
Abstract
Granulosa cells play important roles in ovarian follicular development. To better understand the molecular mechanisms involved in this physiological process in chicken, high-throughput transcriptome analyses were performed to study the expression profiles of granulosa cells harvested from 6 mm white follicles, F5 follicles and F1 follicles. The analyses elucidated a clear tendency of granulosa cells in shifting its expression profile from proliferation to differentiation during follicular development. Transcripts down-regulated during this process were mainly associated with cell division, cell cycle and DNA replication while the up-regulated transcripts were related to ribosomal function, lipid metabolism and protein synthesis. Our study for the first time provides the complete gene expression profiles along follicular development supporting the active involvement of many genes characterized in cell signaling (AMH, Inhibins, Activins, BMPs) and transcription factors (SMAD3, SMAD5, ID1, ID2, ID3). Their temporal expression profiles support the notion of continual cross-talk between granulosa cells and its neighboring cells and shed light on the mechanisms behind avian follicular selection and pave the way to the better understanding of reproductive efficiency.
Similar content being viewed by others

Introduction
Chicken ovarian follicle contains three types of cells, namely oocyte, granulosa cells and theca cells. Granulosa cells, which form a layer surrounding the oocyte, are the main coordinator for follicular development1,2. Granulosa cells synthesize estradiol and progesterone in response to the stimulation of gonadotropins (FSH and LH) respectively, and coordinate follicular maturation and oviposition3. On the other hand, granulosa cell-derived Kit ligand (KL) stimulates oocyte growth and inhibits oocyte-derived BMP-15 expression in an orchestrated manner4. A recent study reported that follicular selection among the cohort of 8–13 prehierarchal follicles (6–8 mm in diameter) is dependent on the up-expression of FSH receptor (FSHR) transcripts in its granulosa cells, substantiating the key role of granulosa cells in follicular selection5.
Within the last few years, high-throughput transcriptome analysis has become a popular and powerful tool to discover novel genes and delineate the complete gene interaction network and has been applied on studies on follicular development. In cows, the transcriptome profile has been analyzed in granulosa cells to study the mechanisms behind dominant follicle selection6. The gene cyclin D2 (CCND2) has been implicated in the regulation of granulosa cell proliferation6,7,8. A recent transcriptome study on the bovine granulosa cells reveal the novel biomarkers of follicular status after FSH decline or withdrawal9. In pigs, the gene expression profiles of granulosa cell harvested from terminal follicles have been analyzed revealing the involvement of novel genes originally thought to be associated with mainly immune response and inflammation10. In horses, the transcriptome analyses reveal the distinct expression profile in granulosa and theca cells from developmental follicles11.
In contrast to its wide application in mammals, the use of high-throughput transcriptome analysis in avian models is limited. So far, studies have mainly used microarray analyses to uncover the genes involved in oocyte maturation and early embryonic development. In broiler breeder ovaries, candidate genes involved in ovarian functions and follicle number decision have been identified, for instance, platelet-derived growth factor receptor-like (PDGFRL) which is believed to play a part in steroid-based feedback12. Another transcriptome analyses on small yellow follicles has highlighted the potential roles of WNT4 in follicular selection13.
The domestic hen ovary is described as a cohort of multiple ovarian follicles, including thousands of primordial follicles, multiple primary follicles (<6 mm), prehierarchal (6–8 mm) and preovulatory follicles (10–40 mm), where in the 3 later stage follicles granulosa and theca cells are enclosed. These ovarian follicles varying in distinct size and developmental stage are excellent research materials for the study of follicular growth, selection and maturation14. In the present study, high-throughput transcriptome analyses were employed to study the differential gene expression profile of chicken granulosa cells in three different stages of developmental follicles, aiming to reveal the active roles of these cells during this significant process often associated with reproductive efficiency. Besides, comparison between our current chicken granulosa cell gene expression profiles and previous bovine studies15 will also pave the way to the better understanding of vertebrate follicular development.
Materials and Methods
Ethics statement
All experiments were performed according to the regulations and guidelines established by the Ministry of Science and Technology of the People’s Republic of China (Approval number: 2006–398). All animal handling procedures followed the animal welfare recommendations and were approved by the Animal Ethics Committee of Sichuan University.
Animal tissues
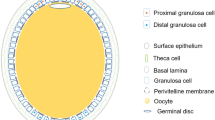
Laying hens with normal follicular hierarchies (Lohmann Layer strain) were kindly provided by MUXING company in Sichuan, China. In this study, follicles were collected from a total of eight hens (n = 8). In general, the hen ovary in peak laying period contains 30–100 small yolky follicles and 4–7 follicles recruited in the hierarchy. Typically, it takes the follicles in average 2 days to grow from 5 mm size to 8 mm size, and another 6 days to develop from 8 mm to pre-ovulatory stage16. Due to the time limit and sequencing cost, only follicles of three developmental stages, namely 6 mm, F5 and F1, were selected for the collection of granulosa cells for further analysis based on the characteristics described as follows (Fig. 1). The 6 mm follicles are white follicles that have yet been selected into the hierarchy17. The F5 follicles are small yellow follicles with diameter ranging from 11 to 12 mm, and upon recruitment into the hierarchy, their granulosa cells are generally believed to enter into an actively proliferative stage in order to accommodate an increasing deposition of egg yolk precursors in the follicles18. The F1 follicles represent the largest follicle found in the hierarchy just before ovulation, with diameter ranging from 38 to 42 mm.

Follicles used for high-throughput RNA analyses. 6 mm: white follicle, or known as prehierarchical follicles prior to the follicular selection; F5, small yellow follicle, also known as early preovulatory follicles after follicular recruitment; F1, the largest yellow follicle, also known as late preovulatory follicles prior to ovulation.
RNA extraction
Each follicle was subjected to RNA extraction independently. The follicle was soaked first in 1 × PBS solution buffer at 4 °C. Based on the established protocols4,19, the granulosa cells were isolated as completely as possible from the rest of the follicular compartment and dispersed in RNAzol reagent (Molecular Research Center, Cincinnati, OH, USA). According to the manufacturer’s instructions, the RNA samples were extracted and then dissolved in diethylpyrocarbonate-treated H2O for quality and quantity evaluation.
RNA-seq library construction and sequencing
RNA-seq libraries were prepared following the standard Illumina protocols by Novogene (Beijing, China). In brief, mRNA (at least 3 μg equally contributed from 8 individuals) were enriched from total RNA by poly-A oligo-attached magnetic beads with an integrity value >8.0. Double-stranded complementary DNAs were synthesized with random hexamer primers and purified with AMPure XP beads. Inserts with expected size were concentrated and subjected for transcriptome analyses.
