
Abstract
Bananas (Musa spp.) are consumed worldwide as dessert and cooking types. Edible banana varieties are for the most part seedless and sterile and therefore vegetatively propagated. This confers difficulties for breeding approaches against pressing biotic and abiotic threats and for the nutritional enhancement of banana pulp. A panel of banana accessions, representative of the diversity of wild and cultivated bananas, was analysed to assess the range of chemotypes available globally. The focus of this assessment was banana leaves at two growth stages (juvenile and pre-flowering), to see when during the plant growth metabolic differences can be established. The metabolic data corresponded to genomic trends reported in previous studies and demonstrated a link between metabolites/pathways and the genomes of M. acuminata and M. balbisiana. Furthermore, the vigour and resistance traits of M. balbisiana was connected to the phenolic composition and showed differences with the number of B genes in the hybrid accessions. Differences in the juvenile and pre-flowering data led to low correlation between the growth stages for prediction purposes.
Similar content being viewed by others

Introduction
The genus Musa originated in Southeast Asia and the western Oceania regions and can be divided into several divisions including Eumusa which comprises most edible bananas1,2,3. The two main seeded ancestors for cultivated bananas are M. acuminata Colla (A genome) and M. balbisiana Colla (B genome). Hybridisation within M. acuminata species and between the two genomes led to diploid, triploid and tetraploid cultivars of different genomic composition (e.g. AA, AB, AAB or ABB), that can be identified based on a set of 15 standard morphological descriptors4,5. Currently, the production of over 300–400 varieties of banana worldwide is over 144 million tonnes annually and included dessert and cooking bananas. The most commercialised bananas, type Cavendish, account for 47% of the production worldwide6.
M. acuminata and M. balbisiana have two very different native habitats, namely tropical rainforests in Southeast Asia and monsoon areas in southern Asia, respectively1,4, and confer different traits to their progenies. The B genome contributes to a more vigorous growth, whereas the A genome gave rise to parthenocarpy – fruit development without fertilisation – and confers fruit flavour and quality7,8. Most banana cultivars are sterile and vegetatively propagated which restricts breeding approaches and most of the natural variation is achieved through spontaneous somatic mutations7,9,10. This complicates the generation of segregating population for (i) biotic resistant bananas against e.g. the reoccurring Panama disease11 and (ii) more nutritious bananas e.g. higher carotenoid content12. On the other side, it emphasises the necessity for banana collections in genebanks e.g. International Musa Transit Centre (ITC) and the accumulation of phenotypical information to establish the diversity in present banana accessions e.g. Musa Germplasm Information System (MGIS)13. Therefore, the responsibility of genebanks involves conservation of collections and assessment of diversity within these collections which is primarily based on molecular studies14,15,16,17,18. These studies in combination with metabolic analysis of the banana plant material can give a better understanding of the value of a variety19. In particular, documentation of the chemotype allows elucidation of biomarkers and general mechanisms to facilitate breeding for desired properties20. Furthermore, the study of plant metabolism at several growth stages can identify those early growth stages when a chemotype/genotype can be measured to shorten breeding cycles.
In the present study, leaf material of a diversity panel was analysed at the juvenile and pre-flowering growth stage of the banana plant (Table 1). This facilitates the comparison of leaf tissue grown under two conditions (in vitro and field) and ascertain the use of in vitro grown plants for reduced selection cycles during breeding. Three analytical platforms/approaches were used to cover a broad range of metabolites defining the chemotypes of banana accessions. The chemotypes observed differed according to genomic composition and gave an overview of the metabolic diversity in the present Musa panel.
Results and Discussion
The biochemical composition of a Musa spp diversity panel (38 accessions) was assessed by screening polar and non-polar extracts of juvenile plantlets and pre-flowering leaves on three different platforms to gain a comprehensive view of the metabolome21. The diversity panel (Table 1) included diploid and triploid genome groups of the section Eumusa: specimen of the two wild species M. acuminata and M. balbisiana, cultivated AA (AAcv), AAA, AAB, AB, ABB and one M. ornata accession of the section Rhodochlamys. The metabolites identified (~105, Supplementary Table S1) comprised a range of intermediates of primary and secondary metabolic pathways (Fig. 1). Relative quantification was carried out and combined into one data set (Supplementary Tables S2 and S3).

Pathway display of metabolites detected for juvenile plantlets and pre-flowering leaves. All metabolites specific to Musa spp identified through LC-MS, GC-MS and UPLC-DAD were highlighted (grey) in the pathway display constructed with BioSynLab software© (RHUL, UK).
Diversity panel – juvenile leaves
Twenty-four accessions were analysed at the juvenile growth stage of the plant (Table 1). The similarities between accessions and genome groups based on the metabolite composition will be discussed in comparison to several genotyping studies, each comprising a majority of the accessions at a given time.
