
Abstract
Vascular endothelial growth factor (VEGF) inhibitors cause glomerular injury. We have recently shown that activation of protease-activated receptor 2 (PAR2) by factor Xa exacerbated diabetic kidney disease. However, the role of PAR2 in glomerular injury induced by VEGF blockade is not known. Herein, we investigated the effect of the lack of PAR2 on VEGF inhibitor-induced glomerular injury. Although administering an anti-VEGF antibody by itself did not show renal phenotype in wild type mice, its administration to mice lacking endothelial nitric oxide synthase (eNOS) caused glomerular injury. Different from what we expected, administration of an anti-VEGF antibody in mice lacking PAR2 and eNOS exacerbated albuminuria and reduced the expression levels of CD31, pro-angiogenic VEGF, and angiogenesis-related chemokines in their kidneys. Podocyte injury was also evident in this model of mice lacking PAR2. Our results suggest that PAR2 is protective against VEGF inhibitor-induced glomerular endothelial and podocyte injury.
Similar content being viewed by others


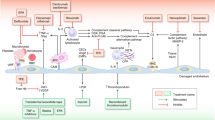
Introduction
Vascular endothelial growth factor (VEGF) inhibitors are used in conjunction with chemotherapy to treat several types of cancer. However, kidney glomerular injury, such as thrombotic microangiopathy (TMA), is observed in a subset of patients and can be a cause of treatment discontinuation1,2. Some preeclamptic patients develop kidney injury and hypertension caused by soluble fms-like tyrosine kinase 1, a decoy of VEGF that suppresses angiogenesis3. Accordingly, there is an increasing interest in exploring novel therapies for VEGF inhibitor-induced kidney injury.
Hypercoagulability is associated with VEGF inhibition. Fibrin deposition is observed within the glomeruli in VEGF inhibitor-induced TMA1. Furthermore, coagulation abnormalities are reported in preeclamptic patients treated with a VEGF inhibitor4,5. Coagulation factors have a pleiotropic effect through the activation of protease-activated receptors (PARs), a G protein-coupled receptor family6. For instance, tissue factor/VIIa complex or factor Xa activates PAR2, which is abundantly expressed in the kidney6,7.
Although several studies, including ours, have shown that PAR2 exacerbates glomerular injury in models of diabetic kidney disease (DKD) or glomerulonephritis7,8, the role of PAR2 in VEGF inhibitor-induced kidney injury is controversial. Tissue factor and PAR2 exacerbate preeclampsia and kidney injury in models of antiphospholipid syndrome9,10. Conversely, PAR2 signaling contributes to endothelial proliferation/migration and increased pro-angiogenic factors11,12. Pro-angiogenic roles of PAR2 on limb ischemia and retinal neovascularization were also shown13,14,15. These findings may indicate that PAR2 protects the glomerular endothelium from damage secondary to VEGF inhibition.
Herein, we demonstrated that a lack of PAR2 in VEGF inhibitor-induced glomerular injury model exacerbated albuminuria, and endothelial and podocyte injury, together with reduced angiogenic markers.
Results
Role of PAR2 in kidney injury in anti-VEGF antibody-induced glomerular injury
To produce a model of mouse kidney injury using an anti-VEGF antibody (Ab), we first tested the effect of anti-VEGF Ab on wild type mice. However, VEGF inhibition did not affect glomerular histology or urinary albumin excretion (Supplementary Fig. 1A,B). Endothelial nitric oxide synthase (eNOS) dysfunction is important in the onset and exacerbation of VEGF inhibitor-induced glomerular injury because eNOS promotes the proliferation and migration of endothelial cells16, and because eNOS derived NO is protective against podocyte injury17. We have previously shown that a lack of eNOS increases endothelin and exacerbates blood coagulation and preeclampsia18,19,20. Furthermore, eNOS polymorphism is associated with a higher risk of preeclampsia21. Accordingly, we next administered an anti-VEGF Ab to eNOS−/− mice. To investigate the role of PAR2 in this model, we produced eNOS−/− mice with or without PAR2 (eNOS−/−; PAR2+/+ and eNOS−/−; PAR2−/−, respectively), and administered them an anti-VEGF Ab. Blood pressure (BP) levels were increased by VEGF inhibition, whereas PAR2 deletion had no effect (Fig. 1A). A lack of PAR2 significantly increased urinary albumin excretion in eNOS−/− mice treated with VEGF inhibitor (58.6 ± 16.4 μg/mg creatinine) compared to that of control groups (Fig. 1B). The level of plasma cystatin C, a marker of renal function, was similar among the groups (Fig. 1C). Two glomerular injury scores (open capillary area and mesangial area) were evaluated as previously demonstrated7,22. Open capillary area was increased in eNOS−/−; PAR2−/− mice that did not receive the Ab as compared to that of control eNOS−/−; PAR2+/+ mice. Anti-VEGF Ab decreased open capillary area in eNOS−/−; PAR+/+ mice compared to that of eNOS−/−; PAR2−/− mice that did not receive the Ab and in eNOS−/−; PAR2−/− mice compared to that of the two control groups. Anti-VEGF Ab increased mesangial area compared to that of control groups (Fig. 1D–F). Other basal characteristics were unremarkable (Supplementary table 1). We concluded that in our model, a lack of PAR2 increases urinary albumin excretion and does not protect against renal injury.