Differentially expressed genes
Using Tophat v2.0.12 as a mapping tool20, clean reads were filtered from raw reads and mapped to the reference genome of Gallus gallus available on ftp://ftp.ensembl.org/pub/release-72/fasta/gallus_gallus/dna. HTSeq v0.6.1 was used to count the reads mapped to each gene. Typically, both FPKM (Fragment reads Per Kilobase per Million mapped reads) and RPKM (Reads Per Kilobase per Million mapped reads) can be employed for the evaluation of the transcripts abundance. Based on the sequencing depth and gene length for the reads count, FPKM was chosen in the present study as the key parameter in gene expression analyses. A q-value of <0.05, which is adjusted from p-value by the Benjamini& Hochberg method, was employed in this study and the 2-fold minimum differential expression was designated as the threshold of differentially expressed genes21.
Functional gene annotation
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed by GOSeq Release2.12 and KOBAS v2.0 respectively22,23. As mentioned above, terms with q-value <0.05 were considered significantly enriched by differential expressed genes. An online database resource search tool for the retrieval of interacting Genes/Proteins (STRING, http://string-db.org/) was employed for the protein-protein interaction analyses. String score higher than 700 was collected and then subjected for the analyses in software Cytoscape v3.3.024.
Reverse transcription and quantitative real-time PCR validation
Oligodeoxythymide (0.5 μg) and total RNA (2 μg) were mixed in a total volume of 5 μL, and incubated at 70 °C for 10 mins, then cooled at 4 °C for 2 mins. 1 μL single strand buffer, 0.5 μL each deoxynucleotide triphosphate, 0.5 μg oligo-deoxythymidine, and 100U moloney murine leukemia virus (MMLV) reverse transcriptase (Promega, Madison, WI, USA) were then added into the reaction mix in a total volume of 10 μL. Reverse transcription (RT) was performed at 42 °C for 90 mins.
To further validate the mRNA level of the many genes including StAR, LHCGR, CYP11A1, CYP51A1, CYP1B1, INHBB, WNT4, SMAD3, SMAD5, ID1, ID2 and ID3, quantitative real-time PCR was performed. Primers used for the amplification of the target genes were listed in Table 1. According to our previously established method25, the real-time PCR was conducted on the CFX96 real-time PCR Detection System (Bio-Rad, Hercules, CA, USA) in a total volume of 20 μL containing 13.2 μL Mili-Q water, 0.5 μL DMSO, 2 μL RT product, 1 μL single PCR buffer, 0.4 μL 2 mM each dNTP, 0.3 μL 20 mM primer, 0.3 μL Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA), and 1 μL EvaGreen (Biotium Inc., Hayward, CA, USA). The specificity of PCR amplification was first checked by agarose gel electrophoresis and the identity of all PCR products were confirmed by sequencing. The mRNA level of the target genes was first normalized as the ratio to that of GAPDH and EF1A, and then expressed as the fold difference to the control group. The qPCR data of three groups (n = 8 each) was analyzed by the comparative Ct method26, and tested for normality using Student’s t test or by one-way ANOVA followed by the Dunnett test using GraphPad Prism 6.01 (GraphPad Software, San Diego, CA). To validate the results, all experiments were repeated at least twice.
Results
Differential Gene Expression Profiles of Granulosa Cells collected from 6 mm and F5 Follicles
In present study, granulosa cells collected from follicles of the three developmental stages, ie 6 mm, F5 and F1 follicles, were subjected to high-throughput RNA analyses (Fig. 1). All sequencing data were deposited in NCBI public database (accession number: GSE112470). The 6 mm follicles and F5 follicles represent the stages before and after follicular selection, thus their expression profiles were compared in this study, in hope to provide some clues to the molecular mechanisms behind the key process of follicular selection.
As shown in Fig. S1, in a total of 963 transcripts showed differential expression profile between granulosa cells harvested from 6 mm follicles and F5 follicles. Among these, 529 transcripts showed up-regulation while the other 434 transcripts showed down-regulation in the stage of F5 follicles when compared with 6 mm follicles. The top 50 up-regulated genes and the top 50 down-regulated genes were listed in Table 2. Multiple genes actively involved in the follicular development showed great variation in their expression level. For example, the gene NR5A2 (or the liver receptor homolog 1, LRH-1), which is a member of the nuclear hormone receptor superfamily and implicated in various physiological processes from bile acid metabolism to steroidogenesis27, showed a significant increase in expression with a Log2 (fold change) value of 9.08. Elevated expression was also observed in RASD1 gene, Ras-related dexamethasone-induced 1 protein [Log2 (fold change) value = 4.19]. Transgelin, which encodes the actin cross-linking protein and has been described as an early and sensitive marker for the onset of transformation28, showed the greatest reduction in expression with Log2 (fold change) value of 11.27. COL3A1 transcript, which encodes the protein found in connective tissues and involved in cell-matrix interactions29, also showed significant down-regulation with Log2 (fold change) value of 7.70.
Besides these genes with significant variation in their expression, numerous genes with increased expression were identified, which can be grouped under similar functions as below: cholesterol metabolism (StAR, CYP11A1 and SORL1), potassium ion transmembrane transport (KCNAB1, KCNJ12, KCNJ5 and SLC8A5), dendrite morphogenesis (ELAVL4 and MAP2), steroid hormone receptor activity (ESRRB, NR5A2 and PPARG) and carboxylic ester hydrolase activity (AADACL2 and CEL). In contrast, the genes demonstrating a reduced expression are mainly implicated in peptide cross-linking (F13A1, TGM2 and TGM4), negative regulation of protein kinase activity (THY1, DCN and HSPB1), extracellular matrix organization (FBLN1, MMP9 and POSTN) and angiogenesis (THY1, ANGPT4 and CCDC80).
In an effort to reveal the relationship between these genes and known biological processes, cellular components and molecular functions, the GO terms of the significantly enrichment of differentially expressed transcripts were summarized. As shown in Fig. 2A, 216 GO terms were categorized into various biological processes including cell adhesion (GO: 0007155), cell communication (GO: 0007154), cell differentiation (GO: 0030154) and system development with 259 genes (GO: 0048731). On the other hand, a total of 36 GO terms were categorized into various cellular components including anchoring junction (GO: 0070161), cell junction (GO: 0030054), extracellular matrix (GO: 0031012) and focal adhesion with 39 genes (GO: 0005925). A total of 17 GO terms were categorized into molecular function including calcium ion binding (GO: 0005509), receptor binding (GO: 0005102), SMAD binding (GO: 0046332) and steroid binding with 13 genes (GO: 0005496).

Gene expression profiles of granulosa cells from 6 mm follilcles and F5 follicles. (A) Gene ontology (GO) functional enrichment of genes differentially expressed in granulosa cells collected from 6 mm and F5 follicles. The y-axis and x-axis indicate the number of genes in each cluster and the names of clusters respectively. (B) Scatter plot of enriched KEGG pathways for differentially expressed genes in granulosa cells collected from 6 mm and F5 follicles. The rich factor is the ratio of the differentially expressed gene number to the total gene number in a certain pathway. The size and color of the dots represent the gene number and the range of the q-value, respectively. (C) The TGF-beta signaling pathway network identified in granulosa cells collected from 6 mm and F5 follicles based on STRING database.
