
Abstract
The EU directive 2001/18/EC requires any genetically modified (GM) event to be stable. In the present work, a targeted Next-Generation Sequencing (NGS) approach using barcodes to specifically tag each individual DNA molecules during library preparation was implemented to detect mutations taking into account the background noise due to amplification and sequencing errors. The method was first showed to be efficient in detecting the mutations in synthetic samples prepared with custom-synthesized mutated or non-mutated P35S sequences mixed in different proportions. The genetic stability of a portion of the P35S promoter targeted for GM detection was then analyzed in GM flour samples. Several low frequency mutations were detected in the P35S sequences. Some mutated nucleotides were located within the primers and probes used in the P35S diagnostic test. If present not as somatic mutations but as the consensus sequence of some individuals, these mutations could influence the efficiency of the P35S real time PCR diagnostic test. This methodology could be implemented in genetic stability studies of GM inserts but also to detect single nucleotide mutant GM plants produced using “new breeding techniques”.
Similar content being viewed by others

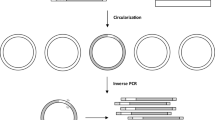
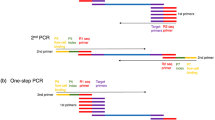
Introduction
Since the first year of commercialization in 1996, the development of genetically modified (GM) crops has constantly grown in terms of cultivated land area, plant species and diversity of GM characteristics. In 2017, 24 countries grew 189.8 million hectares of GM crops worldwide (mostly soybean, maize, cotton, and canola)1. From 1992 to 2017, 4,133 approvals were issued by regulatory authorities for 29 GM crops and 498 events and this number is expected to rise1. Approvals were issued to GM crops for food use, feed use, and environmental release or cultivation.
According to the EU Directive 2001/18/EC on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC2, approval of a new GM crop requires the GM inserts to be genetically characterized and quantified. Applicants must provide information concerning genetic stability of the insert in their notifications. Development, cultivation and propagation of the GM crops should not induce any changes in the introduced DNA sequence3. For plant containing single transformation events, genetic stability encompassing the Mendelian inheritance of the insert and the molecular stability of the event over multiple (usually five) generations or vegetative cycles has to be demonstrated2,4. The Mendelian inheritance is currently checked by segregation analysis and the Chi-square test4. Molecular stability of the event is commonly verified by Southern Blot analyses; however this technique is not appropriate for detecting small DNA changes5 and sequencing approaches represent interesting alternatives4. The detection of small DNA rearrangements is important as one or more nucleotide polymorphisms in the insert could potentially modify transgene expression eventually leading to the loss of the GM characteristics6,7,8. Aguilera et al. (2008) verified the genetic stability of the MON810 trait in seeds of 26 commercial maize varieties by analyzing the intactness of MON810 integration using PCR-RFLP. The MON810 insert was absent in one maize variety suggesting the loss of the transgene locus. In addition to transgene expression alteration or loss, GM detection and quantification methods based on real-time PCR could also be affected by DNA changes. The presence of GMOs is currently assessed by reference laboratory via a screening approach targeting the most common transgenic elements found in GMOs, such as the Cauliflower mosaic virus (CaMV) 35S promoter (P35S) and the nopaline synthase terminator from Agrobacterium tumefaciens (tNOS)9. Once the presence of GM material has been established using screening methods, GMOs are further identified using GM event specific real time PCR methods. Nucleotide polymorphisms within primer and/or probe sequences could alter the method’s reliability, the position of single-nucleotide polymorphisms (SNPs) within oligonucleotides influencing their melting temperature (Tm), the efficiency of polymerase extension, and target specificity10.
