
Abstract
H2O2 is a reactive oxygen species (ROS), which can diffuse away from its site of generation and may act as a cell-to-cell signaling factor. The mechanisms responsible for the generation of H2O2 in human ovarian follicles and possible signaling role(s) of H2O2 are not well known. We identified a source of H2O2, the enzyme NADPH oxidase (NOX) 4, in isolated differentiated, in-vitro fertilisation-derived human granulosa-lutein cells (GCs), in proliferating human granulosa tumour cells (KGN), as well as in situ in cells of growing ovarian follicles. H2O2 was readily detected in the supernatant of cultured GCs and KGN cells. H2O2 levels were significantly lowered by the NOX4 blocker GKT137831, indicating a pronounced contribution of NOX4 to overall H2O2 generation by these cells. We provide evidence that extracellular H2O2 is taken up by GCs, which is facilitated by aquaporins (peroxiporins). We thus conclude that GC-derived H2O2 might act as autocrine/paracrine factor. Addition of H2O2 increased MAPK-phosphorylation in GCs. Moreover, reducing H2O2 production with GKT137831 slowed proliferation of KGN cells. Our results pinpoint NOX4 and H2O2 as physiological players in the regulation of GC functions.
Similar content being viewed by others

Introduction
Reactive oxygen species (ROS), including hydroxyl, peroxyl and other molecules, function as physiological regulators of hormone actions and signal transduction pathways, to affect processes in the female gonad1,2,3. Studies in the rat revealed a role in ovulation4. In the corpus luteum they are beneficial for steroid production5. Furthermore, it is becoming clear that specifically hydrogen peroxide (H2O2) plays fundamental roles in cell proliferation, migration and metabolism, as well as cell death3.
ROS are a double-edged sword and a dysregulation of their levels may also cause or accelerate pathological conditions and oxidative stress, associated, for instance, with impaired oocyte quality, follicular atresia and negative outcome of assisted reproductive techniques (ART)6,7,8.
Several intracellular sources of ROS and modes of their generation exist1,3. However, in the human ovary neither those sources, nor the mechanisms of their actions are well known. It was shown that ROS levels in in-vitro fertilization (IVF)-derived, cultured human granulosa-lutein cells (GCs) are increased upon uptake and intracellular metabolism of the catecholamines dopamine and norepinephrine9,10,11. ROS generation in GCs was also increased upon activation of the epidermal growth factor, as well as of the pigment epithelium derived growth factor12,13.
During these studies it became clear that yet a further ROS source exists in GCs, namely NADPH oxidases (NOX); see e.g.1,14. These are multicomponent, membrane-bound enzymes, of which seven members are known, NOX1-5 and dual oxidases (DUOX) 1 and 215,16. Of those, NOX4 and 5 were previously described in human GCs by RT-PCR12, NOX1-3 were not found and DUOX1 and 2 have not been studied yet.
The main product of NOX4 is H2O2, which is long-lived and the only membrane-permeable ROS17. Therefore, it may serve as an autocrine/paracrine signaling molecule. In accordance with this statement, it was found in human follicular fluid (FF)18. Uptake of extracellular H2O2 can be facilitated by aquaporins (AQPs), besides H2O2 diffusion through the plasma membrane19. Interestingly, a recent study did not only confirm expression of NOX4 in IVF-derived GCs, but demonstrated a decrease of NOX4 levels in GCs from women older than 40 years20. This correlates with the naturally occurring decline in fertility with age and might imply importance of high NOX4 levels in physiological processes in the ovarian follicle. Beneficial and physiological roles of H2O2 and NOX4 have also been reported for the vasculature21.
We examined NOX enzymes in differentiated GCs and in proliferating human granulosa tumor cells, KGN22. We focused on NOX4 as a H2O2-producing enzyme, studied involvement of AQP channels in the cellular uptake of H2O21,19 and explored potential consequences of action.
Results
Identification of NADPH-oxidases in human GCs and ovarian sections
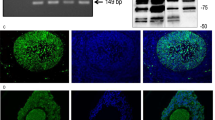
RT-PCR followed by sequencing revealed that GCs on the day of isolation (day 0 = d0) and on culture day 3 (d3) express NOX4, NOX5, DUOX1 and DUOX2 (Fig. 1A). This experiment was repeated using n = 5 independent pooled GCs preparations. NOX4 protein was detected in three GC preparations by Western blotting (Fig. 1B; NOX4: 68 kDa). Immunohistochemistry revealed NOX4 in preantral and antral follicles (Fig. 2A–C) and the corpus luteum of the human ovary (Fig. 2E). Follicular granulosa cells, theca cells and small and large luteal cells were stained (Fig. 2). This staining pattern was also obtained with another antibody (Supplementary Fig. S5). Specificity was shown by pre-adsorption of one of the antibodies with a blocking peptide (Supplementary Fig. S5), or replacing the antibody by serum (Fig. 2D,F).