Principal component analysis (PCA) of the metabolite data showed that the widest metabolic diversity was primarily found in the wild M. acuminata and M. balbisiana accessions (Fig. 2a). This is consistent with genetic diversity studies showing the majority of cytoplastic patterns were found in the wild M. acuminata accessions14,22. Another general trend of the metabolite data was a distinct separation between accessions containing only the A genome and at least one B genome, further referred to as group A and group B (Fig. 2a). M. ornata, the only accession of the section Rhodochlamys, clustered on the far side of group B. Genetic studies are in contradiction whether M. ornata is more similar to M. balbisiana15,23,24 or M. acuminata16,17, but the metabolite profile of this accession suggests a chemotype more similar to the B genome. The wild M. acuminata accessions showed a mixed cluster of malaccensis and burmannica subspecies, which were located closer to the banksii accession than the zebrina and microcarpa accessions. This PCA pattern was also detected based on genotyping data, identifying zebrina as the most different of the wild M. acuminata25. Additionally, the metabolite composition of wild M. acuminata species is a representation of their geographical locations3,26 and of the heterozygosity in M. acuminata species as displayed by the malaccensis accessions8. The exception was the banksii accession (ITC0623), which was located close to Plantains (AAB) and correlates with the strong presence of banksii genes in the putative progeny of Plantains2,26. No definitive separation was observed between the genome groups wild M. acuminata, AAcv and AAA, which might be a result of backcrossing and the dominance of maternal and paternal genes23,27. A comparison of wild and cultivated accessions of banksii, microcarpa and malaccensis showed no common change of metabolite features between the wild and cultivated states (Supplementary Fig. S4). The PCA score plot indicated a different metabolite composition of Tomolo (ITC1187, AAcv) compared to the other two AA cultivars Psiang Jari Buaya (ITC0312) and Psiang Mas (ITC0653), which was visualised in the heatmap (Fig. 3) and coincides with DNA profiling15 and the difference in maternal parents14,22. A similar case was found for Mbwazirume (ITC0084) an East African Highland banana of banksii and zebrina origin, which displayed a different chemotype from other two AAA accessions, related to all other subspecies14,22,28. Additionally, Mbwazirume was located in the upper half of the score plot with group B, indicating that farmers’ selection for a starchy pulp phenotype or the adaptation to a geographical location outside the natural range of the species led to a metabolic phenotype more similar to plantain cultivated in Africa (Figs 2 and 3). The close grouping of the other two AAA Petite naine (ITC0654) and Grande naine (ITC0180), both Cavendish accessions, confirms that an identical genomic background leads to a very similar metabolic composition14. The only diploid hybrid composed of A and B genome, Safet Velchi (ITC0245), was located between the groups A and B and closer to accessions of the genome groups AAB and ABB than wild M. balbisiana17. This indicates a strong influence of the B genome on the metabolome, as Safet Velchi was located with group B despite the prominent genetic background of malaccensis14,15.

PCA analysis of juvenile plantlets. PCA score plot (a) included all accessions analysed. PCA loadings plot (b) included all metabolites identified in the targeted analysis. Abbreviations: Rhodo, Rhodochlamys; AAw, wild M. acuminata; AAcv, cultivated AA; IPP, isopentenyl pyrophosphate derived pigments.

Heatmap of juvenile plantlets. Genotypes were grouped based on the similarity of their metabolite composition. Genome group of the accessions is displayed in the heatmap.
As observed for group A, the accessions of genomic compositions AAB and ABB did not have enough metabolic differences to separate on the score plot. Previous studies showed the close genetic relation between AAB and ABB as well as the different cytoplasmic DNA patterns leading to the observed chemotypes8,14,18. Interestingly, Orishele (ITC1325) was the only Plantain that clustered in the lower half with group A. The different metabolite composition of Orishele and some genetic studies indicate a change in this particular variety compared other Plantains, which are known for their genetic similarities22,29,30. The genetic study by Ude, et al.29 described Agbagba (ITC0111) as the most different Plantain variety as seen from the location of Agbagba in the PCA towards the edge of the plot (Fig. 2a). Within the balbisiana accessions, Pisang Klutuk Wulung (ITC1587, origin Indonesia) clustered towards AAB and ABB accessions, whereas M. balbisiana (ITC1527, origin China) was clustered away the furthest from all accessions analysed. M. balbisiana plants originate in continental Asia and are now widely distributed throughout the tropical and subtropical regions of Asia31. Analysis of cytoplasmic DNA confirmed several patterns unique to the B genome22,23,32,33, which could lead to very different chemotypes as reflected in the PCA analysis of metabolite data. Additionally, the similar metabolite composition between Pisang Klutuk Wulung (ITC1587) and triploids containing a B genome indicates the participation of Pisang Klutuk Wulung or a similar BB accession in the hybridisation of subgroups in the East contact area31. This contradicts Li, et al.22, who concluded that neither Pisang Klutuk Wulung nor Tani (ITC1120, pre-flowering data set) were involved in domestication of cultivated bananas due to the presence of Waxy gene. The similar metabolite composition of the two M. balbisiana accessions to triploids of group B suggests that the presents of some genes might not influence the primary metabolism (Figs 2 and 3). Furthermore, the results emphasise the need for a more detailed study of M. balbisiana accessions throughout tropical and subtropical Asia as the studies from specific regions23,32,33,34 do not provided enough information about the actual diversity of the B genome.