Some published studies have investigated the post-transformational stability of DNA inserts and their flanking regions in Roundup Ready (RR) soybean7, MON810 maize6,8,11,12, stacked maize MON88017 x MON8103 and NK603 x MON8105,12 and GT73 oilseed rape3. In 2005, Ogasawara et al. studied frequency of mutations of the transgene in 72 Roundup Ready (RR) soybean seeds by sequencing 572 bp PCR fragments of the transgene and identified several mutated nucleotides in the 35S promoter region. Ben Ali et al. (2014; 2018) detected some variants in the cry1Ab coding region of seed samples of MON810 maize varieties. They also reported that samples from seeds containing a stacked MON810 event had more variants than MON810 single event varieties12. The more recent studies used a combination of real-time PCR, High-Resolution Melting (HRM) analyses, Sanger sequencing and Illumina sequencing which are appropriate techniques able to identify small DNA rearrangements3,5,12. Real-time PCR coupled with HRM is a rapid, simple, robust and sensitive method for detecting mutation and enabling the screening of many samples13. Nevertheless, this method does not provide information about nucleotide sequences and requires subsequent sequencing. In addition, HRM being based on the melting properties of the double stranded DNA molecules synthetized during PCR amplification, the obtained result is characteristic of the consensus sequence of the DNA molecules present in the sample and does not provide information on the variability of the DNA sequences within the sample. It is therefore only suitable for the analysis of separate individuals (seeds or plants). Rare mutations cannot be detected by analyzing individuals one by one. Their detection requires the simultaneous analysis of a large population of individuals ideally coming from different environments. Next Generation Sequencing (NGS) approaches can provide the sequence of numerous DNA molecules present in an extract. For these reasons, the application of NGS approaches on DNA extracts from flour, which are obtained from populations of seeds from various plants and environments, constitutes a method of choice to characterize the stability of a GM insert.
Currently, the targeted NGS approach is extensively used to identify mutations in various complex samples14,15,16,17,18. However, the detection of rare mutations (frequency below 1%) by NGS remains challenging because of the background noise due to errors introduced by the polymerase during library preparation and to the error rate of sequencing technologies19,20,21. In recent years, effective methods for detecting rare mutations have been developed and investigated for clinical and research applications including non-invasive prenatal diagnostics, early detection of cancer, monitoring of treatment response, detection of infectious disease and food safety19,22,23. Several groups have described barcode-based molecular approach to effectively distinguish amplification and sequencing errors from true mutations19,20,22,23,24,25,26,27. During library preparation, each individual DNA molecule present in the sample is tagged by incorporating a specific molecular barcode14. After amplification of the tagged DNA, multiple copies of each molecule are sequenced and reads with the same molecular barcode are grouped into families to create a high confidence consensus14,23. Mutations found in only a fraction of the reads with the same barcode are amplification or sequencing errors and are eliminated when the consensus is created23. Mutations found in all the reads with the same barcode are true mutations. Stalhberg et al. (2016, 2017) developed a barcode-based approach called “Simple, Multiplexed, PCR-based barcoding of DNA for Sensitive mutation detection using Sequencing” (SiMSen-Seq). Their method uses reduced primer concentration, elongated PCR extension times and hairpin-protected barcode primers to enable simple and short library construction protocol20. SiMSen-Seq can detect variants at or below 0.1% frequency with low DNA input. A detailed protocol for assay development and a bioinformatics package to process the barcoded data are described in Stalhberg et al. (2017). For these reasons, SIMSen-Seq was implemented in our laboratory to study GM insert stability in complex samples. The methodology was first assessed on synthetic samples prepared with custom-synthesized mutated or non-mutated sequences mixed in different proportions. The variability of a portion of the P35S promoter targeted for GM detection was then analyzed in GM flour samples.
Results
Assay quality
In our assay, 1,329,877 (natural sample 1) to 2,095,571 (synthetic sample 10% C4) raw reads were obtained with a mean of 1,883,581 raw reads in synthetic samples and 1,749,361 in natural samples (Tables 1 and 2). Among these raw reads, 1,262,132 (natural sample 1) to 2,080,079 (synthetic sample 10% C4) amplicon reads were identified with a mean of 1,869,150 amplicon reads in synthetic samples and 1,703,026 in natural samples. Amplicon yields (amplicon reads/raw reads) ranged from 0.95 to 0.99, indicating that a very high number of the total data were analyzable. The number of consensus sequences identified per sample is presented according to consensus depth (3, 10, 20 or 30), which corresponds to the number of raw reads with the same barcode used to compute a consensus sequence. The mean number of consensus sequences identified was 199,506 for synthetic samples and 67,541 for natural samples using a consensus(3), 67,857 (synthetic samples) and 39,666 (natural samples) using a consensus(10), 7,187 (synthetic samples) and 24,269 (natural samples) using a consensus(20) and 737 (synthetic samples) and 17,407 (natural samples) using a consensus(30). The distribution of barcode family depths was larger for the natural samples than for the synthetic samples (Supplementary Figure S1). Down-sampling plots (Supplementary Figure S2) showed a saturation of detected barcodes in the synthetic samples (visible horizontal plateau) while few additional barcodes could have been detected in the natural samples (light upward slope) with additional sequencing depth. Overall, summary statistics showed our assay to be of very good quality.