Blood pressure, urinary albumin excretion, and glomerular injury score. (A) Systolic blood pressure (BP). (B) Urinary albumin excretion (U-Alb). (C) Plasma cystatin C (P-cystatin C). (A) lack of PAR2 exacerbates albuminuria. n ≥ 5 each group in panel A–C. (D) Representative photomicrographs of glomeruli in each group, Periodic acid-Schiff stain (PAS) stain, Scale bar indicates 50 μm. (E,F) Quantitative data of open capillary and PAS positive area in the glomeruli. Approximately 100 glomeruli each group from more than five mice were evaluated. Cre, creatinine. Ab, antibody. A.U, arbitrary unit. Data are shown as mean ± s.e.m.
Markers of endothelial cells and podocytes in VEGF inhibitor-treated mice lacking PAR2
Increased urinary albumin excretion could be the result of impaired function of glomerular endothelial cells or podocytes23,24. Because VEGF inhibition causes glomerular endotheliosis1,2, we first tested whether a lack of PAR2 in the eNOS−/− mice receiving anti-VEGF Ab damages glomerular endothelial cells. The result showed that a lack of PAR2 reduced glomerular density of immunopositive CD31 (endothelial marker) in the kidneys of the eNOS−/− mice treated with anti-VEGF Ab (Fig. 2A,B).

Reduced expression of makers of endothelial cell and podocyte. (A) Representative photomicrographs of immunohistochemistry against CD31. Scale bar indicates 50 μm. (B) Density of glomerular CD31 is reduced in the kidneys from eNOS−/−; PAR2−/− with a VEGF inhibitor. (C) Representative photomicrographs of immunohistochemistry against nephrin. Scale bar indicates 50 μm. (D) Density of glomerular nephrin is reduced in the kidneys from eNOS−/−; PAR2−/− with a VEGF inhibitor. Approximately 100 glomeruli each group from 4 to 6 mice were evaluated. Ab, antibody. A.U, arbitrary unit. Data are shown as mean ± s.e.m.
Glomerular endothelial cells communicate with podocytes to maintain their function, and glomerular endothelial injury promotes podocyte injury leading to albuminuria23,24. Because podocyte dysfunction is known as one of the features of VEGF inhibitor - related glomerular injury25,26,27, we measured nephrin level, which is a podocyte-specific protein. A lack of PAR2 reduced the expression of nephrin in eNOS−/− mice receiving anti-VEGF Ab (Fig. 2C,D).
Consistent with these results, electron microscopy showed a loss of endothelial fenestration and podocyte foot process effacement in the glomeruli from eNOS−/−;PAR2−/− mice treated with anti-VEGF Ab (Fig. 3). We concluded that PAR2 deficiency does not cause damage to endothelial cells or podocytes, but does exacerbate the damage induced by anti-VEGF antibody, which is likely responsible for albuminuria secondary to PAR2 deletion in our model of glomerular injury.

Representative transmission electron micrographs in the glomeruli. Arrows indicate loss of endothelial fenestration. Arrow-heads indicate foot process effacement in podocytes. EM, electron microscopy. Ab, antibody. Scale bar indicates 2 μm.
Expression of angiogenic factors in the kidney
Because PAR2 regulates the expression of angiogenic factors, such as VEGF and angiopoietin11,15, we quantified their expression in the kidneys. Among them, the levels of Vegfa and Tie2 were increased in our model, and PAR2 deletion corrected Vegfa level (Fig. 4A). Anti-VEGF Ab reduced the expression of Kdr in the kidneys from eNOS−/−; PAR2−/− mice (Fig. 4A). Consistent with the change in gene expression, the level of glomerular VEGF protein was increased in the kidneys from eNOS−/−; PAR2+/+ mice treated with anti-VEGF Ab, and a lack of PAR2 reduced it (Fig. 4B,C). Taken together, the exacerbation of glomerular injury by a lack of PAR2 was associated with the reduced expression of angiogenic factors in the kidney.

Expression of angiogenic factors in the kidney. (A) Gene expression related to pro-angiogenic factors (Vegfa, Angpt1, Flt1, Kdr, and Tie2). n ≥ 5. (B) Representative photomicrographs of immunohistochemistry against VEGF in the kidneys. Scale bar indicates 50 μm. (C) Quantitative data of glomerular VEGF protein. Approximately 100 glomeruli each group from 4 to 6 mice were evaluated. Ab, antibody. A.U, arbitrary unit. Data are shown as mean ± s.e.m.
