
Abstract
Liver abscesses in feedlot cattle form secondary to high concentrate feeds and rumen acidosis. Antimicrobial drugs are commonly included in cattle feed for prevention of liver abscesses, but concerns regarding antimicrobial resistance have increased the need for alternative treatments. A block randomized clinical trial was conducted to evaluate the effects of a Saccharomyces cerevisiae fermentation product (SCFP) on liver abscesses, fecal microbiomes, and resistomes in cattle raised without antibiotics in a Colorado feedlot. At enrollment, steers (n = 4,689) were sorted, by weight and source, into 2 pens comprising a block (n = 14 blocks, 28 pens); pens were randomly allocated to either the control group or the treatment group, where the diet was supplemented with SCFP. Prior to harvest, composited feces were collected for characterization of the microbiome and resistome using 16S rRNA gene and shotgun sequencing. At harvest, liver abscess severity was quantified for individual cattle. There were no statistical differences detected by treatment group in animal health, liver abscess prevalence or severity. Organisms classified to phylum, Elusimicrobia were more abundant in the feces of treated cattle, however, there were no differences in the resistome by treatment group. Both microbiome and resistome varied significantly among enrollment blocks.
Similar content being viewed by others


Introduction
The occurrence of liver abscesses is a common problem in cattle that is associated with feeding diets containing highly fermentable carbohydrates (i.e., starch), among other factors, during the finishing period. These high grain diets are the primary underlying cause of clinical and subclinical ruminal acidosis, rumenitis, bacterial translocation into the portal circulation, and subsequent liver abscess formation1,2. Consequently, there are detrimental impacts for animal production and welfare, and the diminished value of animals at slaughter result in significant economic loss to the beef cattle industry3,4,5,6. Results of the recent National Beef Quality Audit-2016 demonstrated that the occurrence of liver abscesses in feedlot cattle has increased7. Traditionally, management practices used to control and prevent liver abscessation include nutritional management and inclusion of antibiotics in the feed or water. In the U.S., tylosin phosphate (a macrolide antibiotic) and chlortetracycline are approved for inclusion in cattle diets for the control of liver abscesses, and over 70% of cattle in large commercial beef feedlots have been reported to receive tylosin in diets for this purpose8.
Antimicrobial drugs effectively control liver abscesses, but the impact of antimicrobial exposures on antimicrobial resistance (AMR) is currently of great concern to public health. Antimicrobial resistance in cattle has been traditionally evaluated using culture-based approaches9,10, but the impacts of antimicrobial drug treatments to the microbial communities (the microbiome) or on the entire portfolio of AMR genes (the resistome) have only recently started to be explored11. Due to growing scrutiny regarding the use of medically important antimicrobials in livestock production, the use of tylosin as a feed additive for growth promotion in livestock was banned in the European Union; in the United States, the use of tylosin for this purpose has recently been placed under veterinary oversight with the Veterinary Feed Directive. Future regulatory requirements may restrict the duration for use of tylosin for this purpose in livestock, which has implications for animal health, productivity, and welfare. Thus, reducing the use of antimicrobial drugs and development of alternative treatment strategies are critical areas for feedlot cattle production and an active area of research. These products will be particularly important in treating feedlot cattle that have higher susceptibility to the formation liver abscesses, such as beef breeds raised without antibiotics (‘natural cattle’) and steers originating from dairy farms that are raised for beef production1.
Several non-antimicrobial products and feed additives have been evaluated for their effectiveness in controlling subacute rumen acidosis and liver abscesses, including sunflower seeds12,13, essential oils14,15, and vaccines16,17. Saccharomyces cerevisiae fermentation products (SCFP) have also been evaluated for use as feed additives in dairy and beef production. Some studies have shown that SCFP can positively impact gastrointestinal health for cattle exposed to high-concentrate diets, by providing components including amino acids, peptides, organic acids, and oligosaccharides18,19,20. In some studies, inclusion of SCFP in cattle diets improved performance characteristics, including increased milk production21, increased volatile fatty acid concentration, decreased rumen pH22, efficient feed conversion, and improved carcass traits23. Other studies regarding SCFP supplementation reported reductions in AMR and food safety pathogens detected in the feces, as well as lymph node carriage of virulent Salmonella enterica24. A previous clinical trial found no significant difference in the prevalence of liver abscesses in feedlot cattle fed diets containing SCFP in comparison to cattle fed diets containing tylosin, suggesting there may be similar effectiveness for the control of liver abscesses in cattle25.
We hypothesized that dietary supplementation with SCFP could impact the occurrence of liver abscesses, cattle health and performance, microbial communities, and resistomes through modulation of subacute rumen acidosis in feedlot cattle exposed to a high concentrate diet. A block randomized clinical trial was performed to evaluate these outcomes in the feces of beef cattle raised without exposure to antimicrobial drugs or growth-promoting hormones in a commercial feedlot. The outcomes evaluated in this clinical trial in response to SCFP supplementation included the following: the prevalence and severity of liver abscesses, animal health outcomes and feedlot performance, the fecal microbiome (using 16S rRNA sequencing), and the fecal resistome, including genes related to antimicrobial, metal, and biocide resistance (using shotgun metagenomic sequencing).