NOX expression in GCs in vitro. (A) RT-PCR identified NOX4/5 and DUOX1/2 in cultured GCs after isolation (d0) and on culture day 3 (pooled GCs from 2–5 individuals). Controls including RNA (-RT) and H2O instead of cDNA (H2O) were negative. The displayed figure was cropped from original gel images, which are part of the supplementary data. (B) NOX4 protein was detected in cultured GCs by immunoblotting (anti-NOX4 antibody from ProSci). Controls without primary antibody were negative.

Presence of NOX4 in human ovarian tissue. Immunohistochemistry using human ovarian sections and an anti-NOX4 antibody from ProSci showed positive staining for NOX4 in granulosa (GC) and theca cells (TC) of a secondary follicle (A), of a small antral follicle (B), of a large antral follicle (C) as well as in luteinized GCs (LGC) and luteinized TCs (LTC) of the corpus luteum (E). Serum controls lacked first antibody (D and F). Scale bars: A–E = 30 µm, F = 50 µm.
H2O2 production by NOX4
Fluorometrical measurements of ROS employing H2DCFDA (n = 8 GC pools) and detection of H2O2 by Amplex® Red reagent (n = 4) confirmed basal generation of H2O2 and other ROS (Fig. 3). Superoxide was detected only in small amounts (Supplementary Fig. S1; n = 4). An increase in ROS, and specifically in H2O2 was seen in all independent measurements in the course of 2 h (Fig. 3A,B). The NOX4-blocker GKT137831 significantly reduced ROS generation after 2 h by 55% and H2O2 production by 36%, respectively (Fig. 3C–F). The blocker did not affect cell viability (24 h; confirmed by determination of ATP content, n = 3) and morphology (Supplementary Fig. S2A,B).

GCs are a source of H2O2 and other ROS produced by NOX4. Basal production of ROS (identified by the indicator dye H2DCFDA, n = 8) and specifically of H2O2 (detected by Amplex® Red, n = 4) was measured on culture day 2 to 4 for 2 h (A,B). For clarity, results are presented as mean only (six technical repetitions) and as fold-change relative to the intensity at t = 0. The NOX4 blockerGKT137831 (20 µM) reduced the production of ROS/H2O2. One representative measurement for both is shown in (C,D). Blocking NOX4 significantly reduced production of ROS (E; by 55%; n = 6) and H2O2 (F; by 36%; n = 4) after 2 h (mean ± SEM). One-sample t-tests (two-tailed; *p < 0.05; ***p < 0.001).
Influence of FSH and hCG on NOX enzyme levels
Luteal cells and cultured GCs express receptors for FSH and LH/hCG23. Addition of either hormone for 24 h did not affect levels of NOX4 and NOX5, but significantly increased DUOX1 and DUOX2 (Fig. 4A,B). FSH induced a 1.7-fold increase in DUOX2 expression. DUOX1 was increased 2.2-fold and DUOX2 2.8-fold by hCG. Overall ROS generation after 24 h of stimulation with FSH/hCG was not increased (Fig. 4C).

Hormonal stimulation significantly increases levels of DUOX, but does not elevate basal ROS production in GCs. Results of qRT-PCR experiments showed that mRNA expression levels of NOX5/DUOX1/2 were elevated after hormonal stimulation, but not all reached statistical significance (A,B). Results (mean ± SEM) shown were normalized to the geometric mean of the reference genes RPL19, UBC, PPIA and GAPDH. (C) Measurements of ROS production using H2DCFDA did not show any difference after 2 h measurement when treated with the hormones. Numbers of repeated experiments are shown inside the columns. Mean ± SEM; one-sample t-tests (two-tailed; *p < 0.05)
Uptake of H2O2 by GCs
Several aquaporins (AQP3, 8 and 9) are implicated in the transport of H2O2 and therefore called peroxiporines. The expression of some AQPs in GCs was reported24,25,26,27,28,29,30. We confirmed expression of AQP1-3, 5, and 7–9 by RT-PCR (Fig. 5A). AQP4 and 6 were not found. PO1, a cell permeable fluorescent probe visualizes intracellular H2O2, thereby allows to monitor its cellular uptake. Both, confocal microscopy (n = 3) and quantitative fluorometric evaluations (n = 4) showed that extracellular H2O2 was rapidly taken up by GCs. The increase of intracellular H2O2 was significantly reduced in the presence of AgNO3, which can block AQPs31 (Fig. 5B–D). The blocker used at 500 nM did not affect cell viability (2 h; confirmed by determination of ATP content, n = 3; Supplementary Fig. S3).