Differential expression of the metabolites detected showed that ~37% of the metabolites were significant to distinguish group A and B. These metabolites include isopentenyl pyrophosphate derived pigments such as chlorophylls and carotenoids (IPPs), dopamine, amino acid precursors for phenolics and for fatty acids with a higher mean in group A and amino acids involved in the nitrogen cycle, chlorogenic acids and catechins with a higher mean in group B (Supplementary Table S5). This comparison emphasises the improved vigour and tolerance to stresses associated with the B genome conferred partly by a higher content of phenolics35,36,37. A more detailed differential expression between genomic compositions showed ~75% of metabolites were significantly different. These metabolites comprised all chemical classes detected in the dataset (Supplementary Table S5). The two major chemical classes differentiating group A and B were IPP derived pigments and phenylpropanoids. The levels of most IPPs were higher in group A than group B which indicates differences in the regulation of photosynthetic processes and in the inheritance of photosynthesis related genes in interspecific hybrids. Genes encoding proteins involved in photosynthesis are located in the chloroplastic DNA (cpDNA) as well as the nucleus38. Changes in the regulatory network of photosynthetic processes can overcome climatic challenges39,40. Hence, the differences in the production of photosynthesis related antioxidants is to be expected between M. acuminata, which is prevalent in warmer regions with higher levels of precipitation, and M. balbisiana, cultivated in dryer, moderate warm regions14,41,42,43. Interestingly, the levels of IPPs in AAB were higher than wild M. balbisiana, whereas ABB and AB had up to 20% lower levels of IPPs compared to wild M. balbisiana which suggests that the inherited chloroplastic genome has a prominent role in the composition of photosynthetic metabolites26,27. The detected phenolics participate in structural and chemical barriers against abiotic and biotic stresses. Phenolic acids (e.g. caffeic and coumaric acid) and flavonols (e.g. kaempferol, quercetin and their glycosides) act as phytoanticipins, compounds synthesised before an attack, whereas catechins and chlorogenic acids are involved in the lignin biosynthesis, a physical barrier44,45. The highest levels of phenolics, involved in both structural and chemical barriers, were detected in wild M. balbisiana accessions. Interestingly, chlorogenic acid and dopamine (a tyrosine derived phenol) were detected in wild M. acuminata with similar levels to wild M. balbisiana, which suggests fundamental resistance mechanism are present in the wild M. acuminata species44,46. The burmannica accession Calcutta 4 (ITC0249) is well known for its resistance against several biotic stresses and yet the precise resistance mechanism have not been identified47,48. The metabolite data of Calcutta 4 showed average overall phenolic levels and above average levels of rutin, chlorogenic acid and caffeoyl-malate. This would suggest the resistance traits of Calcutta 4 is based on the regulatory processes engaged during/after the infection. The fact that the metabolite data showed the highest levels of phenolics precursors (phenylalanine and tyrosine) in group A, indicates that the activity of phenylalanine ammonia lyase (PAL), the committed and rate-limiting enzyme in the phenolics pathway, is regulated differently in the A and B genome. Additionally, the presence of the less efficient PAL from the A genome seems to affect accessions containing both genomes. Accessions from AAB, ABB and AB genome groups had significantly lower levels than wild M. balbisiana, which were nevertheless significantly higher than AAA and AAcv.
Diversity panel – Pre-flowering leaves
The accessions analysed at the pre-flowering growth stage of the plant included 29 varieties comprising all genome groups except the Rhodochlamys accession M. ornata (Table 1). PCA analysis of the leaf metabolite profile showed a separation of group A and B along the horizontal axis (Fig. 4), as observed for the data at the juvenile stage. Many other properties of the PCA plot of pre-flowering leaf were similar to the juvenile leaf data and included the location of the AB accession between group A and B, no separation between the genome groups AAA, wild M. acuminata and AAcv and the location of zebrina away from other wild M. acuminata. Despite lack of separation between A groups, the majority of AAA seemed to cluster around two AAcv Pisang Jari Buaya (ITC0312) and Pisang Mas (ITC0653), which might suggest those accessions emerged from the same genepool22,49. A significant difference between the juvenile and pre-flowering stages was the clear separation of AAB and ABB accessions into two cluster, as described by several genotyping studies18,24,50. The wild M. balbisiana Tani (ITC1120) was located between these two genome groups as described for the juvenile data set. Within the ABB accessions, Pelipita (ITC0472) had the lowest allelic contribution of banksii26 and was located away from the other ABB accessions towards the left edge of the score plot. A similar trend could be seen for Pisang Rajah (ITC0587) an AAB accession which was located in the middle of the score plot towards group A. Another difference between the growth stages was that the microcarpa accession was more similar to the malaccensis than the zebrina accession (Fig. 4). This metabolite data matches the chemotypes expected from mitotype14 and geographical origin of microcarpa and malaccensis1,2. Additionally, the pre-flowering leaves showed a metabolic trend in the PCA plot based on consumption type. This was visualised with several group B accessions located towards wild M. acuminata and cooking pulp types of group A towards group B accessions with the same pulp type. Specifically, Tomolo (ITC1187,AAcv) was located in the upper half whereas Namwa Khom (ITC0659, ABB) was located in the lower half of the score plot (Fig. 4). The metabolite profile of Namwa Khom is of special interest as it has the same cytoplasmic background as Simili Radjah (ITC0123)14,26,27, which was clustered towards to top edge of the score plot. Nevertheless, the breeding schemes for these two ABB accessions must have led to different chromosome re-assortment during the exchange of chromosome segments, which resulted in very different regulatory processes for the metabolome27,51. This can also be seen in the metabolite composition (Fig. 5). Furthermore, the remaining genes from the A progenitor in Namwa Khom could be responsible for a metabolic composition of the pulp that is preferably eaten as a dessert banana contrary to other ABB varieties such as Simili Radjah (Table 1)52,53. Previous studies evidenced that the metabolism of the leaf (i) changes slightly before emergence of flowers54,55 and (ii) is connected to the sink organ56,57. In the latter studies, the PCA of leaf and tuber/root showed the same metabolic trends. Furthermore, the metabolite data from the present study emphasises the finding that cooking characteristics are not related to the B genome58.