Results
Animal health, production performance, and liver abscess prevalence
In total, 4,689 steers were randomly enrolled in the study; 2,345 steers housed in 14 pens were administered the control diet, and 2,344 cattle housed in 14 pens were administered a diet supplemented with SCFP. The number of cattle housed in the 28 pens used for this study ranged from 104 to 225 animals per pen (average = 167 cattle). Overall, 278 steers required antimicrobial treatment for disease in compliance with veterinary protocols, and 41 cattle died during the study, including both steers removed from the pen for treatment in addition to cattle found dead within the pen; these animals were removed from further analysis in the trial. Controlling for hierarchical population structure, there were no detectable differences in production (average daily gain (ADG) and dry matter intake (DMI) to gain ratio) or animal health variables (crude or cause-specific morbidity and mortality) between treatment groups receiving the diet containing SCFP or the control ration (Tables 1 and 2; P > 0.05). Across all pens, the duration of days on feed (DOF) ranged from 171 to 262 d, with an average of 205 d; there were no significant differences between SCFP treatment and control groups (P = 0.81).
In total, the livers from 4,324 individual cattle were evaluated at slaughter to assess the presence and severity of liver abscesses using the standardized scoring system. Controlling for randomization block, the adjusted risk for cattle having a detectable liver abscess, of any severity score, was 38.9% (95% CI = 38.0 to 44.5) and 38.1% (95% CI = 31.4 to 46.2) for cattle fed the control ration or cattle fed the SCFP ration, respectively (Table 3). The difference in abscess risk was not statistically significant between treatment groups (P = 0.79). Similarly, there were no statistically detectable differences between treatment groups in the risk for abscesses of different severity, i.e., severe (A-plus), moderate (A) or minor (A-minus); (P > 0.05).
Fecal microbiome composition and diversity
High quality 16S rRNA amplicon sequences totaling 13,749,784 reads were obtained from the 28 composited fecal samples and imported into Qiime2-2017.12 for fecal microbiome characterization26; the median Phred quality score for forward and reverse reads was 38 to 39, respectively (Supplementary Fig. S1). Using the DADA2 pipeline27 and filtering out chloroplasts of plant origin and mitochondrial sequences, 85% of reads passed quality control. Read count summaries, by sample and processing step, are shown in Supplementary Table S1.
The fecal microbiome of composite samples collected from both treatment groups across 28 samples was dominated by the phyla Firmicutes, Bacteroidetes, Proteobacteria, Spirochaetes, and Tenericutes (Fig. 1), and at the family level Ruminococcaceae, Lachnospiraceae, Bacteroidaceae, and Paraprevotellaceae were the predominant taxonomic features in both treatment groups. Additionally, there were 46 rare phyla which accounted for <1% of all reads across 28 samples (Supplementary Table S2). Taxa within the rare phyla category included Actinobacteria, Acidobacter, Fibrobacteres, Verrucomicrobia, Cyanobacteria, Nitrospirae, Chloroflexi, Gemmatimonadetes, Planctomycetes, Euryarchaeota (kingdom Archaea), WS3, Elusimicrobia, Fusobacteria, TM7, WPS-2, Armatimonadetes, Chlamydiae, Chlorobi, AD3, OP3, TM6, Lentisphaerae, Synergistes, FCPU426, GN04, WWE1, Deferribacteres, BRC1, NC10, FBP, Caldithrix, Thermi, OD1, Crenarchaeota (kingdom Archaea), GOUTA4, NKB19, GN02, SC4, OP8, BHI80-139, SBR1093, LCP-89 and OC31.

Percent relative abundance (count within taxonomic group/total count × 100) of composited fecal microbiome at phylum, class, order, and family-levels across 28 samples collected from cattle supplemented with SCFP or the control diet. The top five abundant taxonomic classifications for sequence variants are labelled, and rare taxa (defined as having a percent relative abundance <2%) were summed and put into a distinct category, “rare taxa”, at each level.
Adjusted estimates of abundance revealed that 1 SV classification to the phylum, Elusimicrobia, was present in significantly higher abundance within the composited fecal microbiome from SCFP-fed pens of cattle compared to cattle fed the control ration. However, while Elusimicrobia was detected in 100% of the composited fecal samples, this pattern of higher abundance in SCFP treated cattle was not observed in 4 of 14 blocks. Though the differences were not considered biologically significant due to low relative abundance, SVs that classified to the phylum, Fusobacteria, were present in higher relative abundance within the fecal microbiome of cattle fed the control ration; however, this effect was only present within 3 of 14 blocks. No other SVs were differentially abundant between treatment groups, at any taxonomic level (Supplementary Table S3).
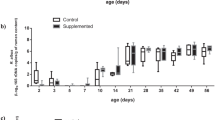
Microbial diversity, estimated using the Shannon diversity index on rarefied counts, did not differ between the SCFP supplemented and control pens of cattle (Fig. 2A; P = 0.43). Though not significant, Shannon alpha diversity varied among blocks, with lower diversity in block 14 (Fig. 2B; P = 0.19). Degree of differentiation in the microbial communities, or beta diversity, was not significantly different by treatment group (Fig. 2C; P = 0.99); however, block-related effects accounted for the significant differences in the composited fecal microbiome (Fig. 2D; P = 0.001). The first axis explained 38.8% of the variation, which was largely driven by differences in the microbiome of the composite fecal sample collected from pens in block 14.

Diversity metrics for the composited fecal microbiome, by treatment group and enrollment group (block). Shannon alpha diversity, or within-sample microbial diversity, did not differ by treatment group (A) or block (B). Statistical differences for alpha diversity were determined using non-parametric Kruskall-Wallis tests. Beta diversity, or microbial community composition, visualized as principal coordinates analysis ordination based on 16S rRNA gene sequence weighted UniFrac distance, differed by treatment group (C) but did not differ by block (D).