Analysis of mutation in synthetic samples
Using Debarcer, raw mutation rates and corrected mutation rates (consensus(3), consensus(10), consensus(20), consensus(30)) were obtained at each position of the P35S sequence studied in the synthetic samples. The raw and corrected consensus(10) mutation rates are presented in Fig. 1. All along the sequence (excluding positions 67 and 71), mutations were detected with a mean mutation rate of 0.165% using raw reads, 0.026% using consensus(3) reads (data not shown) and 0.010% using consensus(10) reads and a maximum error rate of 0.44% using raw reads, 0.1% using consensus(3) reads (data not shown) and 0.05% using consensus(10) reads. Mutations detected at other positions than 67 and 71 correspond to background noise which could be associated with the long oligonucleotide synthesis error rate. The mean mutation rate of 0.010% using consensus(10) reads corresponds to the announced synthesis error rate of 0.050–0.066% (according to the provider, Eurofins).

Raw and corrected mutation rates at each position of the P35S sequence studied in the synthetic samples. 10% C1, 1% C1, 0.1% C1: synthetic samples containing 10%, 1% or 0.1% of the P35S-C1 oligonucleotide mixed with respectively 90%, 99% or 99.9% of the P35S-WT oligonucleotide. 10% C4, 1% C4, 0.1% C4: synthetic samples containing 10%, 1% or 0.1% of the P35S-C4 oligonucleotide mixed with respectively 90%, 99% or 99.9% of the P35S-WT oligonucleotide.
Mutation rates at positions 67 and 71 are presented in Table 3 using raw reads and corrected consensus(3) or consensus(10) reads. Taking into account the maximum background noise (0.44% using raw reads, 0.1% using consensus(3) reads and 0.05% using consensus(10) reads), the mutated nucleotide at position 67 was detected in sample 10% C4 at a rate of 9.04% using raw reads, 1.48% using consensus(3) reads and 0.36% using consensus(10) reads and in sample 1% C4 at a rate of 0.97% using raw reads and 0.14% using consensus(3) reads. Similarly, the mutated nucleotide at position 71 was detected in sample 10% C1 at a rate of 10.65% using raw reads, 1.99% using consensus(3) reads and 0.48% using consensus(10) reads and in sample 1% C1 at a rate of 0.59% using raw reads. The raw mutation rate was close to the real percentage of mutated sequence in the samples, while the corrected mutation rate was under-estimated. At positions 67 and 71, other mutations than the expected mutations were observed at low level (Table 3), corresponding to the synthesis error rates previously observed all along the sequence.
Analysis of mutation in GM flour samples
Using Debarcer, raw mutation rates and corrected error rates (consensus(3), consensus(10), consensus(20), consensus(30)) were obtained at each position of the P35S sequence studied in the natural samples. Results were analyzed using a corrected consensus depth of 10 as recommended previously19 (Fig. 2). At all positions, the corrected mutation rates were lower than the raw mutation rates. The correction applied on raw mutation rates varied depending on the individual nucleotide. In the corrected consensus(10) data, several mutated positions were identified all along the P35S sequence studied in the six samples. Mutation rates varied between the positions and samples, with a maximum of 0.2% at position 58 and 0.17% at position 59 in sample 4. In sample 3, mutation rates were below 0.05%. In other maize samples, mutation rates above 0.05% were observed at positions 12–13 for samples 1, 2 and 4, positions 57-58-59 for sample 1 and positions 58–59 for sample 4. Overall, mutation rates were slightly higher in soybean flours, with 16 positions with mutation rates above 0.05% in samples 5 (at positions 12, 13, 25, 26, 28, 29, 31, 34, 38, 42, 44, 45, 52, 54, 59, 66) and 6 (at positions 12, 13, 25, 26, 28, 29, 31, 32, 33, 34, 38, 44, 45, 59, 62, 80).

Raw and corrected mutation rates at each position of the P35S sequence studied in the natural samples. NS 1, NS 2, NS 3, NS 4, NS 5 and NS 6: Natural GM flour samples 1 to 6. 35SF, 35SR and 35SP: Primers F and R and probe P used to detect P35S using real-time PCR.
Discussion
In this study, a new approach using targeted NGS detected several low frequency mutations in a portion of P35S sequences in complex GM samples. The SiMSen-seq methodology had previously been used for ultrasensitive mutation detection in liquid biopsies20 and tumors28. This was the first time this methodology was successfully implemented to detect mutations in plant materials.