KEGG pathway analysis was also performed to investigate how these differentially expressed genes were involved in different cellular processes. As shown in Fig. 2B, with the threshold of q-value < 0.05, the large dots clearly showed the genes were mainly related to pathways such as focal adhesion, extracellular matrix (ECM)-receptor interaction and endocytosis. Besides pathways mentioned above, these genes also matched to other pathways including steroid biosynthesis, FoxO signaling pathway, PPAR signaling pathway, TGF-beta signaling pathway, arginine and proline metabolism, glycerolipid metabolism and histidine metabolism. To reveal how these genes may interact, protein-protein interaction analyses were carried out based on the STRING database. In particular, 10 genes identified in the present study were found to be associated with TGF-beta signaling pathway (gga04350) (Fig. 2C).
Differential Gene Expression Profiles of Granulosa Cell in F5 and F1 Follicles
The transcriptomes of granulosa cells harvested from F5 and F1 follicles were analyzed in comparison, aiming to reveal the differential gene expression profiles along the large follicle development.
As shown in Fig. S2, 1239 transcripts showed differential expression profile between granulosa cells collected from F5 and F1 follicles. Among them, 529 transcripts showed elevated level in the more developed F1 follicle when compared with F5 follicle, while the remaining 710 transcripts showed reduced level. The top 50 up-regulated genes and top 50 down-regulated genes were presented in Table 3. Some genes showed the continual up-regulation tendency starting from 6 mm follicles. For example, RASD1 transcript showed up-regulation with Log2 (fold change) value of 5.24. Similarly, STAT6 transcript, which plays a central role in the modulation of anti-apoptotic activity of IL430, showed a significant increase with Log2 (fold change) value of 4.72. In contrast, SMAD7a transcript, which is I-SMAD family member and known to compete with SMAD4 and negatively regulate TGF-beta signaling31, showed a substantial down-regulation with Log2 (fold change) value of 9.16. Similar reduction in expression level is also observed in EDN1 transcript with Log2 (fold change) value of 5.63.
Besides genes mentioned above, up-regulation was shown in genes associated with the negative regulation of BMP signaling pathway (GREM1 and VWC2), uterus development (ESR1 and LHCGR) and heart rate regulation (EPAS1 and PDE4D). In contrast, down-regulation was noted in genes related to collagen fibril organization (COL5A1, COL11A1 and LOXL2), response to hypoxia (EGLN3, EDN1 and LOXL2), modulation of synaptic transmission (CEL and NLGN3) and glucose transport (EDN1 and FABP5) and extracellular matrix (OCX36, FBN1 and ZP2).
To reveal the functional aspects of these differentially expressed genes, their GO terms are summarized in Fig. 3A. 281 genes were mapped to 72 GO terms categorized in biological processes including the cell cycle (GO: 0007049), cell division (GO: 0051301), cell proliferation (GO: 0008283) and system development (GO: 0048731). On the other hand, 150 genes were mapped to 67 GO terms categorized in the following cellular components, namely the cell junction (GO: 0030054), cell surface (GO: 0009986), extracellular matrix (GO: 0031012) and cytoskeleton (GO: 0005856). And 89 genes were matched to 4 GO terms categorized in the following molecular function, namely enzyme binding (GO: 0019899), kinase binding (GO: 0019900), structural constituent of ribosome (GO: 0003735) and structural molecule activity (GO: 0005198).

Gene expression profiles of granulosa cells collected from F5 and F1 follicles. (A) Gene ontology (GO) functional enrichment of genes differentially expressed in granulosa cells collected from F5 and F1 follicles. The y-axis and x-axis indicate the number of genes in each cluster and the names of clusters respectively. (B) Scatter plot of enriched KEGG pathways for differentially expressed genes expressed in granulosa cells collected from F5 and F1 follicles. The rich factor is the ratio of the differentially expressed gene number to the total gene number in a certain pathway. The size and color of the dots represent the gene number and the range of the q value, respectively. (C) The steroid biosynthesis network identified in granulosa cells collected from F5 and F1 follicles based on STRING database.
KEGG pathway analysis of these differentially expressed genes was shown in Fig. 3B. Using threshold q-value of <0.05, majority of these genes were associated with pathways such as ribosome function, steroid biosynthesis, cell cycle and DNA replication. Besides these 3 major pathways, other pathways such as ECM-receptor interaction, fanconi anemia pathway, PPAR signaling pathway, lysosome, glycosaminoglycan degradation, metabolic pathways and propanoate metabolism were also identified in our study. Protein-protein interaction analyses based on the STRING database showed 11 of these genes playing role in steroid biosynthesis (gga00100) (Fig. 3C).
Differential Gene Expression Profiles between Chicken and Bovine Granulosa Cells
Vertebrate ovarian follicles share similar developmental stages including primordial follicle initiation, growth, selection, maturation and the final stage, ovulation32,33. In cows, granulosa cells collected from small follicles prior to follicular selection (size <5 mm) and large follicles prior to ovulation (size >10 mm) have been analyzed in previous studies15. In the present study, the developmental stages (prior selection: 6 mm follicles, prior ovulation: F1 follicles) of chicken follicles selected in this study were well in corresponding with the follicles selected in bovine, thus their gene expression profiles were compared which provided us the unique opportunity to reveal the intricate mechanism on follicular development across species.
As shown in Fig. 4, the hierarchical clustering analyses resulted in two differential expression gene clusters between granulosa cells harvested from the two follicular stages which is conserved between bovine and chicken transcriptome findings. Among these, 46 genes showed up-regulation (Fig. 4A) and 36 genes showed down-regulation (Fig. 4B). The FPKM values of these genes from chicken and bovine studies were listed in Table S1.

Heatmap displaying the gene expression profiles of granulosa cells in (A) chicken and (B) bovine. The FPKM values were extracted from the chicken RNA-seq data in present study and the bovine Affymetrix microarrays data (NO:GSE39589). 82 genes were identified to show similar expression pattern in granulosa cells in chicken and bovine follicles.
As shown in Table S1, genes involved in steroid synthesis (NR5A2, RASD1, StAR, CYP11A1, CYP51A1, SCARB1, LHCGR, CYP21A2) were found to show similar expression pattern between the species. Genes associated with intracellular enzymes, protein synthesis and transportation (SORBS2, HERPUD1, SQLE, SEC61A1, SSR1, QSOX1, TRAM2, AOAH, DCTD, EGLN3), and genes encoding the key intracellular signaling proteins (GRK5, SNX17, ARHGAP18, APC, OBSL1, GADD45B, RNF34, PDP1, CHST2, GRAMD4, PTP4A2, STT3B, AHCYL2, PSAP, RAC3, APCDD1, CARHSP1, RGS5, IQCA1, DNAJC6, HOMER2, MTSS1, SMOC2) were noted in the comparison. In addition, genes encoding cytoskeleton constituents and the extracellular matrix components (CDC42EP4, SPTAN1, FLNB, TEX264, EPB41, CDH3, ACTA1, FHL2, EMCN, COL6A1), genes encoding transmembrane transporters, channel proteins and receptors (SLCO3A1, SEMA6A, SLC17A5, ABCA3, FAM174B, ST6GAL1, GPC4, CLIC2, ENTPD1, FXYD6, AQP1, RBP3, PECAM1), genes encoding transcription factors (IDs, GATA6, ZNF462, ZNF609, ANKRD50, MYCBP2, TGIF1, EMX2, TRIM2, FOS, FOXP4, MYC) and genes encoding the signaling molecules (INHBA, IGFBP4, IGF2, BMP15) also shared similar expression profile between the species in two developmental stages.
