloxacin after isolate 4A was collected. Isolate 4B showed increased resistance to beta-lactams and fluoroquinolones, and harboured an insertion in mexR.
Resistance to human serum
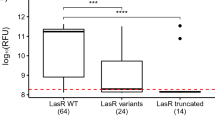
Strains from the 4 patients displayed very different levels of serum resistance (Table 2). In patient 2, all isolates escaped to serum killing, with a 3-hours count >100% of the inoculum whereas in patients 1 and 3, all isolates were very sensitive to serum (1-hour and 3-hours count <10%). In addition, a significant difference was detected between isolates from patient 4. The isolate 4B, a lasR mutant, was more sensitive to serum bactericidal action than isolates 4A and 4C (1-hour and 3-hours counts <100% for isolate 4B vs all counts >100% for isolates 4A and 4C).
Proteolytic activity assay
Important differences were observed between the isolates for 2 patients (Table 2). Proteolytic activity of isolate 3A was higher than isolates 3B and 3C, devoid of protease activity and lasR mutants. Similarly, proteolytic activity of isolates 4A and 4C was higher than isolate 4B, a lasR and rhl mutant which did not show any protease activity. Isolates collected in patient 1 did not show any proteolytic activity, in correlation with a 113-bp deletion in lasR for the 3 isolates (compared to reference strain PAO1).
Pyocyanin quantification assay
Important differences were detected between the isolates for 2 patients (Table 2). Pyocyanin production by isolate 3A was higher than lasR mutants isolates 3B and 3C (although the difference was not statistically significant with p = 0.06). Similarly, isolates 4A and 4C produced more pyocyanin than lasR and rhl mutant isolate 4B (although p = 0.06).
Cytotoxicity assay
Differences in cytotoxic activity were observed, both interpatient and intrapatient (Table 2). ExoU positive isolates had higher levels of cytotoxicity than others (p < 0.01). Besides, a difference appeared between the exoU negative isolates from patient 4. Isolate 4B, lasR and rhl mutant, was less cytotoxic than isolates 4A and 4C (although the difference was not statistically significant with p = 0.07).
Twitching motility assay
No twitching motility was detected for all lasR mutants (Table 2). Based on the 3 experiments performed, isolates 2A and 2B were less mobile than other isolates with intact quorum sensing. However, the isolate 2C, which harboured a nonsense mutation in pilM and an insertion in pilE, recovered twitching motility (with a motility area 3 times larger compared with isolates 2A and 2B).
Biofilm assay
Important differences were detected between the isolates for 2 patients (Table 2). Biofilm production was higher for isolate 3A than for isolates 3B and 3C (p = 0.02), harbouring an insertion in algB. Furthermore, isolate 1C, which exhibited a non-synonymous mutation in gacA, produced more biofilm than isolates 1A and 1B (p = 0.02).
Discussion
In this work, we studied P. aeruginosa isolates collected sequentially in 4 patients with VAP over periods of 12 days to 5 weeks. As shown by whole genome sequencing, each patient was infected with one strain, distinct from strains found in other patients. We highlighted an intrahost diversification of these 4 strains over a short period.
On the one hand, P. aeruginosa developed antibiotic resistance in all patients, correlated with the antibiotics dispensed. The acquisition of resistance mechanisms matched genetic modifications detected by whole genome sequencing. Thus, isolate 1C exhibited increased resistance to beta-lactams, explained by a nonsense mutation in ampD. Indeed, ampD inactivation is known to lead to hyperproduction of beta-lactamase AmpC30. Moreover, strains collected from patients 2 and 3 acquired resistance to imipenem, explained by genetic variations in oprD. Actually, the main mechanism of imipenem resistance in P. aeruginosa is alteration or decreased production of outer membrane porin OprD31. Finally, isolate 4B showed increased resistance to beta-lactams and fluoroquinolones, and harboured an insertion in mexR. It has been shown that premature termination of MexR leads to overexpression of MexAB-OprM efflux pump32, of which beta-lactams and fluoroquinolones are substrates. In a study by Wang et al., an epidemic ST1971 P. aeruginosa strain underwent rapid evolution during VAP infections. They described emergence of mpl mutants under beta-lactam selective pressure in 3 patients. Some of these mutants showed increased resistance to ceftazidime13. In our study, we observed the appearance of a mpl mutant in patient 3 under beta-lactam treatment. However, the V105G substitution in Mpl protein (UDP-N-acetylmuramate:L-alanyl-γ-D-glutamyl-meso-diaminopimelate ligase) was not related to increased resistance to ceftazidime.