In order to test the technique in our laboratory, synthetic samples were prepared and analyzed, containing custom-synthesized mutated or non-mutated P35S sequences mixed in different proportions. The method was efficient in detecting the mutations introduced in the P35S sequence regardless of the position in the sequence or the class of mutation (class I or IV). Custom-synthesized sequences were good materials to test the efficiency of the primers and the method in our laboratory but they could not be used to evaluate the sensitivity of the assay below 1% because of the synthesis error rate creating a background noise. Nevertheless, the method could detect mutations below 0.05% in GM flour samples, indicating a very good sensitivity of the assay.
In our study, hundreds of thousands of P35S sequences extracted from complex GM samples such as maize and soybean seed flours were analyzed individually. This constitutes a major improvement compared to techniques used recently (HRM techniques coupled with sequencing) which analyzed individual seeds one by one3,5,12. By providing the sequence of many individual DNA molecules present in the sample, this methodology allows the analysis of a population of individuals, but it also provides an insight into the mutations which occurred within individuals. The drawback associated with the power of the technique is that some of the mutations observed in the results may have been present in somatic cells without being present in the consensus sequence of any individuals.
Because of its high expression level, the 35S promoter is the most popular and common promoter of transgenes in GM plants7. In transgenic plants, the P35S promoters come from various strains of Cauliflower mosaic virus and have been introduced via different vector constructions29. For these reasons, there may be polymorphisms in the sequences of the P35S promoter. In addition, mutations may have occurred during the breeding process29. This study focused on the conserved region of the P35S promoter targeted by diagnostic methods using real-time PCR. Several mutated nucleotides were identified, with frequency varying between 0.2% to less than 0.05%. By analyzing bibliographic data, 2,746 P35S sequences were retrieved from GenBank database and mapped to our target sequence. At each position of the sequence of interest, mutation frequencies from bibliographic data and from this study are presented in Fig. 3. The frequency of mutations found in the published data does not truly correspond to the mutation rate of the sequence. Indeed this frequency may over represent mutations contained in cloned P35S promoters, which have been extensively sequenced. On the opposite, the data presented by this study truly correspond to the mutation rate in the analyzed samples. Several SNPs were newly identified during this study while some can be retrieved in previously published data. Some of the identified mutated nucleotides are located within the primers and probes used in the P35S diagnostic test, the forward primer being located between nucleotides 28 and 49, the probe between nucleotides 58 and 83 and the reverse primer between nucleotides 85 and 106 (Fig. 2). The mutation frequency (below 0.2%) will likely not influence the efficiency of the P35S real time PCR diagnostic test. Nevertheless, such samples (mixed flour with several GM events) can indicate mutated positions that could be more specifically looked at in flours made with single GM events. Ghedira et al.30 assessed primer/template mismatch effects on real-time PCR amplification for GMO quantification. They showed that a mismatch between the primer and the DNA template can cause partial to complete failure of the amplification of the initial DNA template depending on the type and location of the nucleotide mismatch affecting the estimated total DNA quantity to a varying degree30. Morisset et al. (2009) identified a SNP in the primer binding site of the P35S sequence in maize TC1507, leading to inefficient amplification31. In another study, Lefever et al. (2013) showed that four mismatches in a single primer can block amplification almost completely, whereas three mismatches in one of the primers must be combined with at least two mismatches in the other primer to achieve the same extent of inhibition10.

Mutation frequencies at each position of the P35S sequence in bibliographic data and in this study. In bibliographic data, 2,746 sequences were retrieved from GenBank after blasting our target P35S sequence (nr/nt, megablast, at least 70% coverage and 90% homology). The 2,746 sequences were mapped to our target sequence using Geneious and mutation frequency was calculated at each position of the P35S sequence. In this study, the presented data correspond to the mean mutation frequencies of the 6 natural samples studied.
In addition to an alteration of the real-time PCR diagnostic test, mutated nucleotides in the promoter sequence could potentially influence its activity and consequently the expression rate of the transgene5. Several DNA sequences or motifs are required for its activity32,33,34,35,36. Mutations occurring in essential DNA sequences, such as the boxes for transcription factors CAAT and TATA, could stop or reduce promoter activity, which could potentially lead to the loss of the GM characteristic. Spontaneous mutations can occur in all living organisms during DNA replication, homologous or intrachromosomal recombination, mitosis and meiosis11,12. The mutation rate of maize genes has been estimated at 3 × 10−8 substitutions per site and per generation37. It has been suggested that once integrated into the genome, the rate of mutation of transgenes should be the same as that of internal host genes6,7. Ogasawara et al. (2005) compared the mutation rate of the transgene for RR soybean and an internal host gene, the Cong gene, and obtained almost the same mutation rate. Point mutations such as small deletions or substitutions can also occur as a response of the plants to environmental conditions38.