PCA analysis of leaves of pre-flowering plants. PCA score plot (a) included all accessions analysed. PCA loadings plot (b) included all metabolites identified in the targeted analysis. Abbreviations: AAw, wild M. acuminata; AAcv, cultivated AA; IPP, isopentenyl pyrophosphate derived pigments.

Heatmap of preflowering plants. Genotypes were grouped based on the similarity of their metabolite composition. Genome group of the accessions is displayed in the heatmap.
Differential expression showed that ~27% of the metabolites – less than juvenile data – were significantly different between group A and B and included primarily phenylpropanoids (Supplementary Table S6). Glycosylated phenylpropanoids were in general higher in group A, contrary to the juvenile data, and catechin levels were higher in group B as observed for the juvenile leaves. No dopamine was detected in the leaves at pre-flowering stage. The differential expression between genome groups showed that only ~52% of metabolites were significantly different and mainly comprised phenolics and IPPs. In the pre-flowering leaves higher levels of glycosylated phenolics were detected in group A which might be related to storage in the vacuole rather than transport to further synthesis locations59. Epiafzelechin, a flavonoid monomer unit for tannins60, and the majority of phenolics had the highest means in ABB and wild M. balbisiana accessions. The amount of free phenolics in the ABB accessions indicates a more effective resistance as well as cell wall strengthening through lignification compared to AAB accessions8,60,61. This hypothesis needs confirmation with environmental studies involving biotic and abiotic stresses similar to the effects of Fusarium wilt on banana roots62. Interestingly, accessions of genome groups wild M. acuminata and AAcv had similar levels of phenolics compared to AAB. This data implies dramatic changes in the metabolic regulation from the juvenile to the pre-flowering growth stage, probably related to preparations for flowering as mentioned earlier54,55.
Correlation of juvenile and pre-flowering leaves
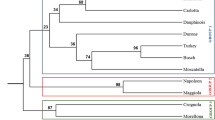
The diversity panels studied at juvenile and pre-flowering growth stages included 15 accessions present in both panels (Table 1). Metabolite composition at both stages were compared to assess the potential of predicting the pre-flowering chemotype from the juvenile plantlet. Two comparisons were conducted to evaluate prediction based on the genome group and on the specific accessions (Fig. 6). The dendrogram based on accessions was similar between juvenile and pre-flowering growth stages in a few aspects, such as grouping of Plantains (Agbagba, Bobby Tannap and Ihitisim) (Fig. 6a,b). Both the RV coefficient (R = 0.784, p = 0.016) and Mantel test were significant (p = 0.02) and ascertain that similar metabolite trends can be observed in the leaves of juvenile and pre-flowering plants. Multifactor analysis combing the PCA plots of the two growth stages (R = 0.577) highlighted that Zebrina, Pisang Jari Buaya, Safet Velchi, Calcutta 4 and Mbwazirume showed the most difference between juvenile and pre-flowering stage causing the low correlation between the growth stages. Based on this data, the correlation between group A and B was significant between the growth stages based on Mantel test (p = 0.008) and RV coefficient (R = 0.53, p = 0.007). The association tests of data summarised by genome groups showed no significant correlation between the juvenile and pre-flowering data sets (RV coefficient: R = 0.288, p = 0.459 and Mantel test p = 0.731).

Hierarchical clustering analysis of juvenile and pre-flowering leaves. Agglomerative hierarchical clustering (AHC) was performed for individual accessions (a,b) at the juvenile (a) and pre-flowering (b) growth stage. Clustering was based on averaged metabolite data detected in accessions present in both growth stages. The AHC of accessions was coloured by accessions contained in group A (blue) and group B (orange).
The present study provides evidence to suggest that the metabolic changes occurring over the two growth stages show a low association on the level of accessions and group A/B and predictions from juvenile plantlets to fully grown plants should be performed with precaution. Several factors could be responsible for the low correlation between the two growth stages. Firstly, the leaves of juvenile plantlets were obtained from tissue culture grown plants. The leaves at this stage are small and hence, during sampling the leaf part of the petiole or even the leaf sheath might have been included. The latter two leaf parts are transport organs with very transient metabolism63 (Drapal et al., submitted) and can affected the overall composition of the juvenile leaves analysed. Secondly, the leaves undergo anatomical and morphological changes during acclimatisation from in vitro to the field64. Lastly, epigenetic changes are known to occur during in vitro propagation on multiple levels and can affect the plant’s regulatory networks (e.g. genome and metabolome) and morphology as reviewed by Us-Camas, et al.65. If the epigenetic effects were temporary, as previously reported, then the metabolism of the in vitro and field grown material will show differences. Hence, to ascertain the usefulness of in vitro plants for prediction in the field a study with a wider variety of banana accessions including genotyping and metabolomics concurrently would be necessary.
