
Abstract
The tea green leafhopper, Empoasca (Matsumurasca) onukii Matsuda, is one of the dominant pests in major tea production regions of East Asia. Recent morphological studies have revealed variation in the male genitalic structures within and among populations. However, the genetic structure of this pest remains poorly understood. This study explores the genetic diversity and population structure of this pest in nineteen populations from the four main Chinese tea production areas using microsatellite markers, with one Japanese population also examined. The results show low to moderate levels of genetic differentiation with populations grouped into four clusters, i.e. the Jiangbei group, the Southwest group 1, the Southwest group 2 and the South China group. Populations from China have a close phylogenetic relationship but show significant isolation by distance. Lower genetic diversity and genetic differentiation of E. (M.) onukii were found in the Kagoshima population of Japan. Evidence for genetic bottlenecks was detected in the South China and Jiangnan populations. Population expansion was found in the Southwest, Jiangbei and Kagoshima populations. This is the most extensive study of the population genetics of this species and contributes to our understanding of its origin and evolutionary history.
Similar content being viewed by others
Introduction
The tea green leafhopper is one of the most dominant pests in the major tea production regions of East Asia1,2. It was first described in 1952 as an Empoasca leafhopper injurious to tea shrubs in Japan3. Although considerable economic losses caused by this pest have drawn substantial attention by farmers and government bodies in China since the 1950s, two incorrect scientific names, Empoasca (s. str.) vitis (Goëthe) and Jacobiasca formosana (Paoli) continued to be applied to the pest in mainland China and Taiwan, respectively, in both basic and applied research for many years4,5,6,7. Recent morphological and molecular evidences reveal this pest in China is the same species that occurs on tea in Japan1,8. Thus, the correct scientific name of the tea green leafhopper in China has been established as Empoasca (Matsumurasca) onukii Matsuda1. Damage caused by adults and nymphs of this pest has been described as the classical symptoms of hopperburn in young tea leaves and shoots. This damage results from the leafhoppers’ piercing-sucking mode of feeding9, leading to yield losses averaging 10–50% in China10,11 and up to 33% in Japan12.
Despite previous studies reporting that tea production originated in Southeast China more than three thousand years ago13, tea trees are now widely cultivated in more than 34 countries across Asia, Africa, Latin America, and Oceania14. However, so far, this pest has been officially reported only from three countries (Japan, Vietnam and China) in Asia1,2,15. In China, tea plantations now account for half the overall tea production by area worldwide, and comprise four tea regions: Jiangbei (North Central), Jiangnan (South Central), South China and Southwest China. These regions are designated based on the ecological conditions, the history of tea production, and the distribution and cultivation characteristics of tea varieties16,17.
Recent morphological study has revealed variation in the structure of the male genitalia (aedeagus) within and among populations of the tea green leafhopper1,18. Thus, it is reasonable to speculate that some genetic differentiation has developed, possibly as a result of restriction of gene flow among populations by geographic barriers (e.g., large rivers and mountains) between these four Chinese tea regions. In Japan, the predominant tea cultivar ‘Yabukita’, is cultivated intensively in Shizuoka Prefecture in southern Honshu and Kagoshima Prefecture in Kyoto19, both well separated from the Chinese tea production areas, leading us to expect the genetic characteristics of Japanese E. (M.) onukii to be different from the Chinese populations.
Previous attempts to explore the genetic structure of this pest include one study that compared samples from a small number of Chinese tea gardens using the RAPD method20 and another study that developed a haplotype network based on several geographic populations using mtDNA COI and 16S rRNA sequence data21,22,23,24,25. However, specimen sampling methods in these studies were not consistent, nor were the male genitalic characters studied to confirm the species identification and population-level variation. Unfortunately, given the past confusion over the identity of the species, it is possible that samples used in previous genetic studies have included other species besides E. (M.) onukii, and this could lead to a misunderstanding of Chinese E. (M.) onukii genetic structure. Because almost all Empoasca species (>200 species occur in the Chinese fauna) can be positively identified only by examining the male genitalia and multiple Empoasca species may coexist in the same habitat, it is crucial to confirm the identity of each individual specimen morphologically before it is incorporated into any population genetics study1,2,3.