In this study, the SiMSen-seq methodology was successfully implemented for sensitive mutation detection in P35S sequences in complex GM samples. The method presents a significant improvement to detect variants in GM inserts due to its sensitivity below 0.05% and because it addresses the main limitation of the use of NGS for mutant detection (i.e. amplification and sequencing errors). In addition, the present study looks for the presence of mutants in a population of DNA extracted from complex samples which seems preferable to the analyses of individual seeds one by one. By analyzing flour complex samples, it proves that the methodology works well and several mutated positions were even reported in the P35S sequence. Because the flour samples contained several GM events, it was not possible to associate some SNPs to a particular GM event. It would be interesting to separately analyze flours made by single GM events to investigate if some events show a more abundant mutation rate compared to others.
In conclusion, the present study focused on a portion of the P35S sequence targeted by screening methods used to undertake a first assessment of the GMO status of samples. Its genetic stability is important for the reliable control of GMOs, food labelling and the traceability of GM plants. This approach could be used to explore the mutation rate in other screening GM targets like the nopaline synthase terminator from Agrobacterium tumefaciens (tNOS), the phosphinothricin acetyltransferase gene from Streptomyces hygroscopicus (bar) or the 5-enolpyruvylshikimate-3-phosphate synthase gene from Agrobacterium tumefaciens sp. strain CP4 (ctp2-cp4epsps). In addition, this method could analyze several GM target at the same time due to its multiplexing capacity. Finally, associated with long-read sequencing instruments, this strategy could cover GM inserts of several kilobases and could be implemented in genetic stability studies or could be used to detect single nucleotide mutant GM plants produced using “new breeding techniques”.
Materials and Methods
Materials
P35S oligonucleotides (5′-TCGTGGAAAAAGAAGACGTTCCAACCACGTCTTCAAAGCAAGTGGATTGATGTGATATCTCCACTGACGTAAGGGATGACGCACAATCCCACTATCCTTCGCAAGACCCTTCCTCTATATAAGGAAGTTCAT-3′; 132 bp), modified or not, were custom synthesized by Eurofins (Luxembourg). The P35S-WT oligonucleotide had no SNP. The P35S-C1 oligonucleotide had a class I SNP (A → G) at position 71 and the P35S-C4 oligonucleotide had a class IV SNP (A → T) at position 67. Oligonucleotides were resuspended in TE buffer (Tris, 10 mM; EDTA, 1 mM; pH 8) at 100 pmol/µL. They were diluted in 0.1X TE buffer in order to prepare synthetic samples by mixing P35S-WT, P35S-C1 and P35S-C4 oligonucleotide solutions in different proportions at a final concentration of 160,000 cp/µL. Six synthetic samples were prepared: 10% C1 (10% of the P35S-C1 oligonucleotide mixed with 90% of the P35S-WT oligonucleotide), 1% C1 (1% of the P35S-C1 oligonucleotide mixed with 99% of the P35S-WT oligonucleotide), 0.1% C1 (0.1% of the P35S-C1 oligonucleotide mixed with 99.9% of the P35S-WT oligonucleotide), 10% C4 (10% of the P35S-C4 oligonucleotide mixed with 90% of the P35S-WT oligonucleotide), 1% C4 (1% of the P35S-C4 oligonucleotide mixed with 99% of the P35S-WT oligonucleotide) and 0.1% C4 (0.1% of the P35S-C4 oligonucleotide mixed with 99.9% of the P35S-WT oligonucleotide). Synthetic samples were analyzed to test the efficiency of the technique for detecting known mutations.
Six natural flour samples (NS1, NS2, NS3, NS4, NS5 and NS6) containing several GM events were obtained from the Joint Laboratories Service Unit in Strasbourg (France). These samples tested positive for P35S using real-time PCR (data not shown). Samples 1 to 4 mainly contained maize while samples 5 and 6 mainly contained soybean. These natural flour samples were analyzed to test the efficiency and sensitivity of the technique for detecting unknown mutations.
In the manuscript, natural samples refer to GM flour samples in opposition to synthetic samples that refer to samples prepared by mixing custom synthesized sequences of P35S.