Validation of gene expression profiles
To confirm the gene expression profiles obtained from the high-throughput RNA sequencing, quantitative real-time PCR analyses were also performed in the present study. Genes showing significant differential expression profiles between the 3 developmental stages including those associated with steroid synthesis (StAR, LHCGR, CYP11A1, CYP51A1, CYP1B1), signaling molecules (INHBB, WNT4), TGF-beta family downstream mediators (SMAD3, SMAD5) and transcription factors (ID1, ID2, ID3) were subjected to qPCR analyses. As shown in Fig. 5, their expression profiles were in line with the high-throughput sequencing data.

Quantitative real-time PCR validation of differentially expressed genes identified in transcriptome sequencing. The results were normalized based on the housekeeping genes GAPDH and EF1A.
Discussion
In the present study, high-throughput transcriptome analyses were employed to study the differential gene expression profiles of granulosa cells collected from three different developmental stages. In addition, the expression profiles of granulosa cells from chicken follicles were compared with that in bovine follicles, in the hope to find some clues to better understand the mechanisms behind vertebrate follicular development.
Comparison of expression profiles between granulosa cells in chicken 6 mm follicles and F5 follicles
In present study, 963 transcripts showed differential expression profiles between granulosa cells harvested from 6 mm follicles and F5 follicles. The 6 mm follicles showed abundance in transcripts mainly associated with cell cycle, cell division and DNA replication. This finding falls in line with previous reports that hen granulosa cells selected at this stage are mitotically active and undifferentiated34,35. In contrast, granulosa cells in F5 follicles were rich in transcripts associated with cell proliferation, cell communication and cell differentiation in general, supporting the phenomena of fast growth with cell differentiation and maturation characteristic in this developmental stage36.
The 529 transcripts showing substantial up-regulation are mainly related to cell communication, developmental processes, multicellular organismal development and cytoplasm in granulosa cells when comparing 6 mm and F5 follicles. On the other hand, GO analyses showed that, the 434 down-regulated transcripts, besides being related to the above processes and cellular components, were also found to be associated with extracellular matrix, extracellular organelles and extracellular regions (Fig. 2A). Different from the mammals, the chicken follicles rely on granulosa cells to transport macromolecules into the oocytes due to the lack of the lumen37. These down-regulation transcripts associated with extracellular matrix likely reflect the changes in cargo and transportation route in granulosa cells between the two developmental stages. In small white follicles, the lipoprotein-rich white yolk is continually transported through tight junctions37. In larger follicles such as F5 follicles, the lipid-rich egg yolk produced from the liver diffuses through the perivitelline membrane to reach the yolk precursor receptor LR838. KEGG analyses in present study also highlighted the focal adhesion and ECM-receptor interaction pathways, supporting the coordination between granulosa cells and its environment in fulfilling the distinct roles they play along follicular development.
Follicular development is an intricate process involving a very refined coordination between different cells, in which granulosa cells undergo cell growth and differentiation under the constant influence of complicated environmental cues. Thus, it is not surprising that 304 differentially expressed transcripts were mapped to the GO category of cell communication in our study. In particular, as shown in the analysis with STRING database (Fig. 2C), the TGF-beta signaling pathway was identified, supporting its active role in follicular development36. For example, Activin A and FSH may control tight junctions through their regulation on occludin expression in granulosa cells37. The role of TGF-beta signaling molecules in the follicular development will be further discussed in later section.
Comparison between expression profiles of granulosa cells in chicken F5 follicles and F1 follicles
In the present study, the F1 and the F5 follicles were subjected for the analyses and compared in an effort to reveal the genes involving the large follicle development. In a total of 1239 transcripts showed the differential expression profile. Along the follicle growth, the up-regulated transcripts were mainly associated in peptide biosynthetic processes, peptide metabolic processes and structural constituents of ribosome reflecting the cell’s state of differentiation. Supporting this hypothesis are the elevated levels of genes encoding LHCGR (luteinizing hormone receptor, LHR) and StAR (steroidogenic acute regulatory protein, StAR), which are markers involved in steroid synthesis39,40. Their up-regulation was also validated by real-time PCR analyses (Fig. 5A). The cell fate of full differentiation was further supported by results of KEGG analyses, in which 11 genes were identified in steroid biosynthesis (gga00100). Among them are CYP11A1, CYP51A1, CYP1B1 which encode key enzymes in steroid synthesis41,42. On the other hand, in coordination with enhanced transcripts associated with steroidogenesis, the transcripts involved in cytoskeleton showed down-regulation. This likely suggests the reorganization of intracellular infrastructure to accommodate for the need in transport of steroids. In addition, many transcripts were identified with cell cycle, cell division and DNA replication in GO analyses showed down-regulation, which falls in line with the physiology of granulosa cells moving away from proliferation in the final stage of follicular development.
Interestingly, the number of down-regulated transcripts (710) is much higher than that of up-regulated transcripts (529) in this study. The reason for this may be related with the significant down-regulation of many signaling molecules such as WNT4, IGF2 and their intracellular mediators. In chicken, the increase in follicular size, especially upon the increase in deposition of yolk protein into the oocytes, may have limited the efficiency of these signaling molecules which have paracrine/autocrine mode of action and rely mainly on simple diffusion for transfer36. Alternatively, steroids and their by-products can diffuse across cell membrane freely thus being efficient (fast) in signal transduction43. The differentiation of granulosa cell and their elevated expression of transcript in steroid synthesis may have allowed the cells to shift to a more effective signaling pathway. Thus, along the follicle growth, the signaling molecules-dependent communication between the granulosa cells may decline.