Expression of chemokines in the kidney
Next, we quantified the levels of angiogenesis-related chemokines (Fig. 5). Anti-VEGF Ab decreased Ccl2 mRNA expression in eNOS−/−; PAR2+/+ mice and there was a greater decrease in eNOS−/−; PAR2−/− mice. The level of Cxcl1 mRNA was similar among the groups. Non-significant reduction of Ccr2 expression was obtained by a lack of PAR2 in the presence and absence of anti-VEGF Ab. The level of Cxcr2 was not affected by the PAR2 genotype, but was reduced in the kidneys from mice receiving anti-VEGF Ab. TLR4 signaling is known to regulate angiogenesis28, and a lack of PAR2 significantly reduced the level of Tlr4 in the kidneys from eNOS−/− mice receiving anti-VEGF Ab. As opposed to the changes in inflammatory genes, the number of infiltrated MAC2 positive cells, a marker of macrophages, was similar among the groups (Supplementary Fig. 2).

Expression of inflammation - related genes in the kidney. The levels of Ccl2, Ccr2, Cxcl1, Cxcr2, and Tlr4 mRNA in the kidney. n ≥ 5. Ab, antibody. A.U, arbitrary unit. Data are shown as mean ± s.e.m.
Effect of PAR2 agonist on human endothelial cells
Because endothelial cells closely interact with coagulation factors and highly express PAR26, we tested the direct effect of the PAR2 agonist, 2f-LIGRLO, on pro-angiogenic factors using a human endothelial cell line (EA.hy926). After treatment of EA.hy926 cells with 2f-LIGRLO (20 μM) for 3 hours, the expression levels of VEGFA, CCL2, CXCL1, and TLR4 mRNA were significantly elevated (Supplementary Fig. 3). Because PAR2 signaling is associated with ERK and PI3K/Akt signaling6, we next tested the effect of MAPK inhibitor (U0126) and PI3K inhibitor (LY294002) on these changes. U0126 significantly reduced elevated VEGFA expression by 2f-LI, whereas LY294002 did not. Elevated CCL2, CXCL1, and TLR4 mRNA were reduced by both U0126 and LY294002 treatments (Fig. 6A–D). The level of VEGF protein in conditioned media harvested from cultured cells was assessed using ELISA. 2f-LIGRLO increased VEGF protein after 24 or 48 hours of treatment with the agonist that was reduced by U0126. On contrary to the changes of mRNA, LY 294002 partially inhibited VEGF production (Fig. 6E). These results show that the expression of VEGF, chemokines, and TLR4 by PAR2 agonist is regulated by both MAPK and PI3K.

Expression of angiogenesis - related genes in human endothelial cell treated with PAR2 agonist. (A–D) Effect of PAR2 agonist (2f-LIGRLO, 20 μM), MAPK inhibitor (U0126, 10 μM), and PI3K inhibitor (LY294002, 10 μM) on expression of VEGFA and inflammatory-related genes. Increased expression of VEGFA is reduced by U0126. Expression levels of CCL2, CXCL1, and TLR4 are reduced by both U0126 and LY294002. Cells were treated for 3 hrs. (E) The protein level of VEGF in conditioned media harvested from cultured endothelial cells. 2f-LIGRLO increases VEGF protein after 24 or 48 hrs treatment. Both U0126 and LY294002 reduce it. #P < 0.01 vs Vehicle, 2f-LI + U0126, and 2f-LI + LY groups. A.U, arbitrary unit. 2f-LI, 2f-LIGRLO. LY, LY294002. Experiments were repeated 3 times. Data are shown as mean ± s.e.m.
Tissue factor expression and fibrin/fibrinogen deposition in the kidney
We next characterized the coagulation abnormalities in our model. Fibrin/fibrinogen thrombi and increased immunoreactive tissue factor were not observed in the glomeruli from eNOS wild type mice regardless of treatment with anti-VEGF Ab or PAR2 expression (Supplementary Fig. 4A). Conversely, glomeruli in which fibrin/fibrinogen thrombi were deposited were easily observed in the kidney from anti-VEGF Ab-treated mice lacking eNOS (Supplementary Fig. 4B). Furthermore, anti-VEGF Ab increased the expression level of tissue factor in mice lacking eNOS, whereas lack of PAR2 did not affect it (Supplementary Fig. 4B,C).
Discussion
We have previously shown that PAR2 exacerbates DKD and adenine-induced chronic kidney disease7,29. Based on these findings, we postulated that a lack of PAR2 would alleviate VEGF inhibitor-induced glomerular injury. However, contrary to our postulation, our results from the present study show that a lack of PAR2 in the eNOS−/− mice receiving anti-VEGF Ab worsens kidney injury (albuminuria) and endothelial and podocyte injury.
Increased urinary albumin excretion secondary to a lack of PAR2 in eNOS−/− mice receiving anti-VEGF Ab is likely caused by endothelial and podocyte injury, which impairs the filtration barrier23. The decreased expression of CD31 and nephrin, and the dysfunction of endothelial cells and podocytes in our model suggest that the damage to the endothelial cells and podocytes is likely responsible for albuminuria.