DNA extraction
Genomic DNA was extracted from flour using a cetyltrimethylammonium bromide (CTAB) method. 250 mg of flour were suspended in 1.2 mL of CTAB extraction buffer (CTAB, 20 g/L; NaCl, 1.4 M; Tris-HCl, 0.1 M; Na2EDTA, 20 mM, pH 8.0) preheated to 65 °C. A volume of 10 µL of α-amylase solution (10 mg/mL) and a volume of 10 µL of RNase (10 mg/mL) were added and the mixture was incubated at 65 °C for 30 min, with vortex mixing every 10 min. A volume of 20 µL of proteinase K (20 mg/mL) was added and the mixture was incubated at 65 °C for another 30 min, with vortex mixing every 10 min. After centrifugation at 12,000 g for 10 min, the supernatant was transferred to a new 2 mL microtube with 240 µL of ammonium acetate (10 M) and 600 µL of isopropanol, mixed by inverting the tube and centrifuged at 12,000 g for 15 minutes. The supernatant was discarded and resulting pellets were dissolved in 200 µL of TE buffer. DNA were purified using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions except that they were eluted in 100 µL of Buffer EB. DNA concentrations were measured with the Qubit 2.0 fluorometer (Life Technologies, Carlsbad, United States) and DNA were stored at −20 °C.
Target primer design
The P35S sequences of several GM plants were retrieved from GenBank or the GMDD database. The sequences were aligned using CLUSTALW available in Geneious 11.0.5. Two sets of primers were designed using PRIMER 3 available in Geneious to amplify the sequence of P35S traditionally used in real time PCR analyses39. The primers were tested as previously recommended19. Primers P35S-F2 (5′-TCGTGGAAAAAGAAGACGTTCC-3′) and P35S-R1 (5′-ATGAACTTCCTTATATAGAGGAAGGG-3′) were selected for the following experiments. The PCR product size was 132 bp. Barcode PCR primers (‘barcode forward primer’ and ‘barcode reverse primer’, see19) were ordered (PAGE-purified, Eurogentec, Liège, Belgium) and stock solutions were prepared as described previously19.
Mutation analysis
DNA barcoding was performed by PCR in 10 µL using 1X AccuPrime PCR Buffer II, 0.2 U AccuPrime Taq DNA Polymerase High Fidelity (Invitrogen, Thermo Fisher Scientific, Waltham, United States), 80 nM of each barcode primer, 0.05 mg/mL of BSA and 800,000 copies DNA (synthetic samples), 25,000 copies DNA for flour sample 1, 194,040 copies DNA for flour sample 2, 83,930 copies DNA for flour sample 3, 117,040 copies DNA for flour sample 4, 25,000 copies DNA for flour sample 5 and 25,000 copies DNA for flour sample 6. The temperature profile was 98 °C for 3 min followed by three cycles of amplification (98 °C for 10 sec, 62 °C for 6 min and 72 °C for 30 sec). A volume of 20 µL of protease (30 ng/µL in TE buffer) was added and the mixture was incubated at 65 °C for 15 min and 95 °C for 15 min. The second PCR was performed in 40 µL using 1X Q5 Hot Start High Fidelity Master Mix (New England BioLabs, Ipswich, United States), 400 nM of each Illumina adaptor primer (NEBNext Multiplex Oligo for Illumina, index primers, New England Biolabs) and 10 µL of PCR products from the first PCR. Sequences of Illumina adaptor primer are presented in Stalhberg et al. (2016; 2017). The temperature profile was 95 °C for 3 min followed by 35 cycles of amplification (98 °C for 10 s, 80 °C for 1 s and 72 °C for 1 min). 40 µL (synthetic sample) or 120 µL (flour sample) of PCR products were purified using the Agencourt AMPure XP beads (Beckman Coulter, Brea, United States) according to the manufacturer’s instructions with a volume ratio between beads and PCR products of 1. The purified products were eluted in 20 µL (synthetic sample) or 45 µL (flour sample) of low TE buffer (10 mM Tris, 0.1 mM EDTA, pH 8.0). Validation of the librairies was obtained by capillary electrophoresis using the Fragment Analyzer and the High Sensitivity NGS fragment analysis kit (Advanced Analyticals, Evry, France). The librairies were stored at −20 °C prior to sequencing. Sequencing was performed on a MiSeq instrument (Illumina, San Diego, United States) at GenoBIRD (Nantes, France) using 150 pb paired-end reads. Raw FastQ files were processed using Debarcer, a package for De-Barcoding and Error Correction of sequencing data containing molecular barcodes, previously developed19,20. Raw data were deposited in the Sequence Read Archive under BioProject ID PRJNA475037.
References
ISAAA Briefs. Global Status of Commercialized Biotech/GM Crops in 2017. No. 53 (2017).
Directive 2001/18/EC of the European Parliament and of the Council of 12 March 2001 on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC, http://www.wipo.int/edocs/lexdocs/laws/fr/eu/eu161fr.pdf (2001).
