
Abstract
Scylla paramamosain is one of the most economically important crabs in China. In this study, the first genome survey sequencing of this crab was performed, and the results revealed that the estimated genome size was 1.21 Gb with high heterozygosity (1.3%). Then, RAD technology was used to construct a high-resolution linkage map for this species. A total of 24,444 single nucleotide polymorphism (SNP) makers were grouped into 47 linkage groups. The total length of the linkage groups was 3087.53 cM with a markers interval of 0.92 cM. With the aid of transcriptome and genome scaffold data, 4,271 markers were linked to genes, including several important growth-related genes such as transforming growth factor-beta regulator I, immune related-gene C-type lectin and ecdysone pathway gene broad-complex-like protein. Further, 442 markers, representing 279 QTLs, associated with 24 traits were identified, and of these markers, 78 were linked to genes. Some interesting genes, such as dedicator of cytokinesis protein 3, tenascin-X and DNA helicase MCM8, were believed to have important relationship with specific traits and merit further exploration. The results of this study will accelerate the genetic improvement and genome sequencing analysis of the mud crab.
Similar content being viewed by others

Introduction
From phenotype breeding and molecular marker-assisted selection (MAS) to genome-wide selection (GS), artificial breeding strategies for economic animals have developed rapidly. Along with technological progress, artificial breeding has become increasingly effective, mainly through a more accurate enrichment of economically important quantitative traits and shorter breeding intervals. In 2001, Meuwissen et al.1 proposed the GS strategy and suggested that genome-wide dense markers should be used to predict genetic value. Further, the GS strategy has been demonstrated to be a popular tool for genetic improvement in livestock, and considerable achievements have been obtained, for example, dairy cattle2, chicken3 and pig4. However, in aquatic economic animals, especially crustacean species, mainstream breeding strategies in many species still consist of phenotype breeding and MAS. Other than unstable breeding and farming processes (compared with those of livestock), the lack of a high-quality genome and linkage map are two of the main problems that constrain the development of this industry.
With the revolution of sequencing technology, it is more effective to obtain a vast number of markers in a specific species, and thus, a high-resolution genetic linkage map becomes possible. Recently, several high-resolution genetic linkage maps were constructed in economic crustacean species, including Penaeus monodon5, E. sinensis6, Litopenaeus vannamei7, Marsupenaeus japonicus8, Portunus trituberculatus9. The average marker distances of these maps were all lower than 1 cM, which greatly assists de novo genome assembly and gene location, and further promotes the animal breeding process. Currently, the only crustacean genomes available are those of the water flea (Daphnia pulex)10, sand flea (Parhyale hawaiensis)11, marbled crayfish (Procambarus virginalis)12 and Chinese mitten crab (Eriocheir sinensis)13. Although most of these genomes were poorly assembled, they provided important insights into the genomic architecture and basic biology of crustacean species.
The mud crab Scylla paramamosain, belongs to the genus Scylla (Family: Portunidae), and is the most common mud crab in China14. As it is one of the most commercially important crustacean species, many research teams have been drawn to investigate the genetic breeding, aquaculture and basic biology of S. paramamosain. We have been devoted to studying the genetic breeding and aquaculture industry for many years, keeping ahead of the artificial breeding of this crab. Currently, there are no genome assembly datasets for this crab, and only several transcriptome libraries have been reported15,16,17,18,19. The only genetic linkage map for the mud crab was constructed by our team using 212 microsatellite and amplified fragment length polymorphism (AFLP) markers, and the mean spacing was 18.68 cM20. These data cannot meet the needs of the modern breeding strategies for this important species. Therefore, a high-resolution genetic linkage map is needed both the artificial breeding industry and genomics research.
The purpose of this study was to construct a high-resolution genetic linkage map for both QTL mapping, locating genes and assisting with further genome assembly and artificial breeding processes in S. paramamosain. Twenty-six traits of 99 F1 generation individuals were measured to obtain the trait-related loci. Genome survey data and transcriptome data were used in the gene location and growth gene identification analysis.
Materials and Methods
Ethics statement
All animal experiments in this study were conducted in accordance with the relevant national and international guidelines. Our project was approved by the East China Sea Fisheries Research Institute. In China, catching wild mud crabs from seawater does not require specific permits. Our study did not involve endangered or protected species.
Genome survey sequencing and analysis
Genomic DNA of S. paramamosain was extracted from muscle for sequencing. Three pair-end DNA libraries, two with insert sizes of 400–500 bp and one with 250 bp, were constructed following standard Illumina operating procedures.