Recently, 1569 microsatellite loci including 87 different repeat units have been found in the genome of the tea green leafhopper, among which triplet repeats (27.84%) and dinucleotide repeats (70.26%) were the most abundant26. Twenty-one microsatellite markers were previously developed and selected for genetic studies of E. (M.) onukii in Chinese tea production areas27. Here we report the results of a comprehensive population genetic study based on these previously identified markers, incorporating specimens confirmed as E. (M.) onukii based on examination of male genitalia and obtained from all major Chinese tea production areas. Similar analyses have been conducted previously to explore the genetic differentiation, population structure and population dynamics of other species of Hemiptera28,29,30,31,32,33,34,35,36.
The purpose of this study was to (i) assess the genetic differentiation and population dynamics of E. (M.) onukii populations through comparative studies of populations in four tea production regions; (ii) examine effects of geographic isolation on the genetic differentiation and observed population genetic patterns of the tea green leafhopper; and (iii) elucidate the origin and evolutionary history of the species. This study represents the most extensive use of microsatellite markers to elucidate the origin and evolutionary history of E. (M.) onukii.
Results
Genetic diversity
Eo-9 exhibited a significant excess of homozygosity. More than a 50% amplified fragment dropout was found for Eo-1-65 and Eo-9. Eo-29, Eo-37, Eo-54, Eo-1-52, Eo-1-57, Eo-83, Eo-1-5, Eo-F-8 and Eo-1-77 deviated from Hardy-Weinberg equilibrium (pHWE < 0.01) across different populations because of null alleles. However, null allele frequencies, lower than 0.2 for 18 markers, yielded little effect on genetic diversity and population structure, excluding Eo-4-5 (null allele frequency > 0.2 in all populations)37,38. The average frequency of null alleles at 18 markers over the populations ranged from 0.027 to 0.075 (Table 1). So, excluding Eo-1-65, Eo-9 and Eo-4-5, the remaining 18 markers were used for population genetic studies. Genotypic linkage disequilibrium was not detected for any pair of markers in any populations.
The number of alleles for the 18 microsatellites ranged from 2 to 18 over the entire population. Fifteen markers (all except Eo-42, Eo-1-57 and Eo-E-12) were highly polymorphic, with polymorphic information content (PIC) values ranging from 0.515 to 0.89239. At the population level, the mean Na and AR ranged from 8.4 (PE, YT and JJ) to 10.4 (CY) and from 6.52 (JJ) to 8.27 (CY), respectively. The mean number of Ne per marker was 5.2, with the lowest value being 4.2 (JJ) and the highest being 5.9 (SX and CY). Only 69 individuals had one or more private alleles across all markers and they were spread across 17 populations. HO had a mean value of 0.679 and was lowest in the ZY population (0.615) and highest in the RZ population (0.737). The mean HE was 0.741, ranging from 0.665 (JJ) to 0.788 (CY) (Table 1). Although there is no significant difference in global mean AR and mean HE among the four tea production areas (all P > 0.05), the mean AR was significantly different between the PE and the other populations in China (t-test: t = 2.395, d.f. = 17. P = 0.028). Similarly, the mean HE was significantly different (t-test: t = 2.55, d.f. = 17. P = 0.021). The genetic diversity was significantly different between the CY population and the other populations (AR: t-test: t = −2.480, d.f. = 17. P = 0.024; HE: t-test: t = −2.172, d.f. = 17. P = 0.044). Overall genetic diversity was explained by 19 populations from China and one population from Japan also differing significantly in AR and HE (China/Japan; AR: 7.419/6.520, P = 0.031; HE: 0.737/0.665, P = 0.002).
Genetic structure
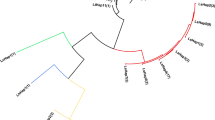
The Bayesian analysis of population structure indicated that 19 populations from China represent four main genetic clusters (K = 4) (Fig. 1). Although ΔK had peaks in K = 2 and K = 4, α was relatively constant at K = 4. Furthermore, individuals were strongly assigned to K = 4. The CX population and PE population remain as a fixed cluster. The numbers of individuals from the CT population assigned to clusters are similar to those of the ZY and CY populations. The XY, RZ, TA and SX populations (all belonging to the Jiangbei tea production area) were assigned to each cluster similarly. The populations from Jiangnan and South China did not show strong population genetic structure.

Structure of E. (M.) onukii populations in China revealed by Bayesian analysis implemented in Structure. Each individual is represented by a vertical bar broken into different colored genetic clusters, with length proportional to probability of assignment to each cluster. Analysis of 19 populations, 570 individuals, with possible numbers of clusters ranging from 2–4, indicated that the most likely number of clusters was 4.