Ben Ali, S. E. et al. Mutation scanning in a single and a stacked genetically modified (GM) event by real-time PCR and high resolution melting (HRM) analysis. International Journal of Molecular Sciences 15, 19898–19923, https://doi.org/10.3390/ijms151119898 (2014).
EFSA Panel on Genetically Modified Organisms. Technical note on the quality of DNA sequencing for the molecular characterisation of genetically modified plants. EFSA Journal 16, 5345, https://doi.org/10.2903/j.efsa.2018.5345 (2018).
Castan, M. et al. Analysis of the genetic stability of event NK603 in stacked corn varieties using high-resolution melting (HRM) analysis and Sanger sequencing. European Food Research and Technology, 1–13, https://doi.org/10.1007/s00217-016-2749-2 (2016).
La Paz, J. L., Pla, M., Papazova, N., Puigdomenech, P. & Vicient, C. M. Stability of the MON 810 transgene in maize. Plant Mol Biol 74, 563–571, https://doi.org/10.1007/s11103-010-9696-2 (2010).
Ogasawara, T. et al. Frequency of Mutations of the Transgene, which might Result in the Loss of the Glyphosate-Tolerant Phenotype, was Lowered in Roundup ReadyⓇ Soybeans. J. Health Sci. 51, 197–201, https://doi.org/10.1248/jhs.51.197 (2005).
Aguilera, M. et al. A Qualitative Approach for the Assessment of the Genetic Stability of the MON 810 Trait in Commercial Seed Maize Varieties. Food Analytical Methods 1, 252–258, https://doi.org/10.1007/s12161-008-9035-2 (2008).
Fraiture, M. A. et al. Current and new approaches in GMO detection: challenges and solutions. BioMed research international 2015, 392872, https://doi.org/10.1155/2015/392872 (2015).
Lefever, S., Pattyn, F., Hellemans, J. & Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem 59, 1470–1480, https://doi.org/10.1373/clinchem.2013.203653 (2013).
Neumann, G., Brandes, C., Joachimsthaler, A. & Hochegger, R. Assessment of the genetic stability of GMOs with a detailed examination of MON810 using Scorpion probes. European Food Research and Technology 233, 19–30, https://doi.org/10.1007/s00217-011-1487-8 (2011).
Ben Ali, S.-E. et al. Genetic and epigenetic characterization of the cry1Ab coding region and its 3′ flanking genomic region in MON810 maize using next-generation sequencing. European Food Research and Technology, https://doi.org/10.1007/s00217-018-3062-z (2018).
Tan, Y.-Y. et al. Development of an HRM-based, safe and high-throughput genotyping system for two low phytic acid mutations in soybean. Molecular Breeding 36, 101, https://doi.org/10.1007/s11032-016-0529-0 (2016).
Christensen, E. et al. Optimized targeted sequencing of cell-free plasma DNA from bladder cancer patients. Scientific Reports 8, https://doi.org/10.1038/s41598-018-20282-8 (2018).
Frawley, T. et al. Development of a Targeted Next-Generation Sequencing Assay to Detect Diagnostically Relevant Mutations of JAK2, CALR, and MPL in Myeloproliferative Neoplasms. Genetic Testing and Molecular Biomarkers 22, 98–103, https://doi.org/10.1089/gtmb.2017.0203 (2018).
Han, X. et al. Gene mutation patterns of Chinese acute myeloid leukemia patients by targeted next-generation sequencing and bioinformatic analysis. Clinica Chimica Acta 479, 25–37, https://doi.org/10.1016/j.cca.2018.01.006 (2018).
Li, S. et al. Targeted Next-Generation Sequencing Reveals Novel RP1 Mutations in Autosomal Recessive Retinitis Pigmentosa. Genetic Testing and Molecular Biomarkers 22, 109–114, https://doi.org/10.1089/gtmb.2017.0223 (2018).
Ihle, M. A. et al. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer 14, https://doi.org/10.1186/1471-2407-14-13 (2014).
Stahlberg, A. et al. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nature Protocols 12, 664–682, https://doi.org/10.1038/nprot.2017.006 (2017).
Stahlberg, A. et al. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Research 44, https://doi.org/10.1093/nar/gkw224 (2016).
Schirmer, M. et al. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Research 43, e37–e37, https://doi.org/10.1093/nar/gku1341 (2015).
Xu, C., Nezami Ranjbar, M. R., Wu, Z., DiCarlo, J. & Wang, Y. Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller. BMC Genomics 18, https://doi.org/10.1186/s12864-016-3425-4 (2017).