All raw data were trimmed to filter out low-quality data and adapter contaminant using NGS QC Toolkit21. De novo assembly was performed on the clean reads using SOAPdenovo software (http://soap.genomics.org.cn/soapdenovo.html) with the following parameters: the k value in k-mer was set at 45, unsolve repeats by reads and fill gaps in scaffolds.
Mapping family and data collection
The F1 full-sib family for linkage map construction was created by two parents from the wild population of Hainan Province, China. The mapping population was reared at Wei-Er-Si Aquafarming Company of Rudong in 2017. In total, 99 progeny were randomly selected after being reared for 100 days in the same pond. Twenty-six traits, representing sex, carapace, cheliped, pereiopod and swimming stroke, were measured for each individual according to the measurement method described by Keenan et al.22. The measurement data are provided in the Supplementary Table 1.
Muscle tissues were sampled and preserved immediately in liquid nitrogen. Total DNA was extracted using a TIANamp Marine Animal DNA Extraction kit (TIANGEN, Beijing, China). DNA concentration was determined using GeneQuant (Amersham Biosciences Ltd., Piscataway, NJ, USA), and integrity was evaluated via electrophoresis in a 1% agarose gel.
RAD library construction and Illumina HiSeq3000 sequencing
Restriction site-associated DNA marker (RAD) library construction, sample indexing and pooling followed for the natural populations23. The restriction enzyme EcoRI was used to cut the DNA. A total of 26 multiplexed sequencing libraries were constructed, in which each DNA sample was assigned a unique nucleotide multiplex identifier (MID) for barcoding. Pair-end (125-bp) sequencing was performed using Illumina HiSeq3000 in a total throughput of six lanes.
SNP discovery, filtering, genotyping and validation
Raw sequence reads were trimmed to 110 nucleotides from the 3′ end to ensure that more than 98% of the nucleotides had a quality value above Q30 (equal to 0.1% sequencing error). The trimmed reads were clustered into read tags (hereafter, RAD-tags) by sequence similarity using ustacks24 to produce unique candidate alleles for each RAD locus. A maximum base-pair mismatch of one was allowed in this step for the genetic mapping population. RAD-tags were then collapsed into clusters using ustacks under the default parameters for SNP calling.
Linkage map construction and QTL mapping
Genotype calling refers to the process of determining the genotypes of the SNP loci of each individual after SNP calling has been performed in accordance with Xu et al.25. Then, customized Perl scripts were applied to generate a “.loc” format file, which was the input file for joinmap 4.026. The genetic map was grouped by joinmap 4.0 with logarithm of the odds (LOD) = 6.0 and the marker order was determined by Lep-MAP 227. Singular markers were added to the established LGs using the joinSingles module with an LOD score limit of 10 and a minimum difference of 3 between the best LG and the second best LG of each joined marker. Then, markers intervals which are larger than 30 cM were removed, except for the LG32, in which the large interval might be caused by the location of centriole.
QTL mapping of the 26 traits was performed with MapQTL 5.028. QTL region detection, the percentage of the phenotypic variance explained, and the genotypic information coefficient (GIC) were calculated with the interval QTL mapping model (IM)29. In the QTL mapping step, the LOD threshold for testing the significance of the QTL peaks was calculated using 1,000 permutations for each of the trait data sets and a genome-wide significant level of 5%. For interval distances >1.0 cM, significant thresholds were estimated every 1.0 cM. In this study, a LOD value of ≥3.0 was set as the minimum threshold to indicate a QTL in the present study.
Gene location and growth-related gene identification
RAD-Tag sequences from the linkage map were BLASTed against genomic scaffolds and transcriptome unigenes, and a reference transcriptome was de novo assembled using the raw reads from our previous work16,17 and some unpublished data using the BLASTn program via an identity value cut-off of 99% and alignment length of 50. For alignment lengths between 30 and 50, manual inspection was performed. The transcriptome data, the genome scaffold data, and tag sequences from linkage map data referred to in this article are provided in Supplementary File 1. RAD-Tag sequences were also annotated via the GenBank nr database, which is hosted by NCBI (http://www.ncbi.nlm.nih.gov/), by blastx with an E-value cut-off of 1.0 × 10−5.
Results
Genome survey of S. paramamosain
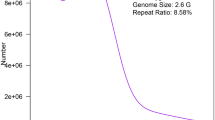
Two 400- to 500-bp and one 250-bp paired-end libraries were constructed for the genome survey analysis, and 147.18 Gb of sequencing data were generated. Raw sequencing data were submitted to the Sequence Read Archive (SRA) database of NCBI with the accession number SRP150472. After QC and filtering, a total of 78.41 Gb of high-quality reads were obtained with a 45.32% GC content. The frequency of 17-mers (nucleotide strings with a length of 17 bp) among the raw sequencing data was calculated, and a k-mer curve was constructed. k-mer analysis revealed that there was a peak at the k-mer length of 60. The genome size was estimated at 1.21 Gb with remarkably high heterozygosity (1.3%), and 55.20% of the genome was estimated to be repetitive30. A total of 16,925,345 contigs with an N50 size of 145 bp were obtained, and of these sequences, 7,027,514 contained at least one simple sequence repeat (SSR), of which 3,900,204 were one-base repeats and 4,446,896 were two-base repeats.