Our finding that healthy endothelial cells cannot be maintained in the lack of PAR2 suggests that it plays a pivotal role in glomerular endothelial protection. Indeed, it was previously reported in the literature that PAR2 directly promotes the expression of angiogenic factors such as VEGF, angiopoietins, and their receptors in several cell lines11,15,30. PAR2 activation promotes endothelial proliferation in primary neuroretinal endothelial cells via pro-inflammatory effect11. PAR2 is indispensable for tissue factor-induced microvessel stabilization12. Moreover, PAR2 is essential in retinal angiogenesis in rodent models13,15. These findings are consistent with our observations. We therefore investigated the protective role of PAR2 in glomerular endothelial injury caused by VEGF inhibition, and several pathways above cooperatively contribute to reno-protection in our model.
PAR2 is known to increase cytokine/chemokine expression6. CCL2–CCR2 and CXCL1–CXCR2 pathways, well-described pro-angiogenic chemokines, mediate corneal neovascularization, hepatic angiogenesis, endothelial recovery in arterial injury, and cancer-related angiogenesis31,32,33,34,35,36. Furthermore, previous reports have shown that TLR4, another inflammatory mediator, contributes to angiogenesis; TLR4 deletion reduced angiogenesis in alkali-induced corneal neovascularization, ischemic neural tissue, and hindlimb ischemia37,38,39. Our experiment using human endothelial cell line demonstrated that PAR2 agonist increased the level of TLR4 expression. Similarly, PAR2 deletion reduced the expression of TLR4 in the kidneys from VEGF inhibitor-treated mice. Although how angiogenesis-related chemokines contribute to repair of glomerular endothelial cells under VEGF inhibition requires further examination, since such inflammatory mediators are known to promote endothelial survival or production of other angiogenic factors32 which were likely protective in our model of glomerular injury.
The main source of glomerular VEGF is podocytes1, however, PAR2 agonist did not increase VEGFA in immortalized murine podocytes in our experiment (data not shown). Interestingly, we found that PAR2 increases production of VEGF in endothelial cells. Based on the literature, endothelial VEGF protects itself by autocrine/paracrine manner40,41, which supports the protective role of PAR2 in our model. A lack of PAR2 did not affect glomerular macrophage infiltration, another source of VEGF42, in anti-VEGF Ab-treated mice. A limitation of this study is that we used systemic knock-out of PAR2. Further studies are required to elucidate how PAR2 is involved in angiogenesis by podocytes, endothelial cells, and macrophages using conditional knock-out of PAR2, which is our future plan.
Podocyte damage is common and an important finding of VEGF inhibitor-induced kidney injury; urinary podocyte excretion is increased in patients treated with bevacizumab or in those with preeclampsia26,27. Endothelial injury is known to promote podocyte injury. Previous papers have shown that endothelial oxidative stress and reactive oxygen species generation is associated with podocyte detachment in a focal segmental sclerosis model23. Other study has demonstrated that exosomes derived from endothelial cells treated with high glucose exacerbate podocyte dysfunction43. Collectively, increased endothelial injury by a lack of PAR2 likely causes secondary podocyte injury, which is likely important in our model of glomerular injury. Although VEGF signaling in podocyte is still controversial44,45, a previous report has shown that VEGFR2 interacts with nephrin, a specific podocyte marker45. How VEGF inhibitor and/or PAR2 directly affect podocyte maintenance and function is still unclear and should be investigated in the future.
A lack of PAR2 worsens glomerular injury in mice lacking eNOS treated with VEGF inhibitor, but not in eNOS wild type mice treated with VEGF inhibitor. It is likely that the lack of eNOS up-regulates coagulation in models of kidney injury19,20, increases TF, and activates coagulation cascade and PAR2. We have shown that VEGF inhibition increased glomerular TF expression in mice lacking eNOS. This finding suggests that a combination of eNOS deficiency and VEGF inhibition may up-regulate TF-PAR2 pathway which has a protective role in VEGF inhibitor-induced glomerular injury.
Both DKD and VEGF inhibitor-induced glomerulopathies are hypercoagulable states. A lack of PAR2 decreases VEGF and pro-angiogenic cytokines in both DKD and VEGF inhibitor-induced glomerular injury (ref.7 and our preliminary observation). However, the lack or inhibition of PAR2 ameliorates DKD7,46, whereas it worsens VEGF inhibitor-induced glomerular injury. DKD is characterized by abnormally high VEGF expression and enhanced angiogenesis47,48. Furthermore, blockade of VEGF signaling ameliorates DKD in rodent models49. The inhibition of factor Xa, which suppresses the activation of PAR2, alleviates DKD7,50. Collectively, these findings indicate that the coagulation and activation of PAR2 promote excessive VEGF production and abnormal angiogenesis, and are pathogenic in DKD. On the contrary, glomerular endothelial injury in our current model is caused by inhibiting the effect of VEGF by anti-VEGF Ab. In this setting, PAR2-driven increase in the production of VEGF and angiogenesis-related chemokines probably maintain healthy glomerular endothelium. The contrasting effect of PAR2 on DKD and anti-VEGF model is consistent with the previous findings that both too high and too low levels of VEGF are pathogenic, and that the window of VEGF level needed to maintain healthy glomeruli/endothelial cells is very narrow51.