H2O2-transporting aquaporins in GCs. (A) RT-PCR identified AQP1, 2, 3, 5, 7, 8 and 9. All controls, including RNA (-RT) and H2O instead cDNA (H2O), were negative. The displayed figure was cropped and the original gel images are part of the supplementary data. (B) Fluorescence image of GCs preloaded with PO1 at the start of the monitoring (t = 0) and 20 min after addition of H2O2 (final concentration: 100 µM). Scale bar = 10 µm. (C,D) The aquaporine blocker AgNO3 (500 nM) significantly decreased H2O2 production by 22 % (after 2 h). Data are shown as mean of six technical repetitions of a single measurement (relative to the intensity at t = 0; n = 1). (D) Percentage decrease of four measurements compared to control. One-sample t-test (two-tailed; *p < 0.05).
Roles of H2O2 in GCs and KGN
To explore specific roles of NOX4-derived H2O2, two strategies were employed. Consequences of the NOX4 blocker GKT137831, thereby lowering production of H2O2, and consequences of direct addition of H2O2 were studied.
H2O2 treatment (2 h) resulted in an increased phosphorylation of MAPK (representative example shown in Fig. 6, n = 2).

H2O2 increases phosphorylation of MAPK. (A) WB membrane shows the expression of MAPK and pMAPK of treated and untreated GCs on culture day 1 of a representative example (arrows show expected size at 42 and 44 kDa; two repetitions). The displayed figure was cropped and the original gel images are part of the supplementary data.
Because primary GCs do not proliferate in vitro, we studied proliferation in a human granulosa tumor cell line (KGN). In KGN, we confirmed expression of NOX4 by RT-PCR and a significant reduction of H2O2 production by NOX4 blocker (Supplementary Fig. S4A,B). Treatment with GKT137831 did not affect KGN viability (Fig. 7D; LDH assay, n = 5), but significantly reduced cell number (Fig. 7A; n = 6) and confluence over 24 h (Fig. 7B; n = 4). Accordingly, expression of the proliferation marker PCNA was reduced, as shown by Western blotting (Fig. 7C; n = 4).

Actions of a NOX4 blocker in KGN. KGN treatment with GKT137831 resulted in a significant reduction in cell number (A; n = 6) and confluence (B; n = 4), as well as reduced PCNA content. The figure was cropped and the original gel images are shown in the supplementary data (C) PCNA in KGN compared to control (blotted membrane of one representative example is shown). Diagram shows PCNA levels of control versus treated cells normalized to β-actin (n = 4). (D) Treatment with GKT137831 had no cytotoxic effect in KGN compared to control (LDH cytotoxicity assay; n = 5). One-sample t-tests (two-tailed; *p < 0.05).
Discussion
The results of this study show that NOX4 is a major producer of H2O2 in human GCs in vitro. NOX4 levels are not under the control of FSH or LH/hCG in GCs. H2O2 can potentially act as a cell-cell signaling factor and AQPs may be involved in facilitating membrane transport of this ROS. The consequences of H2O2 signaling remain to be fully explored, yet expression of NOX4 in situ, and in growing follicles, as well as our results in KGN indicate a role in proliferation of ovarian cells.
There are few studies examining NOX enzymes, their specific ROS products and their roles in the ovary16,32,33,34. NOX1-3 were not found in GCs, but previous studies showed expression of NOX4 in human GCs12,20,34. This was confirmed by our study. NOX4 protein was further detected by immunohistochemistry in granulosa, theca and luteal cells of the human ovary. These results are in line with previous reports35,36,37.
Besides NOX4, we also detected NOX5, DUOX1 and 2. NOX5 was described in a previous study12. This human specific enzyme is absent in mice38, and generates superoxide, which is rapidly dismutated to H2O2. DUOX1/2, as well as NOX4 generate H2O232. This might partially explain why we only found traces of superoxide, but abundant H2O2. H2O2 could be the result of NOX4, and DUOX1 and DUOX2 activities. Experimental studies on NOX5, as well as DUOX1/2 are limited by the lack of specific antibodies and pharmacological tools32, yet such tools exist for NOX4.
In the vascular system NOX4 activity accounts for about one-third of endothelial H2O2 formation17 and a study in mice reported a protective role of NOX4 for vascular function21. We applied the Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit, a purely extracellular H2O2 detection method, and a cell permeable boronate compound (PO1) specifically activated by intracellular H2O2. The measurements confirmed the presence of intra- and extracellular H2O2. The results indicate that GCs, as well as KGN, can produce H2O2, which can leave its cellular sites of generation. In combination with experiments using a NOX4 blocker they revealed a 36% contribution of NOX4 to overall H2O2 generation. Results are in line with a report on the presence of H2O2 in FF39.
As an extracellularly released factor, H2O2 may act on neighboring cells or oocytes. We examined, whether, as a prerequisite for such autocrine/paracrine H2O2 actions, it may be able to enter GCs. This is assumed to be a criterion for interaction with intracellular targets. Our studies using PO1 showed that this is the case. AQP3, 8 and 9 reportedly facilitate transport of H2O2 and are referred to as peroxiporins29,30. In line with previous studies, we identified AQP3, 8 and 9 in GCs24. Blocking AQPs with AgNO3 in the presence of extracellular H2O2 supports a significant contribution of these channels in the uptake of H2O2. Whether they are also involved in the release of this ROS, remains to be studied.