RAD sequencing and genotyping
There were 31 million high-quality reads for the female parent, and on average, 11 million for the offspring. The male parent could not be found because the female parent was captured from the open field sea. The female parent was sequenced at a higher depth (26.90×) than the mean depth of the offspring (8.37×). The statistics for the RAD-tag are summarized in Table 1.
A total of 158,138 heterozygous polymorphic SNP markers were detected initially. A missed genotype number above nine on any SNP was filtered. After filtering, 29,069 high-quality SNP markers, that conformed to the expected Mendelian ratios (P ≥ 0.001) remained and were included in the linkage analysis.
Linkage map construction
At the LOD threshold of 6.0, 24,444 SNP loci were grouped into 47 linkage groups in the merged maps (Fig. 1), and 4,625 SNP markers could not be grouped. The 24,444 SNP markers contained three segregation types, including 13,261 lm x ll (54.3%), 9,125 nn x np (37.3%) and 2,058 hk x hk (8.4%). The genotype information for each SNP locus is provided in Supplementary Table 2, and the paternal information was derived from the maternal and F1 data. Maternal and paternal maps were also constructed, with 46 and 47 linkage groups, respectively. The lengths of the maternal, paternal and merged maps were 3,230.70 cM, 3,334.42 cM and 3,087.53 cM, respectively, and the average marker intervals were 1.87 cM, 1.82 cM and 0.92 cM, respectively. The length of the linkage groups in the maternal map varied from 6.12 cM (LG47) to 279.34 cM (LG2), and the length of those in the paternal map ranged from 6.19 cM (LG47) to 312.65 cM (LG8) (Table 2). The estimated genome size of S. paramamosain is 1.21 Gb; thus, the average recombination rate across all linkage groups was 2.55 cM/Mb.

The 47 linkage groups of the high-density consensus linkage map of S. paramamosain.
QTL mapping
The 26 traits of the 99 mapped filial individuals, which included 78 females and 21 males, are listed in Supplementary Table 3. In total, 442 SNP loci, with a LOD > 3, which representing 279 QTLs, corresponded to 24 traits. We divided these 24 traits into five categories: appearance size, head area, cheliped, pereiopod and swimming stroke, and reproductive ability (Fig. 2). The LOD values of these 442 loci ranged from 3.01 to 12.66. Five markers with a LOD value greater than five were associated with seven traits, including three cheliped traits (markers 11.hk_hk_10468 and 11.hk_hk_11304), three pereiopod and swimming stroke traits (markers 11.hk_hk_7558, 105.nn_np_34985 and 11.hk_hk_11304) and one size trait (11.nn_np_24679). Among the 442 markers, 36 markers were linked to more than one trait, suggesting that these traits might be under the same genetic control. Two to 58 markers distributed in one, two, three or four linkage groups were linked to 24 traits. SW had the most linked markers (58), followed by 2PML (55), 3PML (48), and FW (44), whereas CW and ML had only two markers. The markers associated with the 24 traits are summarized in Table 3.

Measurements traits of S. paramamosain21. (A) Carapace, (B) frontal lobe, (C) sternum, (D) periopods and (E) chelipeds. Appearance size: carapace length (CL), carapace width (CW), carapace width at spine 8 (8CW) and body height(BH); head area: carapace frontal width (FW), distance between frontal median spines (DFMS), frontal median spine height (FMSH), distance between frontal lateral spines(DFLS), distance between the first frontal lateral spines (1DFLS), distance between the second frontal lateral spines (2DFLS); cheliped: dactyl length (DL), merus length (ML), propodus length (PL), propodus width (PW), propodus depth (PD); Periopod and swimming stroke: 1st periopod merus length (1PML), 2nd periopod merus length (2PML), 3st periopod merus length (3PML), swimming stroke dactyl width (5PW), swimming stroke dactyl length (5PL); Reproductive ability: abdomen width (AW).