Although VEGF inhibition caused glomerular endothelial injury, features of platelet activation (glomerular platelet deposition or thrombocytopenia) and hemolytic anemia were unremarkable (data not shown). Furthermore, acute kidney failure that is common in human TMA1,2 was lacking. There was no evidence for the involvement of TMA in our model. VEGF inhibitors cause another form of glomerular injury, minimal change glomerulopathy/focal segmental glomerulopathy, which is characterized by proteinuria with prominent podocyte injury, increased c-mip, and less inflammation52,53. Our model could explain the pathogenesis of renal injury in these patients.
In conclusion, we found that PAR2 promotes pro-angiogenic action and is reno-protective in VEGF inhibitor-induced kidney injury.
Methods
Animal model
All experiments were conducted in compliance with the guidelines of Tohoku University. The Institutional Animal Care and Use Committee at Tohoku University approved the experimental protocol. Ten to fourteen-week-old female eNOS+/+; PAR2+/+, eNOS+/+; PAR2−/−, eNOS−/−; PAR2+/+, or eNOS−/−; PAR2−/− mice with C57BL/6J genetic background7 were used. eNOS−/− or eNOS−/−; PAR2−/− mice were obtained by mating male and female eNOS+/−; PAR2+/− mice. Thereafter, eNOS−/− or eNOS−/−; PAR2−/− littermate colony was individually maintained. These mice were injected with B20–4.1.1 (5 mg/kg), a mouse anti-VEGF Ab, on day 0 and 454. The samples were collected on day 7. B20-4.1.1 was kindly provided by Genentech Inc. for research use (South San Francisco, CA, USA). Control groups received a vehicle. Our preliminary observations demonstrated that IgG isotype does not show apparent kidney injury in our experimental condition (data not shown).
Biochemical measurement
ELISA kits were used to measure plasma cystatin C (R&D Systems, Inc., Minneapolis, MN) and human VEGF protein in conditioned media harvested from cultured cells (R&D Systems, Inc., Minneapolis, MN) according to the manufacturer’s protocol.
Urinary analysis
Spot urine samples were collected on day 7. ELISA kit was used to measure urinary albumin (Exocell Inc., Philadelphia, PA). Urinary creatinine was determined by the method we developed using LC-MS/MS55. Urinary albumin to creatinine ratio was defined as urinary albumin excretion.
BP measurement
BP was measured by the computerized tail-cuff method using CODA system (Kent Scientific Corporation, Torrington, CT) on day 6. All procedures were performed as previously described7,22.
Real-time quantitative PCR
Total RNA from the kidney was extracted using TRI Reagent (Molecular Research Center, Inc., Cincinnati, OH). Reverse transcription reaction and real-time PCR were performed using iScript Advanced cDNA Synthesis kit and SsoAdvanced Universal Probe/SYBR Supermix kit (Biorad, Hercules, CA) according to the manufacturer’s protocol. Hypoxanthine-guanine phosphoribosyltransferase (Hprt) was used as a reference gene as we previously reported7,29. The sequences of primers are available on request.
Histological evaluation
Fixed kidney samples were embedded in paraffin, and sections 1.5 μm in thickness were stained with Periodic acid-Schiff (PAS) stain. Glomeruli with a similar diameter of maximal size containing vascular pole were randomly chosen, so that the glomeruli were all approximately axial. The glomerular open capillary area was expressed as the ratio of the glomerular tuft area. The mesangial matrix score was defined as the ratio of glomerular PAS positive area to glomerular tuft area. All examinations were quantified using ImageJ (National Institute of Health, Bethesda, MD) as we previously described7,18,22.
Immunohistochemistry
For immunohistochemistry, rat anti-mouse CD31 antibody (0.3125 μg/ml, BD Pharmingen, Franklin Lakes, NJ), goat anti-mouse VEGF antibody (0.33 μg/ml, R&D Systems, Inc., Minneapolis, MN), goat anti-human nephrin antibody (0.4 μg/ml, Santa Cruz Biotechnology, Dallas, TX), goat anti-mouse tissue factor (4 μg/ml, R&D Systems, Inc., Minneapolis, MN), rabbit anti-human fibrin/fibrinogen antibody (1.8 μg/ml, Dako, Denmark), and rat anti-human/mouse galectin3 (MAC2) antibody (0.5 μg/ml, ebioscience, San Diego, CA) were used. Heat–induced antigen retrieval was performed using sodium citrate buffer to detect VEGF, fibrin, and MAC2. Proteinase K (Dako, Denmark) was used to detect CD31, nephrin, and tissue factor. Primary antibodies were incubated overnight at 4 °C. N-Histofine simple stain kits (Nichirei biosciences Inc., Tokyo, Japan) were used as a secondary antibody according to the manufacturer’s protocol. We incubated sections with IgG isotype or without primary antibody as a negative control. Glomerular density of each protein was assessed using Image J (National Institute of Health, Bethesda, MD).