On the other hand, we detected genetic events associated with changes in bacterial virulence and especially emergence of quorum sensing deficiency. Indeed, quorum sensing mutants appeared in patients 3 and 4. Isolates 3B and 3C were lasR mutants, while isolate 4B was a lasR and rhl system double mutant (Table 1). Besides, all 3 isolates from patient 1 were lasR mutants. These 6 isolates displayed reduced production of quorum sensing-dependent virulence factors (pyocyanin and protease), but also decreased serum resistance, cytotoxicity on A549 cells (except for exoU positive isolates) and twitching motility (Table 2). It is noteworthy that isolate 4C, collected in a blood culture 2 days after isolate 4B, was not quorum sensing deficient and therefore retained serum resistance.
Quorum sensing mutants have been poorly described apart from chronic P. aeruginosa infections. Three studies have reported such mutants in lung colonisation of mechanically ventilated patients, in highly variable proportions (19 to 76% of P. aeruginosa strains)33,34,35. Quorum sensing mutants were frequently recovered in association with wild-type isolates and their proportion increased in patients over time in one study34. Furthermore, two studies identified quorum sensing deficient isolates in P. aeruginosa acute pneumonia, but in less than one third of patients36,37. As in our work, mutants evolved in parallel towards increased antibiotic resistance in the study by Karatuna et al37. Besides, Wang et al. described quick evolution towards quorum sensing deficiency during VAP, as in our study13. They showed that a deletion causing a LasR deficient phenotype slightly impaired the in vivo virulence of P. aeruginosa in a murine pulmonary infection model. Considering the divergent data found from literature, the virulence of the quorum sensing mutants may actually vary depending on the type of infection. In a study by Köhler et al., VAP occurred less frequently in patients colonised with quorum sensing mutants than in patients colonised with non-deficient P. aeruginosa35. On the opposite, among 100 P. aeruginosa isolates responsible for corneal ulcers, 22% were lasR mutants and were associated with worse patient outcomes38. In our study, among the 3 patients infected with quorum sensing mutants, one died in intensive care unit and 2 could discharge, while the patient without quorum sensing mutant died in intensive care unit. Given the small number of patients, it is here impossible to conclude about the intrahost virulence of lasR mutants. As a matter of fact, it is difficult to define their pathogenicity in vivo since they are probably present in combination with the original non-mutated isolates. Thus, “cheaters” have a fitness advantage as they benefit from quorum sensing-dependent molecules without paying the metabolic cost. The true prevalence of these mutants and their virulence during acute infections in critically ill patients should be further explored, taking into account a potential heterogeneity of the P. aeruginosa population in clinical samples.
In the study by Wang et al., convergent evolution towards attenuated virulence of P. aeruginosa was largely explained by pyoverdine deficiency in 3 VAP patients. They detected mutations in pvdS, which encodes a sigma factor that coordinates the expression of multiple proteins involved in pyoverdine synthesis and many virulence factors13. Pyoverdine-deficient mutants were isolated after 24 to 78 days of mechanical ventilation, at later stages than quorum sensing mutants. We did not find evolution towards pvdS modifications despite an observation period of more than 30 days in some patients.
However, we discovered emergence of a non-synonymous SNP in gacA for patient 1 strain (Table 1) as in the study by Wang et al. GacA is the response regulator of the GacA/GacS two-component system that regulates expression of a large number of genes and especially promotes biofilm formation39. The S201A substitution detected in GacA for isolate 1C was associated with increased biofilm formation (Table 2). Conversely, the strain infecting patient 3 evolved to reduced biofilm production, which was correlated with a 10-bp insertion in algB. Indeed, transcriptional activator AlgB regulates alginate production in P. aeruginosa40. Thus, in our study, biofilm production did not seem to play a relevant role in recurrence of VAP, keeping in mind that its regulation depends on a large number of factors and that its in vitro assessment is quite difficult.
Other variations were detected in genes known to have a crucial role in P. aeruginosa virulence. A non-synonymous SNP in groEL arose in strain from patient 2 (Table 1). GroEL is a chaperonin protein, homolog of heat shock protein 60. Shin et al. demonstrated that secretion of GroEL by P. aeruginosa stimulated the release of pentraxin PTX3 by human monocytes, promoting innate immune response41.