Twelve markers were linked to sex and distributed in LG10 (7), LG12 (2) and LG9 (3) (Fig. 3a). CL (Fig. 3b), CW, 8CW (Fig. 3c) and BH, four important traits representing the appearance size of the crab, were linked to 19 markers distributed in LG11 (8), LG2 (4), LG12 (3), LG29 (2) and LG41 (2). Five traits (DL, ML, PL, PW and PD), indicating the developmental properties of the cheliped, were linked to 37 markers distributed in LG5 (15), LG26 (8), LG13 (4), LG14 (3), LG3 (3), LG11 (2), LG15 (1) and LG22 (1). Five traits (1PML, 2PML, 3PML, 5PW and 5PL) associated with pereiopod and swimming stroke had 168 markers distributed in 11 linkage groups, represented by LG25 (52), LG2 (30), LG12 (26) and LG5 (21). Six traits that might correlate with the development of the head area (FW, DFMS, FMSH, DFLS, 1DFLS and 2DFLS), had 168 markers distributed in 10 linkage groups, represented by LG25 (43), LG13 (33), LG6 (26) and LG15 (18). AW might have some correlation with reproductive ability, with four markers located in LG11 (3) and LG10 (1).

QTL mapping for traits including sex (a), carapace length (CL) (b) and carapace width at spine 8 (8CW) (c).
Gene location and growth gene identification
By means of BLASTing against the nr database, transcriptome and scaffold of the genome survey, 4,271 of the linkage map markers were linked to genes, and the gene numbers distributed in each linkage group ranged from 15 to 210. LG2 has the largest number of located genes, followed by LG3 (204), LG1 (198), LG11(180) and LG4 (171). LG47 has the smallest number of genes, followed by LG44 (16), LG45 (20), LG46 (32), LG41 (37) and LG43 (38) (Supplementary Table 4). Gene numbers were generally positively correlated with the length and markers of linkage groups, with correlation coefficient of 0.818 and 0.823, respectively. Some growth-related genes, including transforming growth factor-beta regulator I, insulin-like androgenic gland hormone and insulin-like growth factor 2 mRNA-binding protein, were located. C-type lectin, which participates in the immune defense process, HR4 nuclear receptor and broad-complex-like protein, which participates in the ecdysone regulation pathway, were also located.
In total, 78 markers have been linked to genes. Among them, 22 markers had annotation information from at least one public database, and others were linked to transcriptome data but lacked annotation information. Of these 22 markers, two were located in the 3′-UTR, seven were in the intron, 11 were in the open reading frame (ORF), and four led to a nonsynonymous mutation; the other two were located in a pseudogene because an abnormal termination codon existed in the translated region (Table 4). 11.hk_hk_4825, a synonymous SNP located in the ORF of a RING finger protein, was a QTL of FW and 5PW. Two RING finger proteins, nhl-1, nesprin-2-like, RNA-directed DNA polymerase from mobile element jockey-like, dedicator of cytokinesis protein 3 isoform X2, carbonic anhydrase 2-like and protein disulfide-isomerase A5-like each had one SNP marker associated with FW (four), DFMS (two) and 2DFLS (one), and these genes or their linked genes might be related to the development of head area. Furthermore, integrator complex subunit 8-like, tenascin-X-like, sialidase-like, BTB/POZ domain-containing protein 3, polypeptide N-acetylgalactosaminyl transferase 1 and cholinephosphotransferase 1 isoform or their linked genes might be related to pereiopod and swimming stroke development. Mitochondrial ribonuclease P protein 3, sedoheptulokinase-like and putative nuclease HARBI1 are three genes with SNP associated with cheliped related traits, and them or their linked genes might be related to cheliped development. In addition, ankyrin repeat and general transcription factor II-I repeat domain-containing protein 2B-like or their linked genes might be related to the appearance size of the crab. GPI mannosyltransferase 4, DNA helicase MCM8-like and kinesin II or their linked genes might be related to reproductive ability, and protein numb isoform X5 or its linked genes might be related to OCS.
Discussion
Mud crabs are euryhaline and widely distributed in tropical, subtropical and temperate waters. In China, S. paramamosain is the dominant species and is mainly cultured in southeastern coastal provinces14. Mud crabs are very popular in China because of their high content of protein, unsaturated fatty acids and trace elements such as vitamins31.
Genome sequencing is an important step for deciphering evolutionary status and molecular mechanisms and accelerating genetic improvements in traits of interest in economically important species. The genome size of S. paramamosain was estimated to be 1.21 Gb with remarkably high heterozygosity (1.3%). This finding is inconsistent with previous studies (1.64 pg), in which flow cytometry was used to assess genome size32. Different results for one species from two methods were also found in P. trituberculatus9 and L. vannamei7. The flow cytometry method is fairly straightforward, but the accuracy is highly dependent on the internal standard and quality of the material used for DNA content measurement33. Additionally, a comparison between flow cytometry and k-mer analysis methods in the estimation of nine insect species suggested that k-mer analysis is more accurate34. Genome survey analysis will be highly useful for the formulation of sequencing strategies for the mud crab.