Comparison between expression profiles of chicken and bovine granulosa cells
Findings in our present study on 6 mm follicles (prior to follicular selection) and F1 follicles (prior to ovulation) in chickens were compared with the bovine findings to uncover the intricate mechanisms on follicular development across species. As shown in Table S1, many transcripts involved in lipid synthesis and metabolism network (NR5A2, RASD1, StAR, CYP11A1, LHCGR, CYP51A1, SCARB1, CYP21A2) were found to be conserved between chicken and bovine expression profiles, reflecting their similar differentiation mechanism leading to fully steroidogenic granulosa cells prior to ovulation15. In addition, cell morphology will change to be in line with the differentiation. Genes associated with cytoskeletal constituents (SPTAN1, EPB41, ACTA1) and genes implicated in their assembly and association with extracellular matrix (CDC42EP4, FLNB, PECAM1, CDH3, EMCN, COL6A1) were found to be conserved in chicken and bovine expression profile, supporting that both might experience similar need for a change in morphology15. To be coordinated with these changes, genes associated with intracellular signaling (SORBS2, IGFBP4, SNX17, ARHGAP18, APC, PDP1, CHST2, GRAMD4, PTP4A2, STT3B, AHCYL2, PSAP, RAC3, FHL2, RBP3, RGS5, IQCA1, AOAH, CARHSP1, MYC, TRIM2, DNAJC6, HOMER2, CLIC2, OBSL1, GADD45B, RNF34, MTSS1), substance transmembrane transportation (SLCO3A1, SLC17A5, ABCA3, ST6GAL1, GPC4, ENTPD1, FXYD6, AQP1) and protein synthesis and secretion (HERPUD1, SEC61A1, SSR1, TRAM2) showed similar up-regulation in both chicken and bovine follicles. The similar trend observed in these species in steroid synthesis, cytoskeleton adjustment and substrate transportation supporting that vertebrate follicles undergo similar developmental processes32,33. In fact, follicular development differs greatly between chicken and bovine. In contrast with granulosa cells lining the antrum in bovine follicles which are responsible for the maintenance of follicular fluid, granulosa cells in chicken engulf the whole oocyte and are responsible for the fast transportation of a large amount of liver-derived yolk protein precursors18. The above transcripts showed similar trend but they differed in FPKM value. For example, INHBA (βA), as the subunit for activin A (βA-βA), showed a high abundance in bovine granulosa cells but a low abundance in chicken granulosa cells. The difference may result from the different methodology employed with differential sensitivity. On the other hand, the differential abundance of INHBA (βA) between species reflect their different strategy pushing follicular development since activin A (βA-βA) is implicated in regulating granulosa cell responsiveness to gonadtropins44.
Expression profiles of the key genes in TGF-beta signaling pathway
TGF-beta superfamily were reported to be actively involved in follicular development36. In the present study, protein-protein interaction analyses on the differential expressed transcripts in F5 and 6 mm follicles highlighted 10 genes involved in TGF-beta signaling pathway (gga04350), emphasizing their importance in this process. Thus, several key members in TGF-beta superfamily were further studied in an effort to provide more insights on their potential reaction time and downstream targets (Table 4).
As a TGF-beta superfamily member, anti-mullerian hormone (AMH) binds its specific receptor to promote cell proliferation36. In female birds, anti-mullerian hormone is known for repressing the development of mullerian duct and the right ovary45. In present study, the FPKM value of AMH in 6 mm follicle was 384.3, while it showed a reduction in F5 and F1 follicles with FPKM values of 18.2 and 0.8 respectively. This drastic decline in its expression level is in line with previous report of abundant AMH expression in small follicles46. AMH is implicated in inhibiting the growth and maintaining the resting phase of remaining primordial follicles. In AMH knockout mice, female mice are fertile while its ovaries showed a significant number of small growing follicles47,48. In chickens, AMH-rich testis-conditioned medium enhances proliferation of granulosa cell in a dose-dependent manner, hinting that AMH may regulate granulosa cells via autocrine/paracrine route49. In cultured granulosa cells harvested from bovine small follicles, AMH mRNAs are actively up-regulated by BMPs secreted from oocytes50. However, treatment of oocyte-conditioned medium inhibited AMH mRNA expression in a dose-related manner in chicken granulosa cells46. The protein factors other than GDF9 and BMP15 from oocyte are suggested to regulate the mRNA expression of AMH46. Since the AMH reduction is reported to relieve its inhibition on FSHR expression, AMH is suggested to play a part in follicular selection17. The finding of potential protein factors will help to reveal the mechanism behind AMH actions in follicular selection.
In mammalian antral follicles, TGF-beta isoforms (TGF-Bs) regulate granulosa and theca cell function in a paracrine/autocrine manner51,52,53. In addition, these molecules show variation in their expression levels in different developmental stages and species specific pattern36. Although TGF-B1 was not detected in the present study, both TGF-B2 (FPKM <4.8) and TGF-B3 (<22.0) showed relatively low level across the three developmental stages studied, suggesting TGF-Bs may not play an important role during avian follicular development.
Belonging to TGF-beta superfamily, inhibin and activin are believed to play important roles in ovarian functions54,55. As a hetero-dimer, inhibin is composed of one α-subunit (INHA) and one of two β-subunits, namely βA (INHBA) and βB (INHBB). The increase in inhibin expression may have helped to maintain androgen synthesis in theca cells, which can serve as a source of substrate for estrogen synthesis during the preovulatory phase56. In present study, based on the transcriptome analyses, the expression levels of the subunits INHA, INHBA and INHBB were summarized which shed light on their intricate time-frame among developmental follicles. The FPKM value of INHA varied from 132 (6 mm follicles), 4631.3 (F5 follicles) to 3013 (F1 follicles) which is in line with previous study using Northern blot57. In accordance with the detection for INHBA transcript58, the FPKM value of INHBA is low, varying from 1.1 (6 mm follicles), 9.5 (F5 follicles) to 53.3 (F1 follicles). Thus, the abundance of inhibin-A (consisted of α/βA subunits) is predicted to be low across the developmental stages with the highest level in the largest follicles. In present study, the FPKM value of the INHBB varied from 321.2 (6 mm follicles), 893.1 (F5 follicles) to 1.7 (F1 follicles), hinting that inhibin-B (consisted of α/βB subunits) may concentrate in F5 follicles, being consistent with the immunoreactive assay59. Taken together, temporal expression of inhibin-A and inhibin-B differ along follicular development.
Activin is a homo-dimer composed of two β subunits (Activin-A: βA/βA, Activin-AB: βA/βB, Activin-B: βB/βB). So, the low level of INHBA mentioned earlier also suggests a low expression of activin-A (βA/βA) across the developmental stages. Activin-A stimulates the expression of FSHR and LHR in large follicles, and is suggested to play an important role in regulating the responsiveness of granulosa cells to gonadotropins44. The relatively low level of activin-A found in the present study supports a former notion that the activin-A from neighboring source other than granulosa cells may participate in this process60. As previously mentioned, INHBB transcript showed the highest expression level in F5 follicles, which is in line with the observation that the expression of βB-mRNA was not detected in the 4 largest follicles (F4-F1)61. In addition, in small yellow follicles, activin-B and FSHR were found to be co-expressed in abundance, and proposed for their role in follicular selection61. However, activin-B showed minimal effect on LHR and FSHR expression in cultured avian granulosa cells61. Thus, activin-B might play a role in proliferation of avian granulosa cells to coordinate the fast growth of the follicle and incorporation of large amount of yolk protein precursors into the oocyte. As TGF-beta superfamily members, activins and inhibins bind their receptors depending on SMAD2/SMAD3 for downstream signaling. As shown in Table 4 and Fig. 5, the abundance of SMAD2/SMAD3 were detected and validated, which supports the active involvement of activins and inhibins.