Culture of human endothelial cells
Human endothelial cells (EA.hy926) were cultured in DMEM-H containing 10% fetal bovine serum56. 2f-LIGRLO was purchased from Tocris Bioscience (Bristol, United Kingdom). U0126 was obtained from Wako Pure Chemical Industries (Osaka, Japan). LY294002 was obtained from Sigma (St. Louis, MO). All experiments were performed after serum starvation for 24 hrs. Both U1026 and LY294002 were administered an hour before 2f-LIGRLO treatment. For quantitative PCR analysis, cells were harvested after 3 hrs of incubation with 2f-LIGRLO. For protein analysis, conditioned media were harvested from cultured endothelial cells after 6, 24, and 48 hrs of incubation with 2f-LIGRLO.
Statistics analysis
Multiple groups were compared using two-way ANOVA with the Tukey-Kramer test for parametric values after checking normality and equal variance. If not pass the tests, log transformation was applied or Kruscal-Wallis test followed by Steel-Dwass test was used for non-parametric values. All analyses were performed using JMP 11.0.0 (SAS Institute Inc., Cary, NC). Values are presented as mean ± s.e.m. Differences were considered statistically significant with P < 0.05.
References
Eremina, V. et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358, 1129–1136, https://doi.org/10.1056/NEJMoa0707330 (2008).
Usui, J. et al. Clinicopathological spectrum of kidney diseases in cancer patients treated with vascular endothelial growth factor inhibitors: a report of 5 cases and review of literature. Hum Pathol 45, 1918–1927, https://doi.org/10.1016/j.humpath.2014.05.015 (2014).
Maynard, S. E. et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111, 649–658, https://doi.org/10.1172/JCI17189 (2003).
Ma, L. et al. In vitro procoagulant activity induced in endothelial cells by chemotherapy and antiangiogenic drug combinations: modulation by lower-dose chemotherapy. Cancer Res 65, 5365–5373, https://doi.org/10.1158/0008-5472.CAN-04-3156 (2005).
Dusse, L. M., Rios, D. R., Pinheiro, M. B., Cooper, A. J. & Lwaleed, B. A. Pre-eclampsia: relationship between coagulation, fibrinolysis and inflammation. Clin Chim Acta 412, 17–21, https://doi.org/10.1016/j.cca.2010.09.030 (2011).
Rothmeier, A. S. & Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol 34, 133–149, https://doi.org/10.1007/s00281-011-0289-1 (2012).
Oe, Y. et al. Coagulation Factor Xa and Protease-Activated Receptor 2 as Novel Therapeutic Targets for Diabetic Nephropathy. Arterioscler Thromb Vasc Biol 36, 1525–1533, https://doi.org/10.1161/ATVBAHA.116.307883 (2016).
Moussa, L., Apostolopoulos, J., Davenport, P., Tchongue, J. & Tipping, P. G. Protease-activated receptor-2 augments experimental crescentic glomerulonephritis. Am J Pathol 171, 800–808, https://doi.org/10.2353/ajpath.2007.061155 (2007).
Redecha, P., Franzke, C. W., Ruf, W., Mackman, N. & Girardi, G. Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest 118, 3453–3461, https://doi.org/10.1172/JCI36089 (2008).
Seshan, S. V. et al. Role of tissue factor in a mouse model of thrombotic microangiopathy induced by antiphospholipid antibodies. Blood 114, 1675–1683, https://doi.org/10.1182/blood-2009-01-199117 (2009).
Zhu, T. et al. Proangiogenic effects of protease-activated receptor 2 are tumor necrosis factor-alpha and consecutively Tie2 dependent. Arterioscler Thromb Vasc Biol 26, 744–750, https://doi.org/10.1161/01.ATV.0000205591.88522.d4 (2006).
Arderiu, G., Espinosa, S., Peña, E., Aledo, R. & Badimon, L. PAR2-SMAD3 in microvascular endothelial cells is indispensable for vascular stability via tissue factor signaling. J Mol Cell Biol 8, 255–270, https://doi.org/10.1093/jmcb/mjv065 (2016).
Uusitalo-Jarvinen, H. et al. Role of protease activated receptor 1 and 2 signaling in hypoxia-induced angiogenesis. Arterioscler Thromb Vasc Biol 27, 1456–1462, https://doi.org/10.1161/ATVBAHA.107.142539 (2007).
van den Hengel, L. G. et al. Protease-activated receptor (PAR)2, but not PAR1, is involved in collateral formation and anti-inflammatory monocyte polarization in a mouse hind limb ischemia model. PLoS One 8, e61923, https://doi.org/10.1371/journal.pone.0061923 (2013).