Lastly, isolate 2C harboured a nonsense mutation in pilM and an insertion in pilE, encoding subunits of type IV pili. This P. aeruginosa major virulence factor contributes to adhesion, biofilm formation, extracellular protein secretion and flagellum-independent twitching motility42. We found that variations in pilM and pilE were correlated with enhanced twitching motility (Table 2). It could be assumed that these modifications in the type IV pili structure improved its flexibility.
In conclusion, we clearly proved that pathoadaptation occurred over a short period (from 10 to 25 days) in 4 different non-hypermutable P. aeruginosa strains. These intrahost genetic and phenotypic modifications under antibiotic selection pressure in the context of VAP combined antimicrobial resistance development and changes in bacterial virulence. We particularly highlighted a convergent trend towards quorum sensing deficiency as in the recent study of Wang et al.13. Further studies are required to investigate the prevalence of quorum sensing mutants in various types of acute infection as well as their link with antibiotic treatment and their pathogenicity.
Data Availability
The whole-genome shotgun sequencing data for P. aeruginosa PA-VAP-1A to PA-VAP-4C have been deposited at Genbank under accession numbers CP028332, CP028331, CP028330 and CP028368.
References
Sader, H. S. et al. Ceftazidime/avibactam tested against Gram-negative bacteria from intensive care unit (ICU) and non-ICU patients, including those with ventilator-associated pneumonia. Int J Antimicrob Agents. 46, 53–9 (2015).
Waters, B. & Muscedere, J. A 2015 update on ventilator-associated pneumonia: new insights on its prevention, diagnosis, and treatment. Curr Infect Dis Rep. 17, 496 (2015).
Safdar, N., Dezfulian, C., Collard, H. R. & Saint, S. Clinical and economic consequences of ventilator-associated pneumonia: a systematic review. Crit Care Med. 33, 2184–93 (2005).
Lee, J. & Zhang, L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell. 6, 26–41 (2015).
Mulet, X. et al. Biological markers of Pseudomonas aeruginosa epidemic high-risk clones. Antimicrob Agents Chemother. 57, 5527–35 (2013).
Juan, C., Peña, C. & Oliver, A. Host and pathogen biomarkers for severe Pseudomonas aeruginosa infections. J Infect Dis. 15, S44–51 (2017).
Giamarellos-Bourboulis, E. J. et al. Experimental sepsis using Pseudomonas aeruginosa: the significance of multi-drug resistance. Int J Antimicrob Agents. 24, 357–61 (2004).
Giamarellos-Bourboulis, E. J. et al. Impact of multidrug resistance on experimental empyema by Pseudomonas aeruginosa. Respir Int Rev Thorac Dis. 82, 46–53 (2011).
Gómez-Zorrilla, S. et al. Impact of multidrug resistance on the pathogenicity of Pseudomonas aeruginosa: in vitro and in vivo studies. Int J Antimicrob Agents. 47, 368–74 (2016).
Skurnik, D. et al. Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc Natl Acad Sci. 110, 20747–52 (2013).
Balasubramanian, D., Kumari, H. & Mathee, K. Pseudomonas aeruginosa AmpR: an acute-chronic switch regulator. Pathog Dis. 73, 1–14 (2015).
Winstanley, C., O’Brien, S. & Brockhurst, M. A. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 24, 327–37 (2016).
Wang, K. et al. The rapid in vivo evolution of Pseudomonas aeruginosa in ventilator-associated pneumonia patients leads to attenuated virulence. Open Biol. 7 (2017).
Lohse, M. et al. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 40, W622–7 (2012).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19, 455–77 (2012).
Dias, Z., Dias, U. & Setubal, J. C. SIS: a program to generate draft genome sequence scaffolds for prokaryotes. BMC Bioinformatics. 13, 96 (2012).
Nadalin, F., Vezzi, F. & Policriti, A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics. 13, S8 (2012).
Angiuoli, S. V. et al. Toward an online repository of standard operating procedures (SOPs) for (meta)genomic annotation. OMICS. 12, 137–41 (2008).
Treangen, T. J., Ondov, B. D., Koren, S. & Phillippy, A. M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15, 524 (2014).
Bruen, T. C., Philippe, H. & Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 172, 2665–81 (2006).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9, 772–772 (2012).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59, 307–21 (2010).
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 12, 402 (2011).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinforma Oxf Engl. 30, 2068–9 (2014).
Li, L. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–89 (2003).
Crémet, L. et al. Innate immune evasion of Escherichia coli clinical strains from orthopedic implant infections. Eur J Clin Microbiol Infect Dis. 35, 993–9 (2016).