How NOX4 expression is regulated also remains to be shown. The human ovarian samples available for immunohistochemistry showed expression in growing follicles, yet NOX4 expression in resting follicles could not be evaluated. The studies in cultured GCs, derived from preovulatory follicles, are in line with the notion that NOX4 is a constitutive enzyme, as our results did not reveal that FSH or hCG affect its level. FSH-, and LH-receptors were present and functional, as concluded from the increase in DUOX1/2 by this treatment. The increase was, however, without measurable consequences with regard to overall ROS or specifically H2O2 production.
The widespread expression of NOX4 in the normal human ovary may imply important physiological roles. As previously described in the rat, H2O2 caused phosphorylation of 42/44 MAPK4. This is also in line with a recently published paper demonstrating the necessity of NOX-derived ROS for the EGF/MAPK signaling pathway in uterine leiomyomas40. We found increased phosphorylation of 42/44 MAPK after direct addition of H2O2 to GCs. Additional studies are required to further examine downstream pathways.
As proliferation is crucial for follicular growth and NOX4 was detected in growing follicles, we studied a possible involvement of NOX4 in GC proliferation by using KGN cells as a model system. These cells, derived from a human GC tumor22, proliferate in contrast to primary GCs. Blocking NOX4 reduced H2O2 production, cell number and confluence. As no signs of cytotoxicity were detected and expression of the proliferation marker PCNA was reduced, our results indicate that NOX4-derived H2O2 is involved in cell proliferation. The precise mechanisms of action and possible other roles remain to be studied.
Materials and Methods
Human GC isolation, culture and treatment, KGN culture
Follicular fluid (FF) containing GCs was derived from more than 300 patients (age range 28 to 40 years) undergoing IVF due to poor quality of sperm or restricted tubal patency. The stimulation protocols (i.e. the “long” protocol), were described9,10,41,42,43,44. The ethics committee of the Ludwig-Maximilians University Munich approved the use of follicular aspirates and GCs for scientific experiments (Project 323-05). A written informed consent of the patients was obtained, samples and clinical information were anonymized. All experiments were performed in accordance with relevant guidelines and regulations (including all laboratory and biosafety regulations). The FF from two to five patients were pooled for each preparation of GCs, following a method45, which utilizes a cell strainer (40 μm; BD, Franklin Lakes, NJ, USA) for filtration, as described previously9,10,46. Cells were cultured in DMEM/Ham’s F12 medium supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml) and 10% fetal calf serum (FCS; all from PAA) for up to 4 days at 37 °C and with 5% CO2. For all experimental treatments, DMEM/Ham’s F12 medium without FCS was used.
The KGN cell line was obtained from RIKEN BioResource Center, and cultured as described22 in DMEM/Ham’s F12 medium supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml) and 10% FCS.
For the experiments 20 µM GKT137831 (Cayman Chemical, Michigan, USA), 10 mM 3-Amino-1,2,4-triazole (3-AT) (Sigma-Aldrich, St. Louis, Missouri, USA), 1 IU/ml human recombinant FSH (Cedarlane, Burlington, NC, USA) or 10 IU/ml hCG (Sigma-Aldrich) were used, together with the appropriate corresponding solvent controls (including DMSO, PBS and 0.01 M NaH2PO4; all from Sigma-Aldrich), diluted in DMEM/Ham’s F12 medium (Thermo Fisher Scientific, Darmstadt, Germany).
Reverse transcription-PCR and quantitative RT-PCR (qRT-PCR)
Total RNA was isolated using the RNeasy Plus Micro Kit (Qiagen, Hilden, Germany). RNA concentration and purity was determined using a NanoDrop® spectrophotometer (Thermo Fisher Scientific). cDNA was produced using random pentadecamer primers (metabion international AG; Munich, Germany) and Superscript II Reverse Transcriptase (Life Technologies). RT-PCR was arranged with different oligomer primers (metabion international AG) listed in Table 1 and GoTaq DNA Polymerase Kit from Promega (Mannheim, Germany), amplicons were visualized by agarose gel electrophoresis and were verified by sequencing.
For qRT-PCR studies duplicates were performed and the QuantiFast SYBR Green PCR Kit (Qiagen) was applied using following protocol in a LightCycler® 96 System (Roche Diagnostics, Penzberg, Germany): heat activation (95 °C, 5 min), 40 cycles denaturation (95 °C, 10 s) and annealing/extension (60 °C, 30 s) followed by melting (95 °C, 10 s; 65 °C, 60 s and 97 °C, 1 s) and cooling-down (37 °C, 30 s). Negative controls included RNA, added instead of cDNA (-RT), and H2O, instead of cDNA. Quantitative results were calculated according to the 2−ΔΔCq method47 and normalized for the geometric mean of the reference genes RPL19, UBC, PPIA and GAPDH, as well as to the respective, untreated control. Results are depicted as means ± SEM.