Among the TGF-beta superfamily ligands, bone morphogenetic proteins (BMPs) are believed to be very important in follicular development62. In present study, the expression profiles of BMP2, BMP4 and BMP6 were investigated. Interestingly, the expression of BMPs is consistently low across the developmental stages. As shown in Table 4, the FPKM value of BMP2 varied from 2.5 (6 mm follicles) to 0.9 (F1 follicles) implying that they may not play a substantial role in follicular development. BMP4, which can reduce FSHR expression and promote the expression of StAR, CYP11A and AMH thus coordinating the follicular growth63, also shared a similar expression profile. Its FPKM value is similar in 6 mm and F1 follicles, at 40.5 and 38.9 respectively. In contrast to BMP2 and BMP4, BMP6 is reported to stimulate FSHR and LHR expression and promote differentiation of granulosa cells64. In present study, BMP6 also showed a low FPKM value, varying from 3.8 (6 mm follicles) to 0.1 (F1 follicles). The BMPs bind their receptor type II (BMPRII) which activates SMAD5 for downstream signaling62. In present study, BMPRII transcript is found to be consistent across different stages, with FPKM varying only slightly from 76.5 (F1 follicles), 96.8 (F5 follicles) to 106.7 (6 mm follicles). This finding, together with the high abundance of SMAD5 with FPKM varied from 70.9 (6 mm follicles), 176.5 (F5 follicles) to 202.4 (F1 follicles), hinted that BMPs may also have sources other than granulosa cells, for instance, neighboring cells. In small follicles, BMPs such as BMP15 were reported to be secreted from the oocytes to promote proliferation of granulosa cells65. In large follicles, owing to the limit of simple diffusion, BMPs may have come from theca cells which are in closer proximity of granulosa cells. To add to this, BMP6 was reported to be expressed in abundance in theca cells and able to influence the granulosa cell proliferation64. In fact, signaling molecules such as BMPs from mesodermal origin cells have been proposed to play key roles in guiding the differentiation of endodermal origin cells such as granulosa cells66.
TGF-beta family members activate the Inhibitor of DNA-binding/differentiation proteins (IDs) in a SMAD-dependent manner thus connecting their role with these ID members67. As transcriptional regulators, IDs negatively regulate a wide range of genes with E boxes and are implicated in the regulation of cell fate68. In present study, multiple ID genes were identified in both chicken and bovine granulosa cell expression profiles, emphasizing their conserved roles along vertebrate follicular development. Our gene expression analyses further validated that ID1, ID2 and ID3 transcripts showed their highest expression level in F5 follicles (Fig. 5E). Such expression profiles are different from previous report that ID1 and ID3 transcripts showed the down-regulation and ID2 transcript showed up-regulation during the follicular development69. The discrepancy may result from the difference in sample numbers (n = 8 in present study) and reference genes (GAPDH and EF1A in present study). The prehierarchical follicles are normally maintained in an undifferentiated state by inhibitory MAP kinase (MAPK) signaling pathway mediated by epidermal growth factor ligands (EGFRLs)70. The high expression of ID1, ID3 and ID4 detected in small follicles were suggested to inhibit MAPK/EGFLs expression71. In addition, the up-regulation of ID2 protein along follicular growth was suggested to be responsible for the up-regulation of FSHR expression, a key step in follicular selection or recruitment5,69. The distinct expression profile of IDs revealed in our study strongly suggests the necessity of tracing the developmental follicles in real time in an effort to reveal the mechanisms of follicular selection.
In summary, high-throughput transcriptome analyses were employed to study the expression profiles of granulosa cells in follicles of three developmental stages in chicken. The analyses elucidated a clear tendency of granulosa cells in shifting its expression profile from proliferation to differentiation during follicular development. Transcripts down-regulated during this process were mainly associated with cell division, cell cycle and DNA replication while the up-regulated transcripts were related to ribosomal function, lipid metabolism and protein synthesis. Both chicken and bovine follicles showed a similar expression profile in steroidogenesis and cytoskeleton adjustment in follicular development. Our study for the first time provided the complete gene expression profiles along chicken follicular development, supporting the active involvement of many genes characterized in cell signaling (AMH, Inhibins, Activins, BMPs) and transcription factors (SMAD3, SMAD5, ID1, ID2, ID3). Their temporal expression profiles supports the notion of continual cross-talk between granulosa cells and its neighboring cells and shed light on the mechanisms behind avian follicular selection and pave the way to the better understanding of reproductive efficiency.
References
Charlier, C. et al. Oocyte-somatic cells interactions, lessons from evolution. BMC Genomics 13, 560, https://doi.org/10.1186/1471-2164-13-560 (2012).
Eppig, J. J. Oocyte control of ovarian follicular development and function in mammals. Reproduction 122, 829–838 (2001).
Pierce, J. G. & Parsons, T. F. Glycoprotein hormones: structure and function. Annu Rev Biochem 50, 465–495, https://doi.org/10.1146/annurev.bi.50.070181.002341 (1981).
Wang, Y., Li, J., Ying Wang, C., Yan Kwok, A. H. & Leung, F. C. Epidermal growth factor (EGF) receptor ligands in the chicken ovary: I. Evidence for heparin-binding EGF-like growth factor (HB-EGF) as a potential oocyte-derived signal to control granulosa cell proliferation and HB-EGF and kit ligand expression. Endocrinology 148, 3426–3440 (2007).
Woods, D. C. & Johnson, A. L. Regulation of follicle-stimulating hormone-receptor messenger RNA in hen granulosa cells relative to follicle selection. Biol Reprod 72, 643–650, https://doi.org/10.1095/biolreprod.104.033902 (2005).
Gupta, P. et al. Regulation and regulatory role of WNT signaling in potentiating FSH action during bovine dominant follicle selection. PloS one 9, e100201 (2014).
Evans, A. et al. Identification of genes involved in apoptosis and dominant follicle development during follicular waves in cattle. Biology of reproduction 70, 1475–1484 (2004).
Mihm, M., Baker, P., Fleming, L., Monteiro, A. & O’Shaughnessy, P. Differentiation of the bovine dominant follicle from the cohort upregulates mRNA expression for new tissue development genes. Reproduction 135, 253–265 (2008).
Landry, D. A. & Sirard, M. A. Follicle capacitation: a meta-analysis to investigate the transcriptome dynamics following follicle-stimulating hormone decline in bovine granulosa cells. Biol Reprod 99, 877–887, https://doi.org/10.1093/biolre/ioy090 (2018).
Bonnet, A. et al. In vivo gene expression in granulosa cells during pig terminal follicular development. Reproduction 136, 211–224 (2008).
Donadeu, F. X. et al. Transcriptome profiling of granulosa and theca cells during dominant follicle development in the horse. Biol Reprod 91, 111, https://doi.org/10.1095/biolreprod.114.118943 (2014).