Yin, H. et al. Tea polyphenols as an antivirulence compound disrupt quorum-sensing regulated pathogenicity of Pseudomonas aeruginosa. Sci Rep. 5, 16158 (2015).
Saurav, K. et al. In search of alternative antibiotic drugs: quorum-quenching activity in sponges and their bacterial isolates. Front Microbiol. 7, 416 (2016).
Leighton, T. L., Yong, D. H., Howell, P. L. & Burrows, L. L. Type IV pilus alignment subcomplex proteins PilN and PilO form homo- and heterodimers in vivo. J Biol Chem. 291, 19923–38 (2016).
Juan, C., Moyá, B., Pérez, J. & Oliver, A. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level beta-lactam resistance involves three AmpD homologues. Antimicrob Agents Chemother. 50, 1780–7 (2006).
Pai, H. et al. Carbapenem resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother. 45, 480–4 (2001).
Choudhury, D. et al. Premature termination of MexR leads to overexpression of MexAB-OprM efflux pump in Pseudomonas aeruginosa in a tertiary referral hospital in India. PloS One. 11, e0149156 (2016).
Denervaud, V. et al. Characterization of cell-to-cell signaling-deficient Pseudomonas aeruginosa strains colonizing intubated patients. J Clin Microbiol. 42, 554–62 (2004).
Köhler, T., Buckling, A. & van Delden, C. Cooperation and virulence of clinical Pseudomonas aeruginosa populations. Proc Natl Acad Sci USA 106, 6339–44 (2009).
Köhler, T., Guanella, R., Carlet, J. & van Delden, C. Quorum sensing-dependent virulence during Pseudomonas aeruginosa colonisation and pneumonia in mechanically ventilated patients. Thorax. 65, 703–10 (2010).
Le Berre, R. et al. Quorum-sensing activity and related virulence factor expression in clinically pathogenic isolates of Pseudomonas aeruginosa. Clin Microbiol Infect. 14, 337–43 (2008).
Karatuna, O. & Yagci, A. Analysis of quorum sensing-dependent virulence factor production and its relationship with antimicrobial susceptibility in Pseudomonas aeruginosa respiratory isolates. Clin Microbiol Infect. 16, 1770–5 (2010).
Hammond, J. H. et al. Environmentally endemic Pseudomonas aeruginosa strains with mutations in lasR are associated with increased disease severity in corneal ulcers. mSphere. 1, e00140–16 (2016).
Parkins, M. D., Ceri, H. & Storey, D. G. Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm formation. Mol Microbiol. 40, 1215–26 (2001).
Damron, F. H., Qiu, D. & Yu, H. D. The Pseudomonas aeruginosa sensor kinase KinB negatively controls alginate production through AlgW-dependent MucA proteolysis. J Bacteriol. 191, 2285–95 (2009).
Shin, H., Jeon, J., Lee, J. H., Jin, S. & Ha, U. H. Pseudomonas aeruginosa GroEL stimulates production of PTX3 by activating the NF-κB pathway and simultaneously downregulating microRNA-9. Infect Immun. 85, e00935–16 (2017).
Burrows, L. L. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu Rev Microbiol. 66, 493–520 (2012).
Acknowledgements
We are most grateful to the Genomics and Bioinformatics Core Facility of Nantes (GenoBiRD, Biogenouest) for its technical support. This work was supported by the french ≪Ministère de l’Enseignement Supérieur et de la Recherche≫.
Author information
Authors and Affiliations
Contributions
E.P., S.D., N.C. and L.C. prepared libraries and performed phenotypic tests. M.S., M.A. and M.B. performed genomic analyses. K.A. contributed to the clinical and therapeutic management of patients. E.P., M.S., M.A., M.B., N.C. and L.C. contributed to writing and reviewing the article.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Persyn, E., Sassi, M., Aubry, M. et al. Rapid genetic and phenotypic changes in Pseudomonas aeruginosa clinical strains during ventilator-associated pneumonia. Sci Rep 9, 4720 (2019). https://doi.org/10.1038/s41598-019-41201-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41201-5
This article is cited by
-
Prevalence of different virulence factors and their association with antimicrobial resistance among Pseudomonas aeruginosa clinical isolates from Egypt
BMC Microbiology (2023)
-
Prediction of Potential Drug Targets and Vaccine Candidates Against Antibiotic-Resistant Pseudomonas aeruginosa
International Journal of Peptide Research and Therapeutics (2022)
-
Rapid expansion and extinction of antibiotic resistance mutations during treatment of acute bacterial respiratory infections
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