Gregory, M. T. et al. Targeted single molecule mutation detection with massively parallel sequencing. Nucleic Acids Research 44, e22–e22, https://doi.org/10.1093/nar/gkv915 (2016).
Kinde, I., Wu, J., Papadopoulos, N., Kinzler, K. W. & Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proceedings of the National Academy of Sciences of the United States of America 108, 9530–9535, https://doi.org/10.1073/pnas.1105422108 (2011).
Kennedy, S. R. et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nature Protocols 9, 2586–2606, https://doi.org/10.1038/nprot.2014.170 (2014).
Schmitt, M. W. et al. Detection of ultra-rare mutations by next-generation sequencing. Proceedings of the National Academy of Sciences 109, 14508 (2012).
Taylor, P. H., Cinquin, A. & Cinquin, O. Quantification of in vivo progenitor mutation accrual with ultra-low error rate and minimal input DNA using SIP-HAVA-seq. Genome Res. 26, 1600–1611, https://doi.org/10.1101/gr.200501.115 (2016).
Fredriksson, N. J. et al. Recurrent promoter mutations in melanoma are defined by an extended context-specific mutational signature. PLoS Genet. 13, e1006773, https://doi.org/10.1371/journal.pgen.1006773 (2017).
Wu, Y. et al. Development of a general method for detection and quantification of the P35S promoter based on assessment of existing methods. Scientific Reports 4, https://doi.org/10.1038/srep07358 (2014).
Ghedira, R. et al. Assessment of Primer/Template Mismatch Effects on Real-Time PCR Amplification of Target Taxa for GMO Quantification. Journal of Agricultural and Food Chemistry 57, 9370–9377, https://doi.org/10.1021/jf901976a (2009).
Morisset, D. et al. Detection of genetically modified organisms–closing the gaps. Nat Biotechnol 27, 700–701, https://doi.org/10.1038/nbt0809-700 (2009).
Lam, E., Benfey, P. N., Gilmartin, P. M., Fang, R. X. & Chua, N. H. Site-specific mutations alter in vitro factor binding and change promoter expression pattern in transgenic plants. Proceedings of the National Academy of Sciences of the United States of America 86, 7890–7894 (1989).
Lam, E. & Chua, N. H. ASF-2: a factor that binds to the cauliflower mosaic virus 35S promoter and a conserved GATA motif in Cab promoters. Plant Cell 1, 1147–1156, https://doi.org/10.1105/tpc.1.12.1147 (1989).
Lam, E. & Chua, N. H. GT-1 binding site confers light responsive expression in transgenic tobacco. Science 248, 471–474 (1990).
Odell, J. T., Nagy, F. & Chua, N. H. Identification of DNA sequences required for activity of the cauliflower mosaic virus 35S promoter. Nature 313, 810–812 (1985).
Bhullar, S. et al. Functional analysis of cauliflower mosaic virus 35S promoter: re-evaluation of the role of subdomains B5, B4 and B2 in promoter activity. Plant Biotechnol J 5, 696–708, https://doi.org/10.1111/j.1467-7652.2007.00274.x (2007).
Clark, R. M., Tavare, S. & Doebley, J. Estimating a nucleotide substitution rate for maize from polymorphism at a major domestication locus. Mol Biol Evol 22, 2304–2312, https://doi.org/10.1093/molbev/msi228 (2005).
Papazova, N. et al. Stability of the T-DNA flanking regions in transgenic Arabidopsis thaliana plants under influence of abiotic stress and cultivation practices. Plant Cell Reports 27, 749–757, https://doi.org/10.1007/s00299-007-0495-4 (2008).
Fernandez, S. et al. Quantification of the 35S promoter in DNA extracts from genetically modified organisms using real-time polymerase chain reaction and specificity assessment on various genetically modified organisms, part I: operating procedure. J AOAC Int 88, 547–557 (2005).
Acknowledgements
The authors thank Benoit Remenant for his help with the bioinformatic analyses.
Author information
Authors and Affiliations
Contributions
A.L.B., A.B., C.D.B. performed the experiments; A.L.B., Y.B., M.R. designed the experiments; A.L.B., A.B., M.R. performed data analysis; A.L.B., M.R. prepared and revised the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boutigny, AL., Barranger, A., De Boisséson, C. et al. Targeted Next Generation Sequencing to study insert stability in genetically modified plants. Sci Rep 9, 2308 (2019). https://doi.org/10.1038/s41598-019-38701-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-38701-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