McDerment, N., Hocking, P. & Dunn, I. Identification and characterisation of alternative transcriptional variants of PDGFRL in two lines of commercial poultry. Animal genetics 46, 498–505 (2015).
Wang, Y. et al. Transcriptome Analysis on Single Small Yellow Follicles Reveals That Wnt4 Is Involved in Chicken Follicle Selection. Front Endocrinol (Lausanne) 8, 317, https://doi.org/10.3389/fendo.2017.00317 (2017).
You, S., Bridgham, J., Foster, D. & Johnson, A. Characterization of the chicken follicle-stimulating hormone receptor (cFSH-R) complementary deoxyribonucleic acid, and expression of cFSH-R messenger ribonucleic acid in the ovary. Biology of reproduction 55, 1055–1062 (1996).
Hatzirodos, N. et al. Transcriptome profiling of granulosa cells of bovine ovarian follicles during growth from small to large antral sizes. BMC Genomics 15, 24, https://doi.org/10.1186/1471-2164-15-24 (2014).
Gilbert, A. B., Perry, M. M., Waddington, D. & Hardie, M. A. Role of atresia in establishing the follicular hierarchy in the ovary of the domestic hen (Gallus domesticus). J Reprod Fertil 69, 221–227 (1983).
Johnson, P. A. Follicle selection in the avian ovary. Reprod Domest Anim 47(Suppl 4), 283–287, https://doi.org/10.1111/j.1439-0531.2012.02087.x (2012).
Perry, M. & Gilbert, A. B. Yolk transport in the ovarian follicle of the hen (Gallus domesticus): lipoprotein-like particles at the periphery of the oocyte in the rapid growth phase. Journal of cell science 39, 257–272 (1979).
Gilbert, A., Evans, A., Perry, M. & Davidson, M. A method for separating the granulosa cells, the basal lamina and the theca of the preovulatory ovarian follicle of the domestic fowl (Gallus domesticus). Journal of reproduction and fertility 50, 179–181 (1977).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578 (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the royal statistical society. Series B (Methodological), 289–300 (1995).
Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome biology 11, R14 (2010).
Mao, X., Cai, T., Olyarchuk, J. G. & Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793 (2005).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504 (2003).
Cai, G., Mo, C., Huang, L., Li, J. & Wang, Y. Characterization of the Two CART Genes (CART1 and CART2) in Chickens (Gallus gallus). PLoS One 10, e0127107, https://doi.org/10.1371/journal.pone.0127107 (2015).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nature protocols 3, 1101–1108 (2008).
Boerboom, D., Pilon, N., Behdjani, R., Silversides, D. W. & Sirois, J. Expression and regulation of transcripts encoding two members of the NR5A nuclear receptor subfamily of orphan nuclear receptors, steroidogenic factor-1 and NR5A2, in equine ovarian cells during the ovulatory process. Endocrinology 141, 4647–4656, https://doi.org/10.1210/endo.141.12.7808 (2000).
Shapland, C., Hsuan, J. J., Totty, N. F. & Lawson, D. Purification and properties of transgelin: a transformation and shape change sensitive actin-gelling protein. J Cell Biol 121, 1065–1073 (1993).
Chen, H. Y. et al. Collagen type 3 alpha 1 polymorphism and risk of pelvic organ prolapse. Int J Gynaecol Obstet 103, 55–58, https://doi.org/10.1016/j.ijgo.2008.05.031 (2008).
Hou, J. et al. An interleukin-4-induced transcription factor: IL-4 Stat. Science 265, 1701–1706 (1994).
Zhang, Y., Feng, X. H. & Derynck, R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 394, 909–913, https://doi.org/10.1038/29814 (1998).
Adams, G. P. Comparative patterns of follicle development and selection in ruminants. J Reprod Fertil Suppl 54, 17–32 (1999).
Johnson, A. L. In Sturkie’s Avian Physiology (Sixth Edition) 635–665 (Elsevier, 2015).
Tilly, J. L., Kowalski, K. I. & Johnson, A. L. Stage of ovarian follicular development associated with the initiation of steroidogenic competence in avian granulosa cells. Biol Reprod 44, 305–314 (1991).
Tilly, J., Kowalski, K., Li, Z., Levorse, J. & Johnson, A. Plasminogen activator activity and thymidine incorporation in avian granulosa cells during follicular development and the periovulatory period. Biology of reproduction 46, 195–200 (1992).
Knight, P. G. & Glister, C. TGF-beta superfamily members and ovarian follicle development. Reproduction 132, 191–206, https://doi.org/10.1530/rep.1.01074 (2006).
Schuster, M. K., Schmierer, B., Shkumatava, A., Kuchler, K. & Activin, A. and follicle-stimulating hormone control tight junctions in avian granulosa cells by regulating occludin expression. Biol Reprod 70, 1493–1499, https://doi.org/10.1095/biolreprod.103.024331 (2004).
Schneider, W. J. Receptor-mediated mechanisms in ovarian follicle and oocyte development. General and comparative endocrinology 163, 18–23 (2009).
Peng, X. R., Hsueh, A. J., LaPolt, P. S., Bjersing, L. & Ny, T. Localization of luteinizing hormone receptor messenger ribonucleic acid expression in ovarian cell types during follicle development and ovulation. Endocrinology 129, 3200–3207, https://doi.org/10.1210/endo-129-6-3200 (1991).
Johnson, A. & Bridgham, J. Regulation of steroidogenic acute regulatory protein and luteinizing hormone receptor messenger ribonucleic acid in hen granulosa cells. Endocrinology 142, 3116–3124 (2001).
Li, Z. & Johnson, A. L. Expression and regulation of cytochrome P450 17 alpha-hydroxylase messenger ribonucleic acid levels and androstenedione production in hen granulosa cells. Biol Reprod 49, 1293–1302 (1993).
Goldstone, J. V. et al. Cytochrome P450 1 genes in early deuterostomes (tunicates and sea urchins) and vertebrates (chicken and frog): origin and diversification of the CYP1 gene family. Mol Biol Evol 24, 2619–2631, https://doi.org/10.1093/molbev/msm200 (2007).
Frye, C. A. Steroids, reproductive endocrine function, and affect. A review. Minerva Ginecol 61, 541–562 (2009).
Davis, A. J., Brooks, C. F. & Johnson, P. A. Activin A and gonadotropin regulation of follicle-stimulating hormone and luteinizing hormone receptor messenger RNA in avian granulosa cells. Biology of reproduction 65, 1352–1358 (2001).
Tran, D. & Josso, N. Relationship between avian and mammalian anti-Mulllerian hormones. Biol Reprod 16, 267–273 (1977).
Johnson, P., Kent, T., Urick, M. & Giles, J. Expression and regulation of anti-mullerian hormone in an oviparous species, the hen. Biology of reproduction 78, 13–19 (2008).
Durlinger, A. L. et al. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology 140, 5789–5796, https://doi.org/10.1210/endo.140.12.7204 (1999).
Behringer, R. R., Finegold, M. J. & Cate, R. L. Mullerian-inhibiting substance function during mammalian sexual development. Cell 79, 415–425 (1994).