Currently, the only reported linkage map of this species was constructed by our team using microsatellite and AFLP markers. In Total, 212 markers were mapped, and the mean spacing of the markers was 18.68 cM20, which cannot fulfill the requirements of the genetic breeding industry. This study reported a high-resolution genetic map of the mud crab with 24,444 SNP markers, a length of 3087.53 cM and a marker interval of 0.92 cM. This map has the highest number of markers compared with that of maps of other crustacean species, including P. trituberculatus (10,963 markers)9, M. japonicus (9289 markers)8 and L. vannamei (6146 markers)7. A large number of markers in the map were at the same location, which means they are completly linked. Therefore, the marker interval should be 0.92 cM instead of 0.13 cM (3087.53/24,444≈0.13). However, the numerous markers will be very useful in QTL mapping, gene location and genome assemble.
Forty-seven linkage groups were obtained in the merged map, and only 46 linkage groups were in the maternal map. The missing linkage group in the maternal map was LG38; this linkage group may not be the sex chromosome because alleles in this linkage were not separated significantly by sex in the F1 offspring. Forty genes were located on LG38, and no sex-determining genes were found in this linkage group. A high-density linkage map suggested a ZW sex determination system in Eriocheir sinensis; but only an ankyrin-2 gene was found on the putative sex chromosome, and the sex determination gene double-sex was located on a putative autosome6. This finding suggested that sex determination genes may not exist on sex-linked chromosomes or that there is a different sex determination system in crustaceans. Further, 12-sex linked QTL were identified, and verification in large numbers of wild populations is needed.
Under the conditions of high marker density (24,444 SNPs), the linkage groups should be consistent with the haploid chromosome number. However, the number of linkage groups in the present map seems to be less than the 49 haploid chromosome numbers reported by Wang et al.35 and Chen et al.36 using karyotype method. In fact, fewer number of linkage groups in the linkage map were also found in M. japonicus8. In this study, the lack of one linkage group in the maternal map and the lower numbers in the merged map might be mostly due to the relatively lower number of mapping populations or the low enzyme-digestion efficiency in the missing chromosome.
Overall, 4,271 markers were linked to genes, and the gene numbers were generally positively correlated with the length and marker numbers of linkage groups, suggesting that genes were almost evenly distributed in the crab genome. Transforming growth factor-beta regulator I, which is also called nuclear interactor of ARF and Mdm2 (NIAM), activates p53/TP53, causes G1 arrest and collaborates with ARF to suppress proliferation, acted as a growth inhibitor37. Insulin-like androgenic gland hormone (IAG), which is produced by androgenic glands in male crustaceans, is regarded as a key regulator of sex differentiation. Interestingly, studies on IAG in Scylla paramamosain suggested that it is involved in regulating ovarian development and somatic growth38. Insulin-like growth factor 2 mRNA-binding protein binds to the 5′-UTR of the insulin-like growth factor 2 (IGF2) mRNA to regulate IGF2 translation, and thus has an important function in animal development39. C-type lectins are a superfamily of proteins that recognize a broad repertoire of ligands and regulate a diverse range of physiological functions40. Indeed, C-type lectins of S. paramamosain were upregulated significantly by Vibrio parahaemolyticus and had an important role in the immune response41,42. Broad-complex-like protein and HR4 nuclear receptor are two ecdysteroidogenic transcription factors that deliver ecdysone signal, which is also important for development43. Knowing the location of these genes will assist the study of genetic breeding.
The head area of the crab contains many organs, such as the cerebral ganglion and antennary glands. Seven linked annotated genes were associated with this area. Among them, nesprin-2 belongs to a novel family of nuclear and cytoskeletal proteins with rapidly expanding roles as intracellular scaffolds and linkers44. Dedicator of cytokinesis protein 3 (DOCK3) is a large protein involved in intracellular signalling networks, which function as activators of small G proteins. DOCK3 is expressed exclusively in the central nervous system of mice and plays an important role in axonal outgrowth and cytoskeleton re-organization45.
Six genes associated with pereiopod and swimming stroke traits were identified. Among them, tenascin-X is a glycoprotein that contributes to matrix stability and is possibly involved in collagen fibril formation46. A SNP in this gene causes a nonsynonymous mutation, which replaces a hydrophilic amino acid Ser with a hydrophobic amino acid Leu. Sialidase-like, which is a glycoside hydrolase enzyme that cleaves the glycosidic linkages of neuraminic acids47, also possesses a nonsynonymous mutation of Ser to Asn. Future studies in large populations are needed to prove the correlation between phenotype and genotype. Polypeptide N-acetylgalactosaminyltransferase 1, which catalyses the initial reaction in O-linked oligosaccharide biosynthesis48, and cholinephosphotransferase 1, which catalyses phosphatidylcholine biosynthesis from CDP-choline and plays a central role in the formation and maintenance of vesicular membranes49, were also suggested to have a relationship with pereiopod and swimming stroke development.