Western blot
The method was performed as described46. Protein samples were analyzed (7,5 µg of GCs on culture day 2 and 10 µg of KGN) with anti-NOX4 rabbit polyclonal antibody (Cat#: 7927, ProSci; Fort Collins, CO, USA), anti-PCNA (Cat#: 610664; BD), anti-MAPK (Cat#: 4695, Cell Signaling), anti-pMAPK (Cat#: 9106, Cell Signaling), as well as anti-β-Actin mouse monoclonal antibody (Cat#: A5441, Sigma-Aldrich). Secondary antibodies were conjugated with horseradish peroxidase (Jackson, Bar Harbor, Maine, USA).
Immunohistochemistry
Immunohistochemistry was performed using the same archival human ovarian sections as described46. A goat polyclonal IgG raised against human NOX4 (NB110-58849, Novus Biologicals, Littleton; CO; USA), and the same antibody used also in Western blot studies, as well as biotinylated secondary antibodies (Jackson, Bar Harbor, Maine, USA) were employed. The primary antiserum was pre-adsorbed by a corresponding blocking peptide (NB110-58849PEP, Novus Biologicals). As a further control the antiserum was replaced by non- immune serum. Hematoxylin-eosin-staining was performed and images were taken with an Axiovert microscope (Zeiss, Jen, Germany).
Measurement of ROS generation
ROS generation was measured with the 2-,7-dichlorodihydrofluorescein diacetate (H2DCFDA) method, as described9,10,11. The cell permeant reagent H2DCFDA is a fluorogenic dye, which measures hydroxyl, peroxyl and other ROS within cells. After diffusion into the cell, it is deacetylated by cellular esterases to a non-fluorescent compound, which is then oxidized by ROS into 2′, 7′ –dichlorofluorescein (DCF). DCF is a highly fluorescent compound, which can be detected by fluorescence spectroscopy. GCs of independent preparations of cells from two to five patients were seeded into black 96-well plates (Nunclon Delta Surface; six technical repetitions for each group), cultured for 24 h, preloaded with H2DCFDA (10 µM) for 30 min and fluorescence levels were measured at 485 nm excitation/ 520 nm emission in a fluorometer (FLUOstar Omega; BMG labtech, Ortenberg, Germany) for 2 h at 37 °C. The final values were statistically analyzed and depicted relative to the value at t = 0.
Measurement of H2O2 generation
The generation of H2O2 was measured using an Amplex® Red Kit (Invitrogen), as described11. Briefly, Amplex® Red reagent (10-acetyl-3,7-dihydroxyphenoxazine) is a colorless substrate, which reacts with H2O2 with a 1:1 stoichiometry to produce fluorescent resorufin. The method is highly sensitive and as little as 10 picomoles of H2O2 in a 100 µL volume (100 nM) can be detected. GCs were seeded as described for ROS measurement and 1.5 × 104 KGNs/ well, respectively. Amplex® Red reagent was used in a final concentration of 2.5 µM and fluorescence levels were measured at 544 nm excitation/590 nm emission in a fluorometer (BMG labtech) for 2 h at 37 °C. The mean values were normalized to the starting point value.
Measurement of H2O2 uptake in GCs
Intracellular H2O2 levels in cultures of human GCs were quantified in black 96-well microplates (Nunclon Delta Surface) using the cell permeable boronate-based fluorescent H2O2-probe Peroxy Orange 1 (PO1) (Tocris, Bristol, UK) by exogenous preloading as described48. Briefly, GCs were seeded one day prior to measurements. Culture medium was replaced with extracellular (EC) fluid buffer (140 mM NaCl, 3 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES and 10 mM glucose; pH 7.4) and cells were preloaded for 20 min with 1 µM PO1 and/or 500 nM AgNO3 (Honeywell, Seelze, Germany). The concentration (500 nM) was chosen in pilot experiments, because it was not toxic to GCs (see Supplementary Figure S3) and effectively blocked the measurable increase in PO1- and DCF-fluorescence. Fluorescence was determined using a microplate reader (BMG labtech) after replacing the staining solution with 100 µM H2O2 in EC buffer (544 nm excitation/590 nm emission).
In addition, fluorescent PO1 was examined using a Leica TCS SP8 confocal microscope (Leica, Wetzlar, Germany). GCs were cultured on a µ-Dish 35mm, high, ibiTreat (ibidi GmbH, Martinsried, Germany) for 2 days. Prior to microscopy, culture medium was replaced by 1 µM PO1 diluted in EC buffer and after 20 min H2O2 (end concentration: 333 µM) was added. Fluorescence imaging was performed at t = 0 and t = 20 min at 37 °C (552 nm excitation/562–650 nm emission via HyD detector).