Resistome composition and diversity
Shotgun sequencing generated 994,459,164 pairs of forward and reverse reads from 28 composite fecal samples. The average quality score across samples was 38 (range 36 to 38), indicating an expected error rate of 1.6 out of 10,000 base calls from sequencing (Supplementary Table S4). Following quality filtering and removal of reads that aligned to the Bos taurus genome, 895,733,367 read pairs remained for downstream analysis. Of these, 1,610,843 read pairs mapped to the gene accessions in the MEGARes and BacMet databases for elements encoding for antimicrobial drug and biocide resistance, respectively. There were no significant differences in the number of raw or trimmed reads or quality scores between the treatment and control groups, suggesting that there was no systematic bias in the sequencing effort between treatment groups.
Overall, 364,482 read alignments aligned to the MEGARes database (i.e., “hits”), and were classified to 35 mechanisms for antibiotic drug, metal, and biocide resistance. Single nucleotide polymorphisms were confirmed as described in the methods for housekeeping genes that confer resistance. At the mechanism level, 0.27% of hits mapped to determinants for metal or biocide resistance and 99.73% of reads mapped to determinants for resistance to antimicrobial drugs (Supplementary Table S5). At the mechanism level, most read alignments were to AMR determinants for tetracycline resistance ribosomal protection proteins (62.3%) and macrolide resistance efflux pumps (25.6%).
Reads aligning to determinants for ribosomal protection protein mechanisms were the most abundant hits within the tetracycline resistance class, including tetQ, tetO, tetW, tet32, tet44, tet40, and tetW. Other hits within the tetracycline drug class included inactivation enzymes (tetX) and the major facilitator superfamily (MFS) efflux pumps (tetA, tetB). Reads aligning to determinants for macrolide efflux pumps (MefA) comprised the majority of hits to the macrolide, lincosamide, and streptogramin resistance class, followed by 23S rRNA methyltransferase target modification genes (ermq), macrolide phosphotransferase genes (mphb), and the lincosamide nucleotidyltransferases (lunc). For hits to determinants in the aminoglycoside class, aminoglycoside O-nucleotidyltransferase mechanisms (ant9) were most abundant. Within the beta-lactam class, alignments to the class A beta-lactamase (cfx) AMR mechanisms were abundant.
Among reads aligning to AMR determinants for biocides, biocide resistance proteins (glpF) and biocide resistance regulators (rpoS, gadA and sugE) were the most abundant. Of alignments that mapped to AMR determinants for metal resistant mechanisms, most of them conferred resistance to multiple metals.
The composition and taxonomic richness of antimicrobial, metal and biocide resistance determinants did not differ significantly between treatment groups at expression levels high enough (the cut-off value was > 1) to infer biological significance at any taxonomic level (Fig. 3A,B). Likewise, NMDS ordination did not show significant separation between the 2 treatment groups, at any taxonomic level. However, the resistome composition and richness differed when evaluating the resistome changes among blocks (Fig. 4A,B), and this shift was observed at the Mechanism and Class levels (average expression levels >1, P < 0.001).

Heatmap showing treatment comparison of the normalized counts of antimicrobial, metal and biocide resistance determinants in the composited fecal resistome by drug class. Each column represents one composited fecal sample, grouped by treatment (A). Boxplot species richness comparison for composited fecal resistome at the class and mechanism levels, measured as unique species using inverse Simpson’s index, by treatment group (B).

Heatmap showing block comparison of normalized counts of antimicrobial, metal and biocide resistance genes, by drug class. Each column represents one composited fecal sample, by enrollment group (block) (A). Boxplot species richness comparison for fecal resistome at the class and mechanism level measured as unique species measured by inverse Simpson’s index, by block (B).
For critically important AMR resistance gene accessions that were specifically screened, only 1 sample collected from a pen of cattle supplemented with SCFP was found to contain alignments to an extended spectrum beta-lactamase (ctx) determinant. However, there were only 23 reads aligning to this determinant (0.038% of the total alignments for AMR determinants in that sample).
Discussion
Results of this block randomized clinical trial showed that supplementation with SCFP did not significantly affect liver abscess prevalence, the fecal microbiome, or the resistome in commercial feedlot cattle raised without antimicrobial drug exposures. There were also no detectable treatment differences in animal health or performance outcomes. Overall, this study population expressed a high prevalence of liver abscesses (38%), which is approximately twice the national average for commercial beef cattle within the United States7. The relatively high liver abscess prevalence in this trial was likely attributable to the absence of antimicrobial inclusion in the trial diets, whereas previous reports regarding liver abscess prevalence were estimated for cattle populations in which tylosin was administered in the feed. This high liver abscess prevalence may have impacted the ability to detect effectiveness of SCFP as a control measure. For example, in a a previous study that demonstrated reduced (although insignificant) numbers of liver abscesses in cattle fed SCFP compared to a diet including tylosin and ionophores, there were overall lower prevalence for both total and severe liver abscesses (10–20%)25.