Three linked annotated genes were suggested to associate with cheliped traits. Among them, mitochondrial ribonuclease P protein 3, which is a part of mitochondrial ribonuclease P (mt-RNase P) that cleaves tRNA molecules at their 5′ -ends50, and a nonsynonymous mutation of Trp to Arg was found in this gene. In addition, the putative nuclease HARBI1, which might promote anti-V. alginolyticus infection by participating in regulating phagocytosis, apoptosis, superoxide dismutase activity, PO activity, and THC in Marsupenaeus japonicus, was another gene associated with cheliped traits51.
The ankyrin repeat was linked to one QTL of 8CW and appeared to be associated with appearance size. Proteins that contain the ankyrin repeat motif can mediate the protein-protein interactions52.
Beneath the abdominal plate is the location in which the eggs are held in female crabs and where the ejaculatory duct is found in male crabs, both of which were possibly associated with reproductive ability. Three genes were linked to QTLs of AW, among which GPI mannosyltransferase 4 is involved in glycosylphosphatidylinositol-anchor biosynthesis53. DNA helicase MCM8, forms a complex with MCM9 and is involved in homologous recombination repair following DNA interstrand cross-links, thus playing a key role during gametogenesis54.
It is likely that some of the association genes seem to have some relationship with specific traits, which indicated by their function studies in other species. However, it should be noted that the association genes do not mean that they are the causative genes, and in-depth studies are still needed.
In summary, this study performed a genome survey analysis, which revealed that S. paramamosain has an estimated genome size of 1.21 Gb with high heterozygosity (1.3%). Then, a high-resolution linkage map was constructed using 24,444 SNP makers. Forty-seven linkage groups were obtained with a total length of 3087.53 cM and a marker interval of 0.92 cM. With the aid of transcriptome and genome scaffold data, 4,271 markers were linked to genes, including some important growth-related, immune-related and hormone pathway genes. Further, 442 QTLs were identified and found to correspond to 24 traits, and of these, 78 QTLs were linked to genes. Some interesting genes, such as dedicator of cytokinesis protein 3, tenascin-X and DNA helicase MCM8, were believed to have important relationships with specific traits and are worth further exploration.
References
Meuwissen, T. H., Hayes, B. J. & Goddard, M. E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829 (2001).
Schaeffer, L. R. Strategy for applying genome-wide selection in dairy cattle. Anim. Breed. Genet. = Zeitschrift fur Tierzuchtung und Zuchtungsbiologie 123, 218–223 (2006).
Muir, W. M. et al. Genome-wide assessment of worldwide chicken SNP genetic diversity indicates significant absence of rare alleles in commercial breeds. P Natl Acad Sci USA 105, 17312–17317 (2008).
Ibáñez-Escriche, N., Forni, S., Noguera, J. L. & Varona, L. Genomic information in pig breeding: Science meets industry needs. Livest Sci 166, 94–100 (2014).
Robinson, N. A. et al. QTL for white spot syndrome virus resistance and the sex-determining locus in the Indian black tiger shrimp (Penaeus monodon). BMC Genomics 15, 731 (2014).
Cui, Z. et al. High-density linkage mapping aided by transcriptomics documents ZW sex determination system in the Chinese mitten crab Eriocheir sinensis. Heredity 115, 206 (2015).
Yu, Y. et al. Genome survey and high-density genetic map construction provide genomic and genetic resources for the Pacific White Shrimp Litopenaeus vannamei. Sci Rep 5, 15612 (2015).
Lu, X. et al. High-resolution genetic linkage mapping, high-temperature tolerance and growth-related quantitative trait locus (QTL) identification in Marsupenaeus japonicus. Mol Genet Genomic: MGG 291, 1391–1405 (2016).
Lv, J., Gao, B., Liu, P., Li, J. & Meng, X. Linkage mapping aided by de novo genome and transcriptome assembly in Portunus trituberculatus: applications in growth-related QTL and gene identification. Sci Rep 7, 7874 (2017).
Colbourne, J. K. et al. The Ecoresponsive Genome of Daphnia pulex. Science 331, 555–561 (2011).
Kao, D. et al. The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. eLife 5, e20062 (2016).
Gutekunst, J. et al. Clonal genome evolution and rapid invasive spread of the marbled crayfish. Nature Ecology & Evolution 2, 567–573 (2018).