Johnson, P., Kent, T., Urick, M. E., Trevino, L. & Giles, J. Expression of anti-Mullerian hormone in hens selected for different ovulation rates. Reproduction 137, 857–863 (2009).
Monniaux, D. et al. Regulation of anti-Mullerian hormone production in domestic animals. Reprod Fertil Dev 25, 1–16, https://doi.org/10.1071/RD12270 (2012).
Nilsson, E. E., Doraiswamy, V. & Skinner, M. K. Transforming growth factor-beta isoform expression during bovine ovarian antral follicle development. Mol Reprod Dev 66, 237–246, https://doi.org/10.1002/mrd.10350 (2003).
Nilsson, E. E. & Skinner, M. K. Bone morphogenetic protein-4 acts as an ovarian follicle survival factor and promotes primordial follicle development. Biol Reprod 69, 1265–1272, https://doi.org/10.1095/biolreprod.103.018671 (2003).
Juengel, J. L. & McNatty, K. P. The role of proteins of the transforming growth factor-beta superfamily in the intraovarian regulation of follicular development. Hum Reprod Update 11, 143–160, https://doi.org/10.1093/humupd/dmh061 (2005).
McNatty, K. P. et al. Growth and paracrine factors regulating follicular formation and cellular function. Mol Cell Endocrinol 163, 11–20 (2000).
Matzuk, M. M. et al. Transgenic models to study the roles of inhibins and activins in reproduction, oncogenesis, and development. Recent Prog Horm Res 51, 123–154; discussion 155–127 (1996).
Johnson, P. A. Inhibin in the hen. Poult Sci 72, 955–958, https://doi.org/10.3382/ps.0720955 (1993).
Johnson, P. A. & Wang, S. Y. Characterization and quantitation of mRNA for the inhibin alpha-subunit in the granulosa layer of the domestic hen. Gen Comp Endocrinol 90, 43–50, https://doi.org/10.1006/gcen.1993.1058 (1993).
Chen, C. C. & Johnson, P. A. Expression and regulation of mRNA for inhibin/activin alpha- and betaA-subunits in the granulosa layer of the two largest preovulatory follicles during the hen ovulatory cycle. Gen Comp Endocrinol 107, 386–393, https://doi.org/10.1006/gcen.1997.6948 (1997).
Hecht, D. J., Davis, A. J., Brooks, C. F. & Johnson, P. A. Molecular cloning and expression analysis of the complementary deoxyribonucleic acid for chicken inhibin/activin beta(B) subunit. Biol Reprod 62, 1128–1134 (2000).
Lovell, T. M., Gladwell, R. T., Cunningham, F. J., Groome, N. P. & Knight, P. G. Differential changes in inhibin A, activin A, and total alpha-subunit levels in granulosa and thecal layers of developing preovulatory follicles in the chicken. Endocrinology 139, 1164–1171, https://doi.org/10.1210/endo.139.3.5813 (1998).
Johnson, P. A., Woodcock, J. R. & Kent, T. R. Effect of activin A and inhibin A on expression of the inhibin/activin beta-B-subunit and gonadotropin receptors in granulosa cells of the hen. Gen Comp Endocrinol 147, 102–107, https://doi.org/10.1016/j.ygcen.2005.12.008 (2006).
Reddi, A. H. & Reddi, A. Bone morphogenetic proteins (BMPs): from morphogens to metabologens. Cytokine Growth Factor Rev 20, 341–342, https://doi.org/10.1016/j.cytogfr.2009.10.015 (2009).
Kim, D., Ocón-Grove, O. & Johnson, A. Bone morphogenetic protein 4 supports the initial differentiation of hen (Gallus gallus) granulosa cells. Biology of reproduction 88 (2013).
Ocon-Grove, O. M., Poole, D. & Johnson, A. Bone morphogenetic protein 6 promotes FSH receptor and anti-Müllerian hormone mRNA expression in granulosa cells from hen prehierarchal follicles. Reproduction 143, 825–833 (2012).
Clelland, E. et al. Bone morphogenetic protein-15 in the zebrafish ovary: complementary deoxyribonucleic acid cloning, genomic organization, tissue distribution, and role in oocyte maturation. Endocrinology 147, 201–209, https://doi.org/10.1210/en.2005-1017 (2006).
Shimasaki, S. et al. A functional bone morphogenetic protein system in the ovary. Proc Natl Acad Sci USA 96, 7282–7287 (1999).
Kowanetz, M., Valcourt, U., Bergstrom, R., Heldin, C. H. & Moustakas, A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol 24, 4241–4254 (2004).
Ruzinova, M. B. & Benezra, R. Id proteins in development, cell cycle and cancer. Trends Cell Biol 13, 410–418 (2003).
Johnson, A., Haugen, M. J. & Woods, D. C. Role for inhibitor of differentiation/deoxyribonucleic acid-binding (Id) proteins in granulosa cell differentiation. Endocrinology 149, 3187–3195 (2008).
Woods, D. C., Haugen, M. J. & Johnson, A. L. Opposing actions of TGFbeta and MAP kinase signaling in undifferentiated hen granulosa cells. Biochem Biophys Res Commun 336, 450–457, https://doi.org/10.1016/j.bbrc.2005.08.107 (2005).
Johnson, A. L. & Woods, D. C. Dynamics of avian ovarian follicle development: cellular mechanisms of granulosa cell differentiation. Gen Comp Endocrinol 163, 12–17, https://doi.org/10.1016/j.ygcen.2008.11.012 (2009).
Acknowledgements
This work was supported by Natural Science Funding of China (31472089) to Juan LI.
Author information
Authors and Affiliations
Contributions
G. Zhu was responsible for the project design, sample collection and the data analysis, C. Fang and C. Mo coordinated the sample collection, Jing Li coordinated the manuscript revision, Y. Wang coordinated the experimental design, Juan Li was responsible for the experimental design, data analysis and manuscript preparation and revision. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, G., Fang, C., Li, J. et al. Transcriptomic Diversification of Granulosa Cells during Follicular Development in Chicken. Sci Rep 9, 5462 (2019). https://doi.org/10.1038/s41598-019-41132-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41132-1
This article is cited by
-
MiR-134-3p targets HMOX1 to inhibit ferroptosis in granulosa cells of sheep follicles
Journal of Ovarian Research (2024)
-
Integrating genomics and transcriptomics to identify candidate genes for high egg production in Wulong geese (Anser cygnoides orientalis)
BMC Genomics (2023)
-
Runs of homozygosity and signatures of selection for number of oocytes and embryos in the Gir Indicine cattle
Mammalian Genome (2023)
-
Dynamic transcriptome and chromatin architecture in granulosa cells during chicken folliculogenesis
Nature Communications (2022)
-
microRNA-194 is increased in polycystic ovary syndrome granulosa cell and induce KGN cells apoptosis by direct targeting heparin-binding EGF-like growth factor
Reproductive Biology and Endocrinology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