Joyal, J. S. et al. Subcellular localization of coagulation factor II receptor-like 1 in neurons governs angiogenesis. Nat Med 20, 1165–1173, https://doi.org/10.1038/nm.3669 (2014).
Cooke, J. P. & Losordo, D. W. Nitric oxide and angiogenesis. Circulation 105, 2133–2135 (2002).
Sun, Y. B. et al. Glomerular endothelial cell injury and damage precedes that of podocytes in adriamycin-induced nephropathy. PLoS One 8, e55027, https://doi.org/10.1371/journal.pone.0055027 (2013).
Li, F. et al. eNOS deficiency acts through endothelin to aggravate sFlt-1-induced pre-eclampsia-like phenotype. J Am Soc Nephrol 23, 652–660, https://doi.org/10.1681/ASN.2011040369 (2012).
Oe, Y. et al. Hepatic dysfunction and thrombocytopenia induced by excess sFlt1 in mice lacking endothelial nitric oxide synthase. Sci Rep 8, 102, https://doi.org/10.1038/s41598-017-18260-7 (2018).
Li, F. et al. Elevated tissue factor expression contributes to exacerbated diabetic nephropathy in mice lacking eNOS fed a high fat diet. J Thromb Haemost 8, 2122–2132, https://doi.org/10.1111/j.1538-7836.2010.03976.x (2010).
Serrano, N. C. et al. Endothelial NO synthase genotype and risk of preeclampsia: a multicenter case-control study. Hypertension 44, 702–707, https://doi.org/10.1161/01.HYP.0000143483.66701.ec (2004).
Li, F. et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc Natl Acad Sci USA 113, 13450–13455, https://doi.org/10.1073/pnas.1614947113 (2016).
Daehn, I. et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest 124, 1608–1621, https://doi.org/10.1172/JCI71195 (2014).
Dimke, H., Maezawa, Y. & Quaggin, S. E. Crosstalk in glomerular injury and repair. Curr Opin Nephrol Hypertens 24, 231–238, https://doi.org/10.1097/MNH.0000000000000117 (2015).
Usui, J. et al. The Detection of Urinary Podocytes from Drug-Induced Glomerular Thrombotic Microangiopathy in Advanced Cancer Patients. Clin Lab 62, 2413–2417 (2016).
Hayman, S. R. et al. Urinary podocyte excretion and proteinuria in patients treated with antivascular endothelial growth factor therapy for solid tumor malignancies. Oncology 86, 271–278, https://doi.org/10.1159/000360180 (2014).
Wang, Y., Zhao, S., Loyd, S. & Groome, L. J. Increased urinary excretion of nephrin, podocalyxin, and βig-h3 in women with preeclampsia. Am J Physiol Renal Physiol 302, F1084–1089, https://doi.org/10.1152/ajprenal.00597.2011 (2012).
Yang, S., Xu, L., Yang, T. & Wang, F. High-mobility group box-1 and its role in angiogenesis. J Leukoc Biol 95, 563–574, https://doi.org/10.1189/jlb.0713412 (2014).
Hayashi, S. et al. Protease-activated receptor 2 exacerbates adenine-induced renal tubulointerstitial injury in mice. Biochem Biophys Res Commun 483, 547–552, https://doi.org/10.1016/j.bbrc.2016.12.108 (2017).
Rasmussen, J. G. et al. Activation of protease-activated receptor 2 induces VEGF independently of HIF-1. PLoS One 7, e46087, https://doi.org/10.1371/journal.pone.0046087 (2012).
Ehling, J. et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 63, 1960–1971, https://doi.org/10.1136/gutjnl-2013-306294 (2014).
Keeley, E. C., Mehrad, B. & Strieter, R. M. Chemokines as mediators of neovascularization. Arterioscler Thromb Vasc Biol 28, 1928–1936, https://doi.org/10.1161/ATVBAHA.108.162925 (2008).
Salcedo, R. et al. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood 96, 34–40 (2000).
Arendt, L. M. et al. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res 73, 6080–6093, https://doi.org/10.1158/0008-5472.CAN-13-0926 (2013).
Hristov, M. et al. Importance of CXC chemokine receptor 2 in the homing of human peripheral blood endothelial progenitor cells to sites of arterial injury. Circ Res 100, 590–597, https://doi.org/10.1161/01.RES.0000259043.42571.68 (2007).
Keane, M. P., Belperio, J. A., Xue, Y. Y., Burdick, M. D. & Strieter, R. M. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol 172, 2853–2860 (2004).
Lin, Q. et al. High-mobility group box-1 mediates toll-like receptor 4-dependent angiogenesis. Arterioscler Thromb Vasc Biol 31, 1024–1032, https://doi.org/10.1161/ATVBAHA.111.224048 (2011).