The results of this study identified significant variation across randomization block with respect to the liver abscess prevalence, fecal microbiome and fecal resistome. It is possible that this significant variation by block may have obscured subtle treatment effects of SCFP supplementation. As cattle were enrolled into the trial, they were randomly sorted into adjacent pens until a randomization block was filled, then randomly assigned to receive either the treatment or control diet. This design ensured that treatment groups of cattle within each block had equivalent distribution of a variety of factors, including cattle origin (i.e., rearing source), weight, pen location, and season of placement. The blocks were intended to control for potential confounding factors, such that they were evenly distributed between treatment groups to reduce the likelihood that differences related to these variable factors would bias the study outcome. However, the significant differences among blocks suggested that these factors could be strong drivers of microbial ecology and resistomes in cattle feces, and potentially liver abscess prevalence. These factors, including cattle source, rearing environments, and influences from within the feedlot may have a greater impact on the microbiome and liver abscess occurrence than has been previously understood, and warrant consideration in the design of future dietary feeding trials and studies designed to understand risk factors for liver abscess occurrence.
Our evaluation of composited feces obtained from the cattle prior to slaughter suggested that overall, there were only minor differences between treatment groups. Only 1 SV classifying to the phylum, Elusimicrobia, had a higher relative abundance in the feces of SCFP supplemented groups compared to the control group. There is limited information in the literature regarding Elusimicrobia in feedlot cattle, however, it has been associated in cattle diagnosed with periodontal disease28. There were no other significant treatment effects detected regarding the resistome or other outcomes measured in this clinical trial, which may be due to several reasons, including the randomization block variation, SCFP dose, sample type, or sample numbers.
While the SCFP was supplemented at the manufacturer’s recommended dosage, the daily exposure dose of SCFP that was used in this clinical trial may have been too low to result in detectable differences in the fecal microbiome or resistome. One study reported linear increases in cellulolytic and lactate-utilizing bacteria associated with increasing concentrations of SCFP in the diet29. However, that study evaluated rumen, not fecal samples, and the dose of SCFP was up to 10 times higher than the levels of SCFP fed in this trial.
Feed additives containing SCFP have been hypothesized to modulate rumen fermentation, yet this trial only characterized fecal composited samples. This limitation was primarily due to several practical limitations associated with the handling of a large populations of owned commercial feedlot cattle. Therefore, the conclusions from this trial regarding impact of SCFP supplementation are restricted to impacts on the fecal microbiome and resistome. The gastrointestinal microbiome of cattle has been previously characterized as heterogeneous with respect to microbiome diversity and composition across distinct anatomical compartments30,31. However, some studies have demonstrated that cattle exposed to grain-induced, subacute rumen acidosis challenges have shifts in both the fecal microbial composition as well as in the rumen30. Interestingly, metagenomic characterizations of the microbiome and resistome of feedlot cattle following exposures to feed additives (tylosin and monensin) have been shown to have minimal to no impact on the in rumen, cecum, and small intestine contents11. Furthermore, epimural rumen microbial communities (bacteria adherent to the rumen epithelial lining) may have greater impact on liver abscess pathogenesis as compared to microbial populations found in liquid or solid fractions of rumen contents2,32,33, and investigation of these microbiome niche communities are warranted. Future work directed towards evaluation of alternative feed additives, including SCFP, would benefit from additional evaluations of other gut compartments, including the rumen and rumen epimural microbial communities, in addition to investigations using fecal samples.
The predominant AMR classes detected within the feces of cattle were tetracycline resistance ribosomal protection proteins, macrolide resistant efflux pumps, beta-lactamases, and aminoglycoside resistance. This resistome profile is consistent with other published metagenomics studies of feedlot and dairy cattle34,35,36,37. The cattle enrolled in this study were not treated with any antimicrobial drugs within their lifetime, and the pens used to house these cattle were only used to manage cattle raised without antimicrobial drug exposures, so cross-contamination with treated populations were unlikely. As such, it is interesting that several AMR mechanisms were detected even though this population of cattle did not have previous antimicrobial exposures. This is highlighted by the identification of small numbers of reads aligning to extended spectrum, beta-lactamase gene accessions (ctx), belonging to the Ambler class A beta-lactamases group, and further highlights the complexity of ecologies related to AMR in feedlot operations and other agricultural production settings. The ctx gene confers resistance to third-generation cephalosporins (e.g., cefotaxime), and is classified as critically important threat to human health when expressed in disease-causing agents, such as Escherichia coli38,39. This AMR gene was previously identified in another study of feedlot cattle that characterized the fecal resistome in populations raised without antibiotics, although the abundance was higher in the conventionally-treated cattle comparison group34.
Despite several advantages, metagenomics approaches are not without limitation. For example, there is a lack of established methods for performing accurate power calculations in metagenomics studies designed for applied research in commercial feedlot settings. This is partly due to lack of baseline information about cattle microbiomes and resistomes exposed to diverse management conditions, including different antimicrobial treatment exposures. As a result, it may be possible that the effects of SCFP treatment may have been obscured by the significant variation in microbiomes across block, driven by one of several potentially confounded factors. Future studies could be designed to evaluate cattle exposed to feed additives through use of more replicates at the pen-level, through use of individuals as the experimental unit, or through collection of other sample types. Additionally, future work should be directed towards investigation of underlying factors that influence the microbiome, independent of dietary treatment groups, which could influence impact the underlying risk factors for formation and severity of liver abscesses in feedlot cattle.
Materials and Methods
Trial design and study population
Methods involving animal care and use were approved by and carried out in accordance with the CSU Research Integrity and Compliance Review Office (Protocol number 102616). In a block randomized clinical trial study design, cattle were allocated to a pen that received either a treatment or control diet; each treatment and control pair made up one randomization block (Fig. 5). The study population was comprised of cattle that were approximately 50% purebred Angus, and crossbreds between Angus and English or continental breeds (i.e., no known Brahman or dairy influence). Yearling steers were purchased directly from domestic producers located across multiple western states during March through June 2016. Eligible cattle were reared through their lives without exposure to antimicrobial drugs or growth promoters to enable marketing of natural beef products. At arrival, cattle were individually weighed and administered injectable and oral anti-parasitic treatments (Noromectin; Synanthic; Standguard), injectable bacterin-toxoid vaccines protecting against respiratory disease complex agents (Titanium-5 + PH-M), and clostridial diseases (Vision-7). The pens used within the feedlot to house these cattle were only used to manage cattle raised without antimicrobial drug exposures.