Cell viability assay
Cellular ATP content49 correlates with viability and was determined using CellTiter-Glo Luminescent Cell Viability Assay (Promega; Mannheim, Germany), as described49. The assay system is based on the properties of a thermostable luciferase to enable reaction conditions, which generate a stable luminescent signal while inhibiting endogenous enzymes released during cell lysis (e.g., ATPases). The luminescence was measured in a luminometer (BMG labtech).
Lactate dehydrogenase (LDH) assay
LDH indicates damaged cell membranes and thus cytotoxicity of GKT137831 was measured in KGN (1.5 × 104 cells/well) using a LDH cytotoxicity assay kit (Thermo Fisher Scientific), as described46. LDH is a cytosolic enzyme released upon plasma membrane damage into the cell culture media. Extracellular LDH can be quantified by a coupled enzymatic reaction, in which LDH catalyzes the conversion of lactate to pyruvate via NAD+ reduction to NADH. Diaphorase then uses NADH to reduce a tetrazolium salt to a red formazan product, which can be measured at 490 nm. The level of formazan formation is directly proportional to the amount of LDH released into the medium, which is indicative of cytotoxicity.
Confluence and cell counting
The methods were described previously46. Briefly, 1.5 × 105 KGNs/dish were monitored for 24 h by taking time-lapse pictures every 20 min under culture conditions. Confluence values were determined by open source software ImageJ (Fiji) and depicted as fold change to control and delta t0 subtracted. Cells were counted before and after treatment (t = 0 and 24 h) by automated cell counter (Biozym).
Statistics
GraphPad Prism 6.0 Software (GraphPad Software Inc., San Diego, CA, USA) was used to perform one-sample t-tests (two-tailed) for comparisons of −ΔΔCq values and for ROS and H2O2 measurements as well as for ATP-, LDH-assay and cell number analysis. If not stated otherwise, all results are presented as means ± SEM. Corresponding p-values that report statistical significance (≤0.05) are individually specified in the captions.
Data Availability
Data generated and analyzed during this study are included in this published article and its Supplementary Information files.
References
Finkel, T. Signal transduction by reactive oxygen species. The Journal of cell biology 194, 7–15, https://doi.org/10.1083/jcb.201102095 (2011).
Reczek, C. R. & Chandel, N. S. ROS-dependent signal transduction. Curr Opin Cell Biol 33, 8–13, https://doi.org/10.1016/j.ceb.2014.09.010 (2015).
Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol 11, 613–619, https://doi.org/10.1016/j.redox.2016.12.035 (2017).
Shkolnik, K. et al. Reactive oxygen species are indispensable in ovulation. Proceedings of the National Academy of Sciences of the United States of America 108, 1462–1467, https://doi.org/10.1073/pnas.1017213108 (2011).
Carlson, J. C., Wu, X. M. & Sawada, M. Oxygen radicals and the control of ovarian corpus luteum function. Free Radic Biol Med 14, 79–84 (1993).
Agarwal, A., Aponte-Mellado, A., Premkumar, B. J., Shaman, A. & Gupta, S. The effects of oxidative stress on female reproduction: a review. Reproductive biology and endocrinology: RB&E 10, 49, https://doi.org/10.1186/1477-7827-10-49 (2012).
Devine, P. J., Perreault, S. D. & Luderer, U. Roles of reactive oxygen species and antioxidants in ovarian toxicity. Biol Reprod 86, 27, https://doi.org/10.1095/biolreprod.111.095224 (2012).
Avila, J., Gonzalez-Fernandez, R., Rotoli, D., Hernandez, J. & Palumbo, A. Oxidative Stress in Granulosa-Lutein Cells From In Vitro Fertilization Patients. Reprod Sci 23, 1656–1661, https://doi.org/10.1177/1933719116674077 (2016).
Saller, S. et al. Dopamine in human follicular fluid is associated with cellular uptake and metabolism-dependent generation of reactive oxygen species in granulosa cells: implications for physiology and pathology. Hum Reprod 29, 555–567, https://doi.org/10.1093/humrep/det422 (2014).
Saller, S. et al. Norepinephrine, active norepinephrine transporter, and norepinephrine-metabolism are involved in the generation of reactive oxygen species in human ovarian granulosa cells. Endocrinology 153, 1472–1483, https://doi.org/10.1210/en.2011-1769 (2012).
Blohberger, J. et al. L-DOPA in the hu man ovarian follicular fluid acts as an antioxidant factor on granulosa cells. Journal of ovarian research 9, 62, https://doi.org/10.1186/s13048-016-0269-0 (2016).
Kampfer, C. et al. Pigment-Epithelium Derived Factor (PEDF) and the human ovary: a role in the generation of ROS in granulosa cells. Life Sci 97, 129–136, https://doi.org/10.1016/j.lfs.2013.12.007 (2014).