In conclusion, the present study revealed that the complex genetic backgrounds of banana accessions have a specific metabolite phenotype. The accession specific chemotypes differ between in vitro and field grown plants, which bears challenges for accelerating breeding programs by screening young plantlets. Nevertheless, both cultivation conditions showed a clear separation between accessions containing solely the A genome and at least one B genome. Furthermore, the metabolite profiles indicated that the combination of M. acuminata and M. balbisiana genome in hybrids can interfere with certain pathways e.g. photosynthetic metabolites and phenolics biosynthesis. As previously reported, the resistance traits associated with the B genome is mediated by phenolic compounds. More specifically, a prominent difference between the two genomes is the ubiquitous presence of elevated phenolics involved in structural and chemical defence in the B genome. Future metabolite analysis of pulp and peel will show whether these metabolite differences can also be detected in the consumed banana product(s) and influence the banana pulp quality.
Material and Methods
Plant material
Banana leaf samples were supplied by Bioversity International Musa Transit Centre (ITC), hosted at KU Leuven (Belgium), and International Institute of Tropical Agriculture (IITA) in Ibadan, Nigeria. The banana accessions analysed comprised diploid and triploid, wild and cultivated accessions (Table 1). The plants obtained from ITC were in vitro propagated plants harvest and present juvenile banana plantlets (JP). The field material obtained from IITA was grown in Ibadan, Nigeria research farm. Field material (third open leaf) was harvest from 6-month old plants, which presents the pre-flowering (PF) growth stage of banana plants. All leaf material was immediately frozen in liquid nitrogen and lyophilised.
Metabolite extraction
Dried tissue was ground up and pooled quality controls (QC) prepared separately for juvenile and pre-flowering leaf sets. Extraction of metabolites followed previously published protocols and included randomisation of samples and extraction in sets of 20 samples with one QC and one extraction blank57,66. The internal standards used were d4-succinic acid (10 µg/extraction) for GC-MS analysis and genistein (1.25 µg/extraction) for LC-MS analysis. Aliquots of the polar and non-polar phase were immediately dried down after extraction. The total number of samples for the juvenile leaf set was 158 and 109 for the pre-flowering leaf set.
Metabolite profiling with GC-MS
Dried aliquots for GC-MS analysis were derivatised as previously described with methoxymation and silylation derivatisation67, injected using a 10:1 split mode and analysed with a heat gradient from 70 to 325 °C66. Automated Mass Spectral Deconvolution and Identification System (AMDIS v2.71, NIST) was used to create an in-house library based on authentic standards and NIST’11 MS library (National Institute of Standards and Technology, USA). Peak convolution was performed with AMDIS in batch mode for each sample set and peak identification conducted according to metabolomics reporting guidelines68,69. Quantification of identified metabolites was expressed relative to the internal standard and sample weight (µg/g dry tissue).
Untargeted analysis with LC-MS
Dried aliquots for LC-MS analysis were resuspended in methanol/water (1:1, v/v) and filtered using syringe filter (nylon, 0.45 µm). Separation of compounds was based on a previously published LC-MS method with modification of solvents A (water and 0.1% formic acid) and B (acetonitrile and 0.1% formic acid)57. Peak picking, alignment and identification was performed based on R package metaMS70,71 with a retention time window match set to 0.5 min. Peak identification was based on m/z difference 0.005 and retention difference 0.3 min to an in-house library with authentic standards. As described for GC-MS analysis, identified metabolites were relatively expressed.
Targeted analysis with UPLC-DAD
For analysis of carotenoids and chlorophylls, the aliquot of the non-polar phase was resuspended in ethyl acetate/acetonitrile (1:9) and analysed as previously described67 with a composition gradient starting at 30% methanol/water (50:50, v/v) to 99% ethyl acetate/acetonitrile (25:75, v/v). The UV/Vis spectrum was continuously monitored from 250–600 nm. Metabolites were identified through retention time and UV/visible light spectrum compared to authentic standards, followed by a total quantification with dose-response curves72.
Data processing and statistical analysis
All data was analysed using XLSTAT add-ins73 within Microsoft Excel, except PCA which was performed and visualised with Simca P 13.0.3.0 (Umetrics, Sweden). Metabolite data was rescaled from 0 to 1 (variable transformation) to remove biased of actual concentration levels (µg/g dry weight), which varied greatly between the different platforms. Differential expression (nonparametric) was performed with Benjamini-Yekutieli post-hoc correction on all replicates. Before the RV coefficient and Mantel test, the groups were averaged based on accession or genome group for each growth stage separately. The contribution of observation from PCAs was transformed with Spearman correlation into a correlation matrix. The p-value computation for the RV coefficient included 5000 permutations and 10000 permutations for the Mantel test with Spearman correlation. Hierarchical clustering was performed via Ward clustering in Simca P 13.0.3.0 (Umetrics, Sweden).