Overview of the block randomized study design. Crossbred steers were purchased in groups and transported to a commercial feedlot in Northern Colorado. Upon arrival, animals from each vendor were processed separately. Cattle were individually weighed using a chute, and heavy or light weight animals (relative to the weight distribution for the source population) were excluded. Remaining cattle were randomly allocated to the 2 treatment groups, which were housed in separated, adjacent pens. Randomized enrollment was blocked by pen, so that cattle were allocated to a pair of pens within a block. Pens within a block had equivalent distributions for cattle weights, breeds, sources, stocking density, and placement and harvest dates. The pens had equivalent dimension, and animals were housed in open-air, dirt-floor pens with a central feed alley. Pens of cattle within each randomization block were managed using identical protocols for cattle movements, timing of feed delivery, feed bunk management, and daily health inspection. Cattle housed together in a pen were shipped for harvest based on standard procedures to determine when they have reached market weight (1400–1500 lbs). Treatment and control pens within each block were shipped for slaughter on the same day, in the same order that blocks were randomly enrolled into the trial.
Except for SCFP supplementation included in the diet of treated cattle, the diets fed to both treatment groups were identical, and were formulated to meet the National Research Council nutritional requirements40. The SCFP product (NaturSafe, Diamond V) was supplemented to treated cattle diets at the manufacturer’s recommended dose of 17.8 g/d (Supplementary Table S6). Cattle started receiving the treatment diets when they were transitioned to the finishing ration starting week 2 to 3 following placements. Cattle were fed twice per day. Separate equipment was used for preparation and delivery of treatment diets, and the mill and trucks were sanitized between diets to minimize cross-contamination.
Trained personnel, masked to treatment assignment, evaluated cattle for signs of illness at arrival and daily under the supervision of a veterinarian. When identified, ill or injured animals were moved for examination, and primary illness was classified by body system (i.e., respiratory, digestive, or other diseases). Cattle requiring treatment with antibiotics, as per trial protocols, were moved and permanently housed elsewhere. Cattle that died underwent a necropsy by a veterinarian for cause of death classification. The cattle were harvested between October to December 2016.
Sample and Data collection
One day prior to harvest, a composited sample of fresh feces was collected from the pen floor for each pen within a block (n = 28 fecal samples), using previously described methods37,41. Briefly, composites were created by combining several samples of freshly voided feces (~10 g each) taken using a gloved hand from the top and center of individual fecal pats from 20 locations along crossing diagonals of each pen of cattle. The fecal composites were transported on ice within insulated containers within 1 h of collection to the laboratory at Colorado State University for further processing (described below). Animal health and weight data were collected, and individual animal weights were summarized through calculation of arithmetic mean, by pen. The ADG was calculated using the difference of pen-averaged slaughter weight and starting weight divided by the DOF. The DMI to gain ratio was calculated by dividing daily DMI of feed by ADG. Crude morbidity attack rate was calculated as the number of sick animals, whether they received treatment or not, divided by the initial number of animals assigned to each pen. Crude mortality attack rate was calculated as the number of animals that died, whether they received treatment or not, divided by the initial total number of animals assigned to each pen. Cause-specific morbidity attack rates were calculated as the number animals treated and divided by the initial number of animals in the pen. Cause-specific mortality attack rates for disease categories were calculated as the number of animals that died, whether they died in the pen or after removal for antimicrobial treatment, divided by the total number of animals assigned to each the pen.
The cattle were euthanized using approved procedures at a commercial abattoir. Identities of feedlot pens (not treatment group assignments as all personnel remained masked to treatment group throughout the study) were tracked in the slaughter plant to allow for determination of liver abscess occurrence by pen and treatment group. After evisceration, the presence and severity of liver abscesses for individual animals were classified by a trained observer using a modified Elanco Liver Check System, as previously described6. Briefly, the external surfaces of livers were observed and palpated. Livers without visible abscesses were scored as normal, and livers were classified as A-minus, A or A-plus to denote increasing severity of abscessation. Other pathological liver abnormalities were not scored.
Sample processing and sequencing
Within the collection bags, fecal composites were manually homogenized for 1 minute each, and aliquots were weighed and stored at −80 °C. DNA was isolated from samples using commercial kits (PowerMax Soil DNA Isolation Kit) with modifications as previously described42. Briefly, 10 g of sample aliquots were thawed at 4 °C, and a sedimentation step was performed to remove large particulates and sediment layers. The resulting pellet was resuspended in PowerBead solution, and the remainder of the standard DNA extraction kit protocol followed. DNA concentration and quality were evaluated using a NanoDropTM spectrophotometer (Thermo Fisher Scientific, Inc.). Samples with 260:280 nm ratios >1.8 and DNA concentrations >20 ng/μl were sent for sequencing; samples that did not meet the concentration threshold were concentrated by ethanol precipitation before sequencing.