Song, L. et al. Draft genome of the Chinese mitten crab. Eriocheir sinensis. GigaScience 5, 5 (2016).
Ma, L.-B., Zhang, F.-Y., Ma, C.-Y. & Qiao, Z.-G. Scylla paramamosain (Estampador) the most common mud crab (Genus Scylla) in China: evidence from mtDNA. Aquac Res 37, 1694–1698 (2006).
Xie, C. P. et al. Transcriptome and Expression Profiling Analysis of the Hemocytes Reveals a Large Number of Immune-Related Genes in Mud Crab Scylla paramamosain during Vibrio parahaemolyticus Infection. PloS one 9, e114500, https://doi.org/10.1371/journal.pone.0114500 (2014).
Ma, H. et al. Transcriptome analysis of the mud crab (Scylla paramamosain) by 454 deep sequencing: assembly, annotation, and marker discovery. PloS one 9, e102668, https://doi.org/10.1371/journal.pone.0102668 (2014).
Ma, H. et al. Identification of transcriptome-derived microsatellite markers and their association with the growth performance of the mud crab (Scylla paramamosain). PloS one 9, e89134, https://doi.org/10.1371/journal.pone.0089134 (2014).
Gao, J. et al. Transcriptome analysis of the differences in gene expression between testis and ovary in green mud crab (Scylla paramamosain). Bmc Genomics 15, 585 (2014).
Zhu, F., Qian, X. & Ma, X. Comparative transcriptomic analysis of crab hemocytes in response to white spot syndrome virus or Vibrio alginolyticus infection. Fish Shellfish Immun 80, 165–179 (2018).
Ma, H. Y. et al. First genetic linkage map for the mud crab (Scylla paramamosain) constructed using microsatellite and AFLP markers. Genet Mol Res: GMR 15, gmr. 15026929 (2016).
Patel, R. K. & Jain, M. NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PloS one 7, e30619, https://doi.org/10.1371/journal.pone.0030619 (2012).
Keenan, C. P., Davie, P. J. & Mann, D. A revision of the genus Scylla de Haan, 1833 (Crustacea: Decapoda: Brachyura: Portunidae). Raffles B Zool 46, 217–245 (1998).
Baird, N. A. et al. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PloS one 3, e3376, https://doi.org/10.1371/journal.pone.0003376 (2008).
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W. & Postlethwait, J. H. Stacks: Building and Genotyping Loci De novo From Short-Read Sequences. G3: Genes|Genomes|Genetics 1, 171–182 (2011).
Pei, X. et al. Population genomic analyses from low‐coverage RAD‐Seq data: a case study on the non‐model cucurbit bottle gourd. Plant J 77, 430–442 (2014).
Van Ooijen, J. W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genetics Res 93, 343–349 (2011).
Rastas, P., Calboli, F. C. F., Guo, B., Shikano, T. & Merilä, J. Construction of Ultradense Linkage Maps with Lep-MAP2: Stickleback F 2 Recombinant Crosses as an Example. Genome Biol Evol 8, 78–93 (2016).
Ooijen, V. MapQTL, Software for the mapping of quantitative trait loci in experimental populations, in Kyazma B. V. Wageningen, (Netherlands, 2004).
Jansen, R. C. & Stam, P. High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136, 1447–1455 (1994).
Zeng, X. et al. The draft genome of Tibetan hulless barley reveals adaptive patterns to the high stressful Tibetan Plateau. P Natl Acad Sci USA 112, 1095–1100 (2015).
Li, Y., Ai, C. & Liu, L. Mud Crab, Scylla paramamosain China’s Leading Maricultured Crab. Aquaculture in China: Success Stories and Modern Trends, Chapter 3.4 (2018).
Liu, L. et al. Flow cytometric analysis of DNA content for four commercially important crabs in China. Acta Oceanologica Sinica 35, 7–11 (2016).
Dolezel, J. & Greilhuber, J. Nuclear genome size: are we getting closer? Cytometry. Part A: the journal of the International Society for Analytical Cytology 77, 635–642 (2010).
He, K., Lin, K., Wang, G. & Li, F. Genome Sizes of Nine Insect Species Determined by Flow Cytometry and k-mer Analysis. Front Physiol 7, 569 (2016).
Wang, G., Chen H., Li, S., Guan W. & Li, Q. Studies on the Chromosomal Karyotype of Mud Crab, Scylla serrata (Forskal). Marine Sci, 1, 9–13 (2002) (In Chinese with English Abstract).
Chen, X., Wang G., Chen, L. & Li, S. Methodologoical improvement and its application effect in chromosome study of mud crab, Scylla serrata. J Oceanography in Taiwan Strait, 3, 347–353 (2004). (In Chinese with English Abstract).