Adam, M. et al. Decorin is a part of the ovarian extracellular matrix in primates and may act as a signaling molecule. Hum Reprod 27, 3249–3258, https://doi.org/10.1093/humrep/des297 (2012).
Wong, J. L., Creton, R. & Wessel, G. M. The oxidative burst at fertilization is dependent upon activation of the dual oxidase Udx1. Dev Cell 7, 801–814, https://doi.org/10.1016/j.devcel.2004.10.014 (2004).
Guo, S. & Chen, X. The human Nox4: gene, structure, physiological function and pathological significance. J Drug Target 23, 888–896, https://doi.org/10.3109/1061186x.2015.1036276 (2015).
Sirokmany, G., Donko, A. & Geiszt, M. Nox/Duox Family of NADPH Oxidases: Lessons from Knockout Mouse Models. Trends Pharmacol Sci 37, 318–327, https://doi.org/10.1016/j.tips.2016.01.006 (2016).
Schroder, K. et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circulation research 110, 1217–1225, https://doi.org/10.1161/circresaha.112.267054 (2012).
Elizur, S. E., Lebovitz, O., Orvieto, R., Dor, J. & Zan-Bar, T. Reactive oxygen species in follicular fluid may serve as biochemical markers to determine ovarian aging and follicular metabolic age. Gynecol Endocrinol 30, 705–707, https://doi.org/10.3109/09513590.2014.924100 (2014).
Bienert, G. P. et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem 282, 1183–1192, https://doi.org/10.1074/jbc.M603761200 (2007).
Maraldi, T. et al. NADPH oxidase-4 and MATER expressions in granulosa cells: Relationships with ovarian aging. Life Sci 162, 108–114, https://doi.org/10.1016/j.lfs.2016.08.007 (2016).
Sies, H., Berndt, C. & Jones, D. P. Oxidative Stress. Annu Rev Biochem 86, 715–748, https://doi.org/10.1146/annurev-biochem-061516-045037 (2017).
Nishi, Y. et al. Establishment and characterization of a steroidogenic human granulosa-like tumor cell line, KGN, that expresses functional follicle-stimulating hormone receptor. Endocrinology 142, 437–445, https://doi.org/10.1210/endo.142.1.7862 (2001).
Simoni, M., Gromoll, J. & Nieschlag, E. The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev 18, 739–773, https://doi.org/10.1210/edrv.18.6.0320 (1997).
Lee, H. J. et al. Expressions of aquaporin family in human luteinized granulosa cells and their correlations with IVF outcomes. Hum Reprod 31, 822–831, https://doi.org/10.1093/humrep/dew006 (2016).
Sales, A. D., Lobo, C. H., Carvalho, A. A., Moura, A. A. & Rodrigues, A. P. Structure, function, and localization of aquaporins: their possible implications on gamete cryopreservation. Genet Mol Res 12, 6718–6732, https://doi.org/10.4238/2013.December.13.5 (2013).
Thoroddsen, A. et al. The water permeability channels aquaporins 1-4 are differentially expressed in granulosa and theca cells of the preovulatory follicle during precise stages of human ovulation. J Clin Endocrinol Metab 96, 1021–1028, https://doi.org/10.1210/jc.2010-2545 (2011).
Rodgers, R. J. & Irving-Rodgers, H. F. Formation of the ovarian follicular antrum and follicular fluid. Biol Reprod 82, 1021–1029, https://doi.org/10.1095/biolreprod.109.082941 (2010).
Huang, H. F. et al. Function of aquaporins in female and male reproductive systems. Hum Reprod Update 12, 785–795, https://doi.org/10.1093/humupd/dml035 (2006).
Miller, E. W., Dickinson, B. C. & Chang, C. J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proceedings of the National Academy of Sciences of the United States of America 107, 15681–15686, https://doi.org/10.1073/pnas.1005776107 (2010).
Watanabe, S., Moniaga, C. S., Nielsen, S. & Hara-Chikuma, M. Aquaporin-9 facilitates membrane transport of hydrogen peroxide in mammalian cells. Biochem Biophys Res Commun 471, 191–197, https://doi.org/10.1016/j.bbrc.2016.01.153 (2016).
Niemietz, C. M. & Tyerman, S. D. New potent inhibitors of aquaporins: silver and gold compounds inhibit aquaporins of plant and human origin. FEBS letters 531, 443–447 (2002).
Altenhofer, S., Radermacher, K. A., Kleikers, P. W., Wingler, K. & Schmidt, H. H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid Redox Signal 23, 406–427, https://doi.org/10.1089/ars.2013.5814 (2015).
Brandes, R. P., Weissmann, N. & Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med 76, 208–226, https://doi.org/10.1016/j.freeradbiomed.2014.07.046 (2014).
Bedard, K. & Krause, K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87, 245–313, https://doi.org/10.1152/physrev.00044.2005 (2007).