The DNA quantification results for negative control samples were evaluated following extraction with only kit reagents (blank extractions). Sample concentrations and DNA quantification results following PCR amplification were assessed using agarose gel electrophoresis of the genomic DNA and PCR products following amplification. The PCR products from blank samples extracted during this study’s sample extractions were obtained only after several rounds of amplification and were of insufficient quantity for sequencing.
The 16S rRNA gene amplification and sequencing for composited fecal sample DNA was performed by a commercial sequencing company (Novogene Corporation). Following isolation, DNA aliquots (1500 ng, 30 µl) were delivered to the sequencing facility. DNA from the V4 region of the 16S rRNA gene was amplified using the primer set 515F/806R43, with reverse primers containing unique barcodes. Library sequencing (paired-end, 2 × 250 bp) was performed on a HiSeq. 2500 Sequencing System (Illumina).
Fecal samples were also sequenced using shotgun sequencing. DNA aliquots (3 µg, 50 µl) were delivered to the University of Colorado Genomics and Microarray Core for sequencing. Genomic libraries for all samples were prepared using the TruSeq DNA PCR-Free Library Prep Kit (Illumina) following the manufacturer’s protocol to obtain an average insert size of 350 bp. Library sequencing was completed on the HiSeq 4000 Sequencing System (Illumina) with 17 samples loaded into each lane, v4 chemistry and paired-end reads of 150 nucleotides in length.
Bioinformatics and statistical analysis
Treatment group assignments were masked during data analysis. The 16S rRNA gene forward and reverse reads were imported into Qiime2-2017.1226. The DADA2 pipeline27 was used for detecting and correcting Illumina amplicon sequences, removal of primers and chimeric reads, and assembly into sequence variants (SV). Quality filtering included primer trimming and sequence truncation to remove low quality sequences. Taxonomy was assigned using a naïve Bayes classifier44 trained on the Greengenes 13_8_99% database. Sequences classified as chloroplasts, mitochondria, and sequences with a count less than 10 were filtered. A rooted phylogenetic tree was created through de novo multiple sequence alignment using MAFFT, vs.745, highly variable positions were removed to decrease noise in the tree, and FastTree-2 was applied to generate a tree from the masked alignment46. The feature table was rarefied to 65,000 sequences, allowing retention of all samples. Beta diversity analysis using the distance matrix generated from the weighted UniFrac phylogenetic metric42 was visualized in a principle coordinates analysis (PCoA), using Emperor47. Alpha diversity was estimated using the Shannon diversity metric.
The SV count data were extracted from Qiime2, and imported with metadata into the “metagenomeSeq” R package (v1.20.1)48. Taxonomic lineage for each SV was identified and alignments summarized by phylum, class, order, and family. The count data were normalized using cumulative sum scaling (using default percentile of 0.5 for normalization) and analyzed using zero-inflated Gaussian mixed-model regression, with treatment as a fixed effect and block as a random effect. Log-fold changes and P-values for regression coefficients were calculated by calculating pair-wise contrasts between treatment levels using Limma’s “makeContrasts” function49. P-values were corrected for multiple testing using the Benjamini-Hochberg false discovery rate50. Testing for significant effects of categorical metadata, including treatment group, for alpha and beta diversity metrics were performed with rarefied feature tables (as described above).
The AmrPlusPlus pipeline v.1.20.051 was used to characterize AMR at the class, mechanism and group levels from the shotgun metagenomics data. The pipeline was executed using Nextflow (v.0.26.0)52. The sequence data were filtered and quality controlled using Trimmomatic (v0.36)53. Host (bovine) genomic DNA contamination was removed by aligning the quality controlled reads to the Bos taurus genome (NCBI accession AC_000158.1) using the Burrows Wheeler Aligner (BWA-MEM)54, then removing the B. taurus genome using SAMtools55. Remaining reads were aligned to the MEGARes AMR resistance database (v1.01) and hand-annotated genes from the BacMet database using BWA-MEM56. An 80% gene fraction threshold (i.e., 80% of the full length of each AMR gene accession within each sample) was applied to identify potential positive AMR gene accessions.
The count data for AMR determinants were normalized using cumulative sum scaling and analyzed using zero-inflated Gaussian mixed-model regression in the “metagenomeSeq” R package (v1.20.1)48, with treatment as a fixed effect and block as a random effect. Measures for alpha diversity were calculated on data normalized to the lowest sample size using the vegan package (v.2.4-6)57. Ordination was performed using non-metric multidimensional scaling (NMDS) with Bray-Curtis distance. Log-fold changes, expression levels, and P-values for regression coefficients were calculated with pair-wise contrasts between treatment levels. P-values were corrected for multiple testing using the Benjamini-Hochberg false discovery rate50. Differential abundance testing for AMR mechanism and class were only considered biologically significant at expression levels >1. As previously described37, the resistome from composited fecal samples in this study was specifically screened for selected high profile resistance genes and drugs of concern to public health.
All animal health and performance were analyzed using SAS, (SAS Institute, release 9.4) using pen as the experimental unit. Animal health variables were compared between treatment groups using mixed-effects Poisson regression, with block specified as a random effect. Performance variables were compared between treatment groups in a generalized linear model adjusting for pen and block structures. Least squares means were significantly different if P was ≤0.05.
Data Availability
Quality-trimmed, concatenated sequencing reads for the 28 shotgun metagenomic sequencing reads described have been uploaded to the NCBI collection of biological data (BioProject) in Accession PRJNA453374 ID 453374. Raw, paired end reads for the 28 16S rRNA gene V4 region sequence sets have been uploaded to the NCBI collection of biological data (BioProject) in Accession PRJNA472376 ID 4058025.