Tompkins, V. S. et al. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J Biol Chem 282, 1322–1333 (2007).
Huang, X., Ye, H., Huang, H., Yang, Y. & Gong, J. An insulin-like androgenic gland hormone gene in the mud crab, Scylla paramamosain, extensively expressed and involved in the processes of growth and female reproduction. Gen Comp Endocrinol 204, 229–238 (2014).
Nielsen, J. et al. A Family of Insulin-Like Growth Factor II mRNA-Binding Proteins Represses Translation in Late Development. Mol Cell Biol 19, 1262–1270 (1999).
Brown, G. D., Willment, J. A. & Whitehead, L. C-type lectins in immunity and homeostasis. Nat Rev Immunol 18, 374–389 (2018).
Wei, X. et al. C-type lectin B (SpCTL-B) regulates the expression of antimicrobial peptides and promotes phagocytosis in mud crab Scylla paramamosain. Dev Comp Immunol 84, 213–229 (2018).
Jiang, K. et al. Isolation of the C-type lectin like-domain cDNAs from the mud crab Estampador, 1949, and its expression profiles in various tissues, during larval development, and under challenge. Crustaceana 85, 817–834 (2012).
Niwa, Y. S. & Niwa, R. Transcriptional regulation of insect steroid hormone biosynthesis and its role in controlling timing of molting and metamorphosis. Dev Growth Differ 58, 94–105 (2016).
Rajgor, D. & Shanahan, C. M. Nesprins: from the nuclear envelope and beyond. Expert Reviews in Molecular Medicine 15, e5 (2013).
Helbig, K. L., Mroske, C., Moorthy, D., Sajan, S. A. & Velinov, M. Biallelic loss-of-function variants in DOCK3 cause muscle hypotonia, ataxia, and intellectual disability. Clin genet 92, 430–433 (2017).
Egging, D., van den Berkmortel, F., Taylor, G., Bristow, J. & Schalkwijk, J. Interactions of human tenascin-X domains with dermal extracellular matrix molecules. Arch Dermatol Res 298, 389–396 (2007).
von Itzstein, M. The war against influenza: discovery and development of sialidase inhibitors. Nature reviews. Drug Discov 6, 967–974 (2007).
Wandall, H. H. et al. Substrate specificities of three members of the human UDP-N-acetyl-alpha-D-galactosamine:Polypeptide N-acetylgalactosaminyltransferase family, GalNAc-T1, -T2, and -T3. J Biol Chem 272, 23503–23514 (1997).
Henneberry, A. L., Wistow, G. & McMaster, C. R. Cloning, genomic organization, and characterization of a human cholinephosphotransferase. J Biol Chem 275, 29808–29815 (2000).
Holzmann, J. et al. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135, 462–474 (2008).
Sun, B., Qian, X. & Zhu, F. Molecular characterization of shrimp harbinger transposase derived 1 (HARBI1)-like and its role in white spot syndrome virus and Vibrio alginolyticu sinfection. Fish Shellfish Immun 78, 222–232 (2018).
Li, J., Mahajan, A. & Tsai, M. D. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochem 45, 15168–15178 (2006).
Taron, B. W., Colussi, P. A., Wiedman, J. M., Orlean, P. & Taron, C. H. Human Smp3p adds a fourth mannose to yeast and human glycosylphosphatidylinositol precursors in vivo. J Biol Chem 279, 36083–36092 (2004).
Nishimura, K. et al. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol Cell 47, 511–522 (2012).
Acknowledgements
This work was supported by grants from the Program of Science and Technology Commission of Shanghai (No. 15391900900), China Agriculture Research System-48 (No. CARS-48), the Basic Research Fund for State-level Nonprofit Research Institutes of ESCFRI (No. 2014Z01) and Special Program on Agricultural Aspect of Science and Technology Commission of Ningbo (2016C11003).
Author information
Authors and Affiliations
Contributions
L.M. conceived and supervised the project. L.M., C.M., F.Z. and K.J. supplied the experimental animals. W.W., W.C., M.Z., J.L., L.D., H.Q., J.Z. and T.W. measured the traits and sampled the tissues. M.Z., L.M., W.W. and W.C. contributed to the genome survey analysis, linkage map construction and QTL mapping. M.Z. contributed to the gene location analysis. M.Z., L.M., W.W. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, M., Wang, W., Chen, W. et al. Genome survey, high-resolution genetic linkage map construction, growth-related quantitative trait locus (QTL) identification and gene location in Scylla paramamosain. Sci Rep 9, 2910 (2019). https://doi.org/10.1038/s41598-019-39070-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39070-z
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