Graham, K. A. et al. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol Ther 10, 223–231 (2010).
Koziel, R. et al. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem J 452, 231–239, https://doi.org/10.1042/BJ20121778 (2013).
Kuroda, J. et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes to cells: devoted to molecular & cellular mechanisms 10, 1139–1151, https://doi.org/10.1111/j.1365-2443.2005.00907.x (2005).
Bedard, K., Jaquet, V. & Krause, K. H. NOX5: from basic biology to signaling and disease. Free Radic Biol Med 52, 725–734, https://doi.org/10.1016/j.freeradbiomed.2011.11.023 (2012).
Hennet, M. L., Yu, H. Y. & Combelles, C. M. Follicular fluid hydrogen peroxide and lipid hydroperoxide in bovine antral follicles of various size, atresia, and dominance status. Journal of assisted reproduction and genetics 30, 333–340, https://doi.org/10.1007/s10815-012-9925-5 (2013).
Mesquita, F. S. et al. Reactive oxygen species mediate mitogenic growth factor signaling pathways in human leiomyoma smooth muscle cells. Biol Reprod 82, 341–351, https://doi.org/10.1095/biolreprod.108.075887 (2010).
Bulling, A. et al. Identification of an ovarian voltage-activated Na+ -channel type: hints to involvement in luteolysis. Mol Endocrinol 14, 1064–1074, https://doi.org/10.1210/mend.14.7.0481 (2000).
Mayerhofer, A. et al. FSH regulates acetycholine production by ovarian granulosa cells. Reproductive biology and endocrinology: RB&E 4, 37, https://doi.org/10.1186/1477-7827-4-37 (2006).
Mayerhofer, A., Fohr, K. J., Sterzik, K. & Gratzl, M. Carbachol increases intracellular free calcium concentrations in human granulosa-lutein cells. J Endocrinol 135, 153–159 (1992).
Mayerhofer, A., Sterzik, K., Link, H., Wiemann, M. & Gratzl, M. Effect of oxytocin on free intracellular Ca2+ levels and progesterone release by human granulosa-lutein cells. J Clin Endocrinol Metab 77, 1209–1214, https://doi.org/10.1210/jcem.77.5.8077313 (1993).
Ferrero, H. et al. Efficiency and purity provided by the existing methods for the isolation of luteinized granulosa cells: a comparative study. Hum Reprod 27, 1781–1789, https://doi.org/10.1093/humrep/des096 (2012).
Blohberger, J. et al. Readthrough acetylcholinesterase (AChE-R) and regulated necrosis: pharmacological targets for the regulation of ovarian functions? Cell death & disease 6, e1685, https://doi.org/10.1038/cddis.2015.51 (2015).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry 55, 611–622, https://doi.org/10.1373/clinchem.2008.112797 (2009).
Dickinson, B. C., Huynh, C. & Chang, C. J. A palette of fluorescent probes with varying emission colors for imaging hydrogen peroxide signaling in living cells. Journal of the American Chemical Society 132, 5906–5915, https://doi.org/10.1021/ja1014103 (2010).
Saller, S. et al. Oxytocin receptors in the primate ovary: molecular identity and link to apoptosis in human granulosa cells. Hum Reprod 25, 969–976, https://doi.org/10.1093/humrep/dep467 (2010).
Acknowledgements
The authors thank Carola Herrmann, Kim Dietrich, Astrid Tiefenbacher, Daniel Aigner and Karin Metzrath for excellence technical and editorial assistance, Verena Kast for performing some of the KGN experiments, Anna Klemm and the other members of the Core Facility Bioimaging at the BMC, Munich, for assistance, especially with open source software ImageJ (Fiji). We further thank Prof. Dr. Toshihiko Yanase, Fukuoka University, Japan for supplying KGN cells. The study was supported by grants from Deutsche Forschungsgemeinschaft (DFG), DFG MA1080/26-1, KU1282/7-1. This work was done in partial fulfilments of the requirements for a “Dr. rer. nat.” degree (T.B.) and a “Dr. med.” degree (C.T.H.) at LMU, Munich.
Author information
Authors and Affiliations
Contributions
T.B. performed the majority of the cellular experiments, C.T.H., performed studies with PO1, and together they evaluated the results. D.B. and U.B. provided FF and GCs, as well as conceptual input, L.K. and A.M. conceived of the study, directed the work and supervised the experiments. T.B. and A.M. drafted the manuscript. All authors contributed to and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Buck, T., Hack, C.T., Berg, D. et al. The NADPH oxidase 4 is a major source of hydrogen peroxide in human granulosa-lutein and granulosa tumor cells. Sci Rep 9, 3585 (2019). https://doi.org/10.1038/s41598-019-40329-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40329-8
This article is cited by
-
Implications of obstructive sleep apnea on reproductive health: a study on anti-Mullerian hormone levels
Sleep and Breathing (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