References
Amachawadi, R. G. & Nagaraja, T. G. Liver abscesses in cattle: A review of incidence in Holsteins and of bacteriology and vaccine approaches to control in feedlot cattle. Journal of Animal Science 94, 1620–1632 (2016).
Nagaraja, T. G. & Chengappa, M. M. Liver abscesses in feedlot cattle: a review. Journal of animal science 76, 287–298 (1998).
Brown, T. R. & Lawrence, T. E. Association of liver abnormalities with carcass grading performance and value. Journal of Animal Science 88, 4037–4043 (2010).
Nagaraja, T. G. & Lechtenberg, K. F. Liver abscesses in feedlot cattle. Veterinary Clinics of North America: Food Animal Practice 23, 351–369 (2007).
Reinhardt, C. D. & Hubbert, M. E. Control of liver abscesses in feedlot cattle: A review. Contribution no. 10-205-J from the Kansas Agric. Exp. Stn., Manhattan 66506. The Professional Animal Scientist 31, 101–108 (2015).
Rezac, D. J. et al. A survey of gross pathologic conditions in cull cows at slaughter in the Great Lakes region of the United States. Journal of Dairy Science 97, 4227–4235 (2014).
Eastwood, L. C. et al. National Beef Quality Audit-2016: Transportation, mobility, and harvest-floor assessments of targeted characteristics that affect quality and value of cattle, carcasses, and by-products. Translational Animal Science 1, 229–238 (2017).
USDA–APHIS–VS–CEAH–NAHMS. Feedlot 2011 ‘Part IV: Health and Health Management on US Feedlots with a Capacity of 1,000 or More Head’.
Beukers, A. G. et al. Effect of in-feed administration and withdrawal of tylosin phosphate on antibiotic resistance in enterococci isolated from feedlot steers. Front. Microbiol. 6 (2015).
Amachawadi, R. G. et al. Effects of in-feed copper and tylosin supplementations on copper and antimicrobial resistance in faecal enterococci of feedlot cattle. J Appl Microbiol 118, 1287–1297 (2015).
Thomas, M. et al. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci Rep 7 (2017).
Mir, P. S., Dugan, M. E. R., He, M. L., Entz, T. & Yip, B. Effects of dietary sunflower seeds and tylosin phosphate on production variables, carcass characteristics, fatty acid composition, and liver abscess incidence in crossbred steers. Journal of Animal Science 86, 3125–3136 (2008).
Gibb, D. J. et al. Value of sunflower seed in finishing diets of feedlot cattle. J. Anim. Sci. 82, 2679–2692 (2004).
Elwakeel, E. A. et al. In vitro degradation of lysine by ruminal fluid-based fermentations and by Fusobacterium necrophorum. J. Dairy Sci. 96, 495–505 (2013).
Meyer, N. F. et al. Effect of essential oils, tylosin, and monensin on finishing steer performance, carcass characteristics, liver abscesses, ruminal fermentation, and digestibility. J. Anim. Sci. 87, 2346–2354 (2009).
Checkley, S. L., Janzen, E. D., Campbell, J. R. & McKinnon, J. J. Efficacy of vaccination against Fusobacterium necrophorum infection for control of liver abscesses and footrot in feedlot cattle in western Canada. Can Vet J 46, 1002–1007 (2005).
Fox, J. T., Thomson, D. U., Lindberg, N. N. & Barling, K. A comparison of two vaccines to reduce liver abscesses in natural-fed beef cattle. Bovine Practitioner 43, 168–174 (2009).
Callaway, E. S. & Martin, S. A. Effects of a Saccharomyces cerevisiae culture on ruminal bacteria that utilize lactate and digest cellulose. Journal of Dairy Science 80, 2035–2044 (1997).
Mullins, C. R. et al. Analysis of rumen microbial populations in lactating dairy cattle fed diets varying in carbohydrate profiles and Saccharomyces cerevisiae fermentation product. Journal of Dairy Science 96, 5872–5881 (2013).
Yang, C., Guan, L., Liu, J. & Wang, J. Rumen fermentation and acetogen population changes in response to an exogenous acetogen TWA4 strain and Saccharomyces cerevisiae fermentation product. J Zhejiang Univ Sci B 16, 709–719 (2015).
Poppy, G. D. et al. A meta-analysis of the effects of feeding yeast culture produced by anaerobic fermentation of Saccharomyces cerevisiae on milk production of lactating dairy cows. Journal of Dairy Science 95, 6027–6041 (2012).
Yoon, I. K. & Stern, M. D. Effects of Saccharomyces cerevisiae and Aspergillus oryzae cultures on ruminal fermentation in dairy cows. Journal of Dairy Science 79, 411–417 (1996).
Wagner, J. J., Engle, T. E., Belknap, C. R. & Dorton, K. L. Meta-analysis examining the effects of Saccharomyces cerevisiae fermentation products on feedlot performance and carcass traits. Prof Ani Sci 32, 172–182 (2016).
Feye, K. M. et al. Abrogation of Salmonella and E. coli O157:H7 in Feedlot cattle Fed a proprietary Saccharomyces cerevisiae Fermentation Prototype. Journal of Veterinary Science & Technology 7 (2016).
Scott, M. F. et al. Effects of feeding a Saccharomyces cerevisiae fermentation prototype on performance, carcass characteristics, and liver abscess prevalence of beef heifers at a commercial feedlot. The Professional Animal Scientist 33, 320–326 (2017).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336 (2010).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Meth 13, 581–583 (2016).