References
Janssens, S. B. et al. Evolutionary dynamics and biogeography of Musaceae reveal a correlation between the diversification of the banana family and the geological and climatic history of Southeast Asia. The New Phytologist 210, 1453–1465 (2016).
Perrier, X. et al. Multidisciplinary perspectives on banana (Musa spp.) domestication. Proc Natl Acad Sci USA 108, 11311–11318 (2011).
De Langhe, E., Vrydaghs, L., de Maret, P., Perrier, X. & Denham, T. Why Bananas Matter: An introduction to the history of banana domestication. Ethnobotany Research & Applications 7, 13 (2009).
Ploetz, R., Kepler, A., Daniells, J. & Nelson, S. Banana and plantain – an overview with an emphasis on Pacific island cultivars [Musacea (banana family)]. Species Profiles for Pacific Island Agroforestry (Permanent Agriculture Resources, Ho-lualoa, Hawaii) (2007).
Simmonds, N. W. & Shepherd, K. The taxonomy and origins of the cultivated bananas. Journal of the Linnean Society of London, Botany 55, 302–312 (1955).
Nations, F. A. O. o. t. U. In Banana facts and figures. (FAO, Rome, 2014).
Heslop-Harrison, J. S. & Schwarzacher, T. Domestication, Genomics and the Future for Banana. Annals of Botany 100, 1073–1084 (2007).
de Jesus, O. N. et al. Genetic diversity and population structure of Musa accessions in ex situconservation. BMC Plant Biology 13, 41 (2013).
Ortiz, R. & Swennen, R. From crossbreeding to biotechnology-facilitated improvement of banana and plantain. Biotechnology Advances 32, 158–169 (2014).
Padmesh, P. et al. Exploring wild genetic resources of Musa acuminata Colla distributed in the humid forests of southern Western Ghats of peninsular India using ISSR markers. Plant cell reports 31, 1591–1601 (2012).
Ordonez, N. et al. Worse Comes to Worst: Bananas and Panama Disease—When Plant and Pathogen Clones Meet. PLOS Pathogens 11, e1005197 (2015).
Englberger, L. et al. Carotenoid content and flesh color of selected banana cultivars growing in Australia. Food and nutrition bulletin 27, 281–291 (2006).
Ruas, M. et al. In Database Vol. 2017 (2017).
Carreel, F. et al. Ascertaining maternal and paternal lineage within Musa by chloroplast and mitochondrial DNA RFLP analyses. Genome 45, 679–692 (2002).
Youssef, M., James, A. C., Rivera-Madrid, R., Ortiz, R. & Escobedo-GraciaMedrano, R. M. Musa Genetic Diversity Revealed by SRAP and AFLP. Molecular Biotechnology 47, 189–199 (2011).
Christelová, P. et al. A platform for efficient genotyping in Musa using microsatellite markers. AoB PLANTS 2011 (2011).
Čížková, J. et al. Molecular and Cytogenetic Characterization of Wild Musa Species. PLOS ONE 10, e0134096 (2015).
Ude, G., Pillay, M., Nwakanma, D. & Tenkouano, A. Genetic Diversity in Musa acuminata Colla and Musa balbisiana Colla and some of their natural hybrids using AFLP Markers. Theoretical and Applied Genetics 104, 1246–1252 (2002).
Kumar, R., Bohra, A., Pandey, A. K., Pandey, M. K. & Kumar, A. Metabolomics for Plant Improvement: Status and Prospects. Frontiers in Plant Science 8 (2017).
Pandey, A. et al. Genome-wide Expression Analysis and Metabolite Profiling Elucidate Transcriptional Regulation of Flavonoid Biosynthesis and Modulation under Abiotic Stresses in Banana. Scientific Reports 6, 31361 (2016).
Halket, J. M. et al. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J Exp Bot 56, 219–243 (2005).
Li, L.-F. et al. Origins and Domestication of Cultivated Banana Inferred from Chloroplast and Nuclear Genes. PLOS ONE 8, e80502 (2013).
Ude, G., Pillay, M., Nwakanma, D. & Tenkouano, A. Analysis of genetic diversity and sectional relationships in Musa using AFLP markers. Theoretical and Applied Genetics 104, 1239–1245 (2002).
Muiruri, K. S. et al. Dominant Allele Phylogeny and Constitutive Subgenome Haplotype Inference in Bananas Using Mitochondrial and Nuclear Markers. Genome Biology and Evolution 9, 2510–2521 (2017).
Martin, G. et al. Evolution of the Banana Genome (Musa acuminata) Is Impacted by Large Chromosomal Translocations. Molecular biology and evolution 34, 2140–2152 (2017).
Perrier, X. Combining Biological Approaches to Shed Light on the Evolution of Edible Bananas. 2009 7, 18, (2009).
De Langhe, E., Hřibová, E., Carpentier, S., Doležel, J. & Swennen, R. Did backcrossing contribute to the origin of hybrid edible bananas? Annals of Botany 106, 849–857 (2010).
Boonruangrod, R., Fluch, S. & Burg, K. Elucidation of origin of the present day hybrid banana cultivars using the 5′ETS rDNA sequence information. Molecular Breeding 24, 77–91 (2009).