Analysis of molecular variance (AMOVA) indicated that 2.43% of the genetic variation was partitioned among groups (P < 0.001) and 2.14% among populations (P < 0.001) (Table 2). The majority of the genetic variance originated from variation among individuals within populations (P < 0.001). The FST ranged from 0.0012 to 0.0981 among the populations. FST values between the JJ population and the other populations ranged from 0.0267 to 0.0981, revealing moderate differentiation. The largest range of FST was found in Southwest group 2, ranging from 0.0286 to 0.0981 (Supplementary Table S1). The mean FST values show moderate differentiation between Southwest group 2 and the other groups.
The NJT (Fig. 2) based on Nei’s genetic distance clustered the Chinese populations into 4 major groups, consistent with the results of both the Bayesian analysis and AMOVA. The populations from the Southeast tea production area were divided into two groups. The populations from the Jiangnan and South China tea production areas were clustered into one group. Nei’s genetic distances between Chinese populations and the Kagoshima population were 0.119–0.255 (Supplementary Table S2).

Neighbor-joining tree based on Nei’s genetic distances for 20 populations of E. (M.) onukii with allelic frequencies obtained from 18 microsatellite markers. Numbers on nodes represent bootstrap support values (values below 50% not shown). The colors indicate the major clusters inferred by Structure analysis when K = 4.
Similar results were obtained in PCoA for these populations, as shown in Fig. 3. The mean factor scores for the 20 populations are shown in the first two principal component axes, which account for 33.54% and 14.76% of the total variance. The CX population is clearly distant from the PE population. This analysis shows conspicuous divergence of populations in the Jiangnan and South China tea production areas from the other populations.

PCoA at population level generated from 18 microsatellite markers in 20 populations in China and Japan. The first two principle component factors, PC1 and PC2, account for 33.54% and 14.76% of total variance. Colors are coded as in Fig. 2.
Analyses of microsatellite data consistently indicate that Chinese E. (M.) onukii populations have differentiated into four groups: the Jiangbei group (purple populations), the Southwest group 1 (blue populations), the Southwest group 2 (orange populations) and the South China group (yellow populations) (Fig. 2).
Isolation by distance
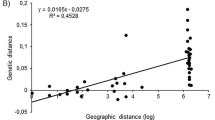
Mantel tests of genetic and geographical distances over all populations revealed that there was a significant correlation between these two variables (RXY = 0.549, R2 = 0.301, P = 0.000, Supplementary Fig. S1a). However, it was not significant when only the Jiangbei populations (RXY = 0.495, R2 = 0.245, P = 0.318) and Southwest populations (RXY = 0.609, R2 = 0.371, P = 0.062) were analyzed. There was a significant correlation when the South China group (including Jiangnan and South China populations) (RXY = 0.539, R2 = 0.291, P = 0.0001, Supplementary Fig. S1b) was analyzed.
Bottleneck analysis
Significant heterozygosity excess was detected in 4 of 20 populations (HS, YT, GL, FZ) under the SMM model. The TPM model revealed significant evidence of heterozygosity excess (P < 0.05) in XY, South China and most of the Jiangnan populations except for the CD (P = 0.054) and JH (P = 0.142) populations, which may indicate a recent genetic bottleneck40 in these populations. In contrast, most of the Jiangbei, Southwest and Kagoshima populations show significant heterozygosity deficiency, indicating that these populations have been expanding40.
Discussion
Our analysis of microsatellite data reveals that genetic differentiation of E. (M.) onukii within a particular region is mainly reflected in the differences among individuals within a population. This is consistent with previous results from AMOVA based on mtDNA COI and 16S rRNA21,22,23,24,25. Analysis of microsatellite markers detected low to moderate levels of genetic differentiation among populations within the main tea growing regions of China, and much more genetic variation within populations (95.43%) than among populations (2.18%). This suggests substantial amounts of gene flow and a more homogeneous gene pool across different geographical populations. Measured genetic and geographical distances are correlated. Within a population, genetic mutation and drift are expected to yield genetic differentiation among individuals, whereas geographic isolation (such as geographic distance and geographic barriers) mainly influences genetic differentiation between populations.