He, C. et al. Angiogenesis mediated by toll-like receptor 4 in ischemic neural tissue. Arterioscler Thromb Vasc Biol 33, 330–338, https://doi.org/10.1161/ATVBAHA.112.300679 (2013).
Sachdev, U. et al. TLR2 and TLR4 mediate differential responses to limb ischemia through MyD88-dependent and independent pathways. PLoS One 7, e50654, https://doi.org/10.1371/journal.pone.0050654 (2012).
Xu, H. et al. Protein kinase C alpha promotes angiogenic activity of human endothelial cells via induction of vascular endothelial growth factor. Cardiovasc Res 78, 349–355, https://doi.org/10.1093/cvr/cvm085 (2008).
Imaizumi, T. et al. Expression of vascular endothelial growth factor in human umbilical vein endothelial cells stimulated with interleukin-1alpha–an autocrine regulation of angiogenesis and inflammatory reactions. Thromb Haemost 83, 949–955 (2000).
Xiong, M., Elson, G., Legarda, D. & Leibovich, S. J. Production of vascular endothelial growth factor by murine macrophages: regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am J Pathol 153, 587–598, https://doi.org/10.1016/S0002-9440(10)65601-5 (1998).
Wu, X. et al. Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci Rep 7, 9371, https://doi.org/10.1038/s41598-017-09907-6 (2017).
Sison, K. et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 21, 1691–1701, https://doi.org/10.1681/ASN.2010030295 (2010).
Bertuccio, C., Veron, D., Aggarwal, P. K., Holzman, L. & Tufro, A. Vascular endothelial growth factor receptor 2 direct interaction with nephrin links VEGF-A signals to actin in kidney podocytes. J Biol Chem 286, 39933–39944, https://doi.org/10.1074/jbc.M111.241620 (2011).
Kumar Vr, S. et al. Cathepsin S Cleavage of Protease-Activated Receptor-2 on Endothelial Cells Promotes Microvascular Diabetes Complications. J Am Soc Nephrol, https://doi.org/10.1681/ASN.2015020208 (2015).
Nakagawa, T., Kosugi, T., Haneda, M., Rivard, C. J. & Long, D. A. Abnormal angiogenesis in diabetic nephropathy. Diabetes 58, 1471–1478, https://doi.org/10.2337/db09-0119 (2009).
Nakagawa, T. et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol 18, 539–550, https://doi.org/10.1681/ASN.2006050459 (2007).
Sung, S. H. et al. Blockade of vascular endothelial growth factor signaling ameliorates diabetic albuminuria in mice. J Am Soc Nephrol 17, 3093–3104, https://doi.org/10.1681/ASN.2006010064 (2006).
Sumi, A. et al. Roles of coagulation pathway and factor Xa in the progression of diabetic nephropathy in db/db mice. Biol Pharm Bull 34, 824–830 (2011).
Eremina, V. & Quaggin, S. E. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens 13, 9–15 (2004).
Izzedine, H. et al. Expression patterns of RelA and c-mip are associated with different glomerular diseases following anti-VEGF therapy. Kidney Int 85, 457–470, https://doi.org/10.1038/ki.2013.344 (2014).
Ollero, M. & Sahali, D. Inhibition of the VEGF signalling pathway and glomerular disorders. Nephrol Dial Transplant 30, 1449–1455, https://doi.org/10.1093/ndt/gfu368 (2015).
Fuh, G. et al. Structure-function studies of two synthetic anti-vascular endothelial growth factor Fabs and comparison with the Avastin Fab. J Biol Chem 281, 6625–6631, https://doi.org/10.1074/jbc.M507783200 (2006).
Takahashi, N., Boysen, G., Li, F., Li, Y. & Swenberg, J. A. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int 71, 266–271, https://doi.org/10.1038/sj.ki.5002033 (2007).
Edgell, C. J., McDonald, C. C. & Graham, J. B. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA 80, 3734–3737 (1983).
Acknowledgements
We thank members of Tohoku University, Faculty of Pharmaceutical Sciences, for their assistance, Dr. Nobuyo Maeda (The University of North Carolina at Chapel Hill) for her helpful comment, and Genentech Inc. for providing anti-VEGF antibody. Our work was supported by Grants-In-Aid from the Japan Society for Promotion of Science (JSPS 16J03192, 18K15993) and Kanzawa Medical Research Foundation, Japan.
Author information
Authors and Affiliations
Contributions
Y.O., T.F., E.S., A.S. and K.K. performed experiments. Y.O. and N.T. analyzed data and co-wrote manuscript. H.S., J.S. and S.I. interpreted data and edited manuscript. N.T. contributed to conception of research.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oe, Y., Fushima, T., Sato, E. et al. Protease-activated receptor 2 protects against VEGF inhibitor-induced glomerular endothelial and podocyte injury. Sci Rep 9, 2986 (2019). https://doi.org/10.1038/s41598-019-39914-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39914-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