Ude, G., Pillay, M., Ogundiwin, E. & Tenkouano, A. Genetic diversity in an African plantain core collection using AFLP and RAPD markers. Theoretical and Applied Genetics 107, 248–255 (2003).
Risterucci, A. M. et al. Development and assessment of Diversity Arrays Technology for high-throughput DNA analyses in Musa. Theor Appl Genet 119, 1093–1103 (2009).
De Langhe, E., Perrier, X., Donohue, M. & Denham, T. P. The Original Banana Split: Multi-disciplinary implications of the generation of African and Pacific Plantains in Island Southeast Asia. Ethnobotany Research & Applications 14, 14 (2015).
Wang, X.-L., Chiang, T.-Y., Roux, N., Hao, G. & Ge, X.-J. Genetic diversity of wild banana (Musa balbisiana Colla) in China as revealed by AFLP markers. Genet Resour Crop Evol 54, 1125–1132 (2007).
Ahmad, F., Megia, R. & Poerba, Y. S. Genetic Diversity of Musa balbisiana Colla in Indonesia Based on AFLP Marker. HAYATI Journal of Biosciences 21, 39–47 (2014).
Crouch, J. H. et al. Comparison of PCR-based molecular marker analyses of Musa breeding populations. Molecular Breeding 5, 233–244 (1999).
Nayar, N. M. In Horticultural Reviews. 117–164 (John Wiley & Sons, Inc., 2010).
Kundapura Venkataramana, R., Hastantram Sampangi-Ramaiah, M., Ajitha, R. N., Khadke, G. & Chellam, V. Insights into Musa balbisiana and Musa acuminata species divergence and development of genic microsatellites by transcriptomics approach. Plant. Gene 4, 78–82 (2015).
Carpentier, S. C. et al. The use of 2D-electrophoresis and de novo sequencing to characterize inter- and intra-cultivar protein polymorphisms in an allopolyploid crop. Phytochemistry 72, 1243–1250 (2011).
Berry, J. O., Yerramsetty, P., Zielinski, A. M. & Mure, C. M. Photosynthetic gene expression in higher plants. Photosynthesis Research 117, 91–120 (2013).
Sugiura, M. The chloroplast genome. Essays in biochemistry 30, 49–57 (1995).
Dutta, S., Mohanty, S. & Tripathy, B. C. Role of Temperature Stress on Chloroplast Biogenesis and Protein Import in Pea. Plant Physiology 150, 1050–1061 (2009).
Peel, M. C., Finlayson, B. L. & McMahon, T. A. Updated world map of the Köppen-Geiger climate classification. Hydrol. Earth Syst. Sci. 11, 1633–1644 (2007).
Li, L.-F., Häkkinen, M., Yuan, Y.-M., Hao, G. & Ge, X.-J. Molecular phylogeny and systematics of the banana family (Musaceae) inferred from multiple nuclear and chloroplast DNA fragments, with a special reference to the genus Musa. Molecular Phylogenetics and Evolution 57, 1–10 (2010).
Nwakanma, D. C., Pillay, M., Okoli, B. E. & Tenkouano, A. Sectional relationships in the genus Musa L. inferred from the PCR-RFLP of organelle DNA sequences. Theoretical and Applied Genetics 107, 850–856 (2003).
Ewané, C. A., Lepoivre, P., de Lapeyre de Bellaire, L. & Lassois, L. Involvement of phenolic compounds in the susceptibility of bananas to crown rot. A review. BASE [En ligne] 16, 393–404 (2012).
Gall, H. L. et al. Cell Wall Metabolism in Response to Abiotic Stress. Plants 4, 112–166 (2015).
Guidotti, B. B., Gomes, B. R., Siqueira-Soares, R. D. C., Soares, A. R. & Ferrarese-Filho, O. The effects of dopamine on root growth and enzyme activity in soybean seedlings. Plant Signaling & Behavior 8, e25477 (2013).
Sanchez Timm, E. et al. Identification of Differentially-Expressed Genes in Response to Mycosphaerella fijiensis in the Resistant Musa Accession ‘Calcutta-4’ Using Suppression Subtractive Hybridization. PLoS One 11, e0160083 (2016).
Miller, R. N. G. et al. Analysis of non-TIR NBS-LRR resistance gene analogs in Musa acuminata Colla: Isolation, RFLP marker development, and physical mapping. BMC Plant Biology 8, 15–15 (2008).
Teo, C. H. et al. Genome constitution and classification using retrotransposon-based markers in the orphan crop banana. Journal of Plant Biology 48, 96–105 (2005).
Gawel, N. & Jarret, R. L. Cytoplasmic genetic diversity in bananas and plantains. Euphytica 52, 19–23 (1991).
Jeridi, M. et al. Homoeologous chromosome pairing between the A and B genomes of Musa spp. revealed by genomic in situ hybridization. Ann Bot 108, 975–981 (2011).
Daniells, J., Jenny, C., Karamura, D. & Tomekpe, K. Musalogue: a catalogue of Musa germplasm. Diversity in the genus Musa. (International Network for the Improvement of Banana and Plantain, 2001).
Valmayor, R. V. et al. Banana cultivar names and synonyms in Southeast Asia. (INIBAP Regional Office for Asia and the Pacific, 2000).