Borsanelli, A. C. et al. Microbiomes associated with bovine periodontitis and oral health. Veterinary Microbiology 218, 1–6 (2018).
Zhu, W. et al. Effects of Saccharomyces cerevisiae fermentation products on performance and rumen fermentation and microbiome in dairy cows fed a diet containing low quality forage. J Anim Sci Biotechnol 8 (2017).
Plaizier, J. C. et al. Changes in Microbiome in Rumen Digesta and Feces Due to a Grain-Based Subacute Ruminal Acidosis (SARA) Challenge. Microb Ecol 74, 485–495 (2017).
Mao, S., Zhang, M., Liu, J. & Zhu, W. Characterizing the bacterial microbiome across the gastrointestinal tracts of dairy cattle: membership and potential function. Scientific Reports 5, srep16116 (2015).
Narayanan, S. et al. Biochemical and ribotypic comparison of Actinomyces pyogenes and A. pyogenes-like organisms from liver abscesses, ruminal wall, and ruminal contents of cattle. Am. J. Vet. Res. 59, 271–276 (1998).
Rezac, D. J. et al. Prevalence, severity, and relationships of lung lesions, liver abnormalities, and rumen health scores measured at slaughter in beef cattle. Journal of Animal Science 92, 2595–2602 (2014).
Vikram, A. et al. Impact of “Raised without Antibiotics” beef cattle production practices on occurrences of antimicrobial resistance. Appl. Environ. Microbiol. 83, e01682–17 (2017).
Noyes, N. R. et al. Characterization of the resistome in manure, soil and wastewater from dairy and beef production systems. Sci Rep 6 (2016).
Weinroth, M. D. et al. Effects of ceftiofur and chlortetracycline on the resistome of feedlot cattle. Appl. Environ. Microbiol. AEM.00610–18, https://doi.org/10.1128/AEM.00610-18 (2018).
Noyes, N. R. et al. Resistome diversity in cattle and the environment decreases during beef production. eLife 5, e13195 (2016).
WHO. WHO | Critically important antimicrobials for human medicine. WHO Available at, http://www.who.int/foodsafety/publications/antimicrobials-fifth/en/. (Accessed: 15th February 2018) (2017).
Courpon-Claudinon, A. et al. Bacteraemia caused by third-generation cephalosporin-resistant Escherichia coli in France: prevalence, molecular epidemiology and clinical features. Clinical Microbiology and Infection 17, 557–565 (2011).
National Academies of Sciences, E. and Medicine. Nutrient Requirements of Beef Cattle: Eighth Revised Edition, https://doi.org/10.17226/19014 (The National Academies Press, 2016).
Yang, X. et al. Use of metagenomic shotgun sequencing technology to detect foodborne pathogens within the microbiome of the beef production chain. Appl Environ Microbiol 82, 2433–2443 (2016).
Lozupone, C., Hamady, M. & Knight, R. UniFrac – An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7, 371 (2006).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624 (2012).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLOS One 5, e9490 (2010).
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. GigaScience 2, 16 (2013).
Paulson, J. N., Stine, O. C., Bravo, H. C. & Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat Meth 10, 1200–1202 (2013).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B 57, 289–300 (1995).
Lakin, S. M. et al. MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res 45, D574–D580 (2017).
Tommaso, P. D. et al. Nextflow enables reproducible computational workflows. Nature Biotechnology, https://doi.org/10.1038/nbt.3820 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio] (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Pal, C., Bengtsson-Palme, J., Rensing, C., Kristiansson, E. & Larsson, D. G. J. BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res 42, D737–D743 (2014).
Dixon, P. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science 14, 927–930 (2003).
Acknowledgements
We thank Five Rivers Cattle Feeding, LLC, and Drs Tony Bryant and John Ruby for providing access to the commercial abattoirs and feedlots. Thank you to the undergraduate and graduate students of Colorado State University for assistance with sample collection and processing. Thank you to the Computer Assisted Teaching Support Lab of the College of Veterinary Medicine and Biomedical Sciences for assistance with figure quality and design. This work was funded by the National Cattleman’s Beef Association Beef Checkoff.
Author information
Authors and Affiliations
Contributions
K.L.H. was responsible for coordinating sample collection, data analysis, and writing the manuscript. C.W., K.H., M.W., J.P., J.M. and I.G. assisted with sample collection and sample processing. J.P. led DNA isolation and sequencing methods and the writing of these methods. Z.A., J.M., E.D. and S.L. assisted with data analysis and writing of these methods. P.M., J.M., T.B., D.W. participated in the study design and coordination, and T.B. was responsible for management of trial cattle at the feedlot and animal health data collection. All authors assisted with writing and review of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huebner, K.L., Martin, J.N., Weissend, C.J. et al. Effects of a Saccharomyces cerevisiae fermentation product on liver abscesses, fecal microbiome, and resistome in feedlot cattle raised without antibiotics. Sci Rep 9, 2559 (2019). https://doi.org/10.1038/s41598-019-39181-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39181-7
This article is cited by
-
Mock community as an in situ positive control for amplicon sequencing of microbiotas from the same ecosystem
Scientific Reports (2023)
-
Resistomes and microbiome of meat trimmings and colon content from culled cows raised in conventional and organic production systems
Animal Microbiome (2022)
-
Administration of probiotic lactic acid bacteria to modulate fecal microbiome in feedlot cattle
Scientific Reports (2022)
-
A review of the resistome within the digestive tract of livestock
Journal of Animal Science and Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