Urban, L., Lu, P. & Thibaud, R. Inhibitory effect of flowering and early fruit growth on leaf photosynthesis in mango. Tree Physiol 24, 387–399 (2004).
Grainger, J. METABOLISM AND FLOWERING. Annals of Applied Biology 27, 311–322 (1940).
Price, E. J., Bhattacharjee, R., Lopez-Montes, A. & Fraser, P. D. Metabolite profiling of yam (Dioscorea spp.) accessions for use in crop improvement programmes. Metabolomics 13, 144 (2017).
Drapal, M. et al. Identification of metabolites associated with water stress responses in Solanum tuberosum L. clones. Phytochemistry 135, 24–33 (2017).
Nayar, N. The Bananas: Botany, Origin, Dispersal.In Horticultural Reviews (ed. Janick, J.) Ch. 2, 117-164 (John Wiley & Sons, Inc., 2010).
Gutierrez, E. et al. Transcriptomics, Targeted Metabolomics and Gene Expression of Blackberry Leaves and Fruits Indicate Flavonoid Metabolic Flux from Leaf to Red Fruit. Frontiers in Plant Science 8, 472 (2017).
Shahat, A. A. & Marzouk, M. S. In Medicinal Plant Research in Africa (ed. Kuete, V.) Ch. 13, 479–555 (Elsevier, 2013).
Hu, W. et al. Comparative physiological and transcriptomic analyses provide integrated insight into osmotic, cold, and salt stress tolerance mechanisms in banana. Scientific Reports 7, 43007 (2017).
Li, C.-Y. et al. Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genomics 13, 374 (2012).
Singh, P. et al. Spatial transcriptome analysis provides insights of key gene(s) involved in steroidal saponin biosynthesis in medicinally important herb Trillium govanianum. Scientific Reports 7, 45295 (2017).
Sandoval, J., Muller, L. & Weberling, F. Foliar morphology and anatomy of Musa cv. Grande Naine (AAA) plants grown in vitro and during hardening as compared to field-grown plants. Fruits 49, 37–46 (1994).
Us-Camas, R., Rivera-Solís, G., Duarte-Aké, F. & De-la-Peña, C. In vitro culture: an epigenetic challenge for plants. Plant Cell, Tissue and Organ Culture (PCTOC) 118, 187–201 (2014).
Price, E. J., Wilkin, P., Sarasan, V. & Fraser, P. D. Metabolite profiling of Dioscorea (yam) species reveals underutilised biodiversity and renewable sources for high-value compounds. Scientific Reports 6, 29136 (2016).
Nogueira, M., Mora, L., Enfissi, E. M., Bramley, P. M. & Fraser, P. D. Subchromoplast sequestration of carotenoids affects regulatory mechanisms in tomato lines expressing different carotenoid gene combinations. Plant Cell 25, 4560–4579 (2013).
Fernie, A. R. et al. Recommendations for reporting metabolite data. Plant Cell 23, 2477–2482 (2011).
Sumner, L. W. et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 3, 211–221 (2007).
Shahaf, N. et al. Constructing a mass measurement error surface to improve automatic annotations in liquid chromatography/mass spectrometry based metabolomics. Rapid Commun Mass Spectrom 27, 2425–2431 (2013).
Wehrens, R., Bloemberg, T. G. & Eilers, P. H. Fast parametric time warping of peak lists. Bioinformatics 31, 3063–3065 (2015).
Fraser, P. D., Pinto, M. E. S., Holloway, D. E. & Bramley, P. M. Application of high-performance liquid chromatography with photodiode array detection to the metabolic profiling of plant isoprenoids. The Plant Journal 24, 551–558 (2000).
Addinsoft. XLSTAT statistical and data analysis solution (2019).
Acknowledgements
The authors would like to thank Chris Gerrish for his excellent technical assistance. This work was supported by a CGIAR Research Program on Roots, Tubers and Bananas (RTB) grant to PDF.
Author information
Authors and Affiliations
Contributions
All authors (M.D., E.B.C., M.R., D.A., J.S., I.V.H., A.B., N.R., R.S. and P.D.F.) contributed to the design of the study. D.A. and I.V.H. provided the samples. M.D. and E.B.C. performed the experiments. M.D. performed data analysis and wrote the manuscript. All authors revised, edited and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Drapal, M., de Carvalho, E.B., Rouard, M. et al. Metabolite profiling characterises chemotypes of Musa diploids and triploids at juvenile and pre-flowering growth stages. Sci Rep 9, 4657 (2019). https://doi.org/10.1038/s41598-019-41037-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41037-z
This article is cited by
-
Assessment of genetic diversity and volatile content of commercially grown banana (Musa spp.) cultivars
Scientific Reports (2022)
-
Transcriptome and metabolome profiling provide insights into molecular mechanism of pseudostem elongation in banana
BMC Plant Biology (2021)
-
Elicitation of antioxidant metabolites in Musa species in vitro shoot culture using sucrose, temperature and jasmonic acid
Plant Cell, Tissue and Organ Culture (PCTOC) (2021)
-
Ripe pulp metabolite profiling of ten Indonesian dessert banana cultivars using UHPLC-Q-Orbitrap HRMS
European Food Research and Technology (2021)
-
Safeguarding and using global banana diversity: a holistic approach
CABI Agriculture and Bioscience (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