The low to moderate levels of genetic differentiation and subdivision are concordant with geographic distribution. The genetic structure based on 18 microsatellite markers confirms the genetic difference between Southwest groups 1 and 2 in the topographically complex Southwest tea production area, similar to the result obtained previously27. Furthermore, different allelic richness and mean expected heterozygosity were observed in the PE and CY populations of the Southwest tea production area (Tables 1 and 2). There was moderate differentiation between populations of Southwest group 2 and the other populations. Genetic diversity differs significantly between the CY and CT populations and other populations, which are isolated by the Sichuan Basin (AR: t-test: t = −2.944, d.f. = 17. P = 0.009; HE: t-test: t = −2.582, d.f. = 17. P = 0.019). These subdivisions in the Southwest tea production area populations are probably due to different combinations of geographic isolation and climatic variation. Substantial climatic differences between the Southwest tea production area and the other tea production areas and the existence of geographic barriers in this area, such as mountain ranges, basins and large rivers, may account for the genetic differentiation among E. (M.) onukii populations that have limited dispersal capacity22,23.
Fossil evidence suggests that tea trees originated 60–70 Mya (million years ago) in the Paleogene of the Yunnan-Guizhou plateau41. Terpene index analysis has shown that the original tea cultivars are from Yunnan Province42. This Southwest tea production area was previously suggested as the area in which tea was first cultivated. The “original” haplotypes of 16S rRNA of E. (M.) onukii (based on a median joining haplotype network) were obtained from Yunnan and Sichuan populations22, suggesting that the tea green leafhopper expanded its range from the Southwest tea production area into other areas, following the spread of tea cultivation. Based on this evidence, as well as the genetic diversity and structure inferred in this study, we propose the following scenario for the spread of E. (M.) onukii populations in Chinese tea production areas: (1) the tea green leafhopper spread from Yunnan to Guangxi and Guangdong, and subsequently to Jiangnan and the South China tea production areas; (2) this pest then expanded to the Jiangbei tea production area from Yunnan through Shaanxi; and (3) the leafhopper was introduced to Jiangnan and the South China tea production areas from Yunnan through Guizhou, Chongqing and Sichuan provinces. The genetic pattern of E. (M.) onukii is similar to the historical propagation path of tea in China42. Low genetic diversity and moderate genetic differentiation of E. (M.) onukii observed in the Kagoshima population of Japan may have resulted from a bottleneck caused by anthropogenic transport of tea plants, geographical isolation and restricted genetic resources43,44. Based on Nei’s genetic identity >0.85 the Kagoshima population is closest to the populations in Jiangnan and South China. Similar tea germplasms in these populations also provide similar biotopes for this pest45.
This study suggests that different populations of E. (M.) onukii are currently undergoing population expansion in the South China and Jiangnan tea production areas but are experiencing bottlenecks in other tea production areas. Mating interference and trapping of males may be used to keep E. (M.) onukii from spreading into surrounding tea production areas beyond Kagoshima, Southwest China and most of the Jiangbei populations. The South China and most of the Jiangnan populations, may show the effect of a recent genetic bottleneck, possibly resulting from heavy use of chemical pesticides. In these regions, only a few resistant individuals may have survived, leading to the genetic bottleneck effects, similar to those previously reported for Bemisia tabaci and Myzus persicae46,47. In these cases, pesticides may have stimulated the reproduction and expansion of pest populations, promoting gene exchange and homogenizing the genetic structure48. Integrative pest management incorporating biocontrol or organic production methods may be needed to build ecologically sustainable tea plantations in these areas49,50.
Microsatellite markers revealed lower genetic differentiation between Sichuan and Yunnan populations than was shown in a previous study on this same species in the same region27. This may be because the present study used samples obtained from multiple localities in Sichuan. To provide more robust results, future studies should employ larger numbers of genetic markers and more individuals within the same locations to more fully represent the diversity of local E. (M.) onukii populations. The area of origin of E. (M.) onukii should be further explored by more intensive sampling of populations in the Southwest tea production area, where the level of genetic differentiation is highest. The dispersal routes of E. (M.) onukii among Chinese tea production areas should also be analyzed in more detail using more genetic loci (such as SNPs). Use of SNPs that have a high abundance throughout the genome, predictable mutation modes and low back mutation rates51,52 could reduce error in estimation of population parameters (such as FST and migration rates) compared to our use of microsatellite markers.
This study found low to moderate levels of genetic differentiation and segregation in E. (M.) onukii populations among the main Chinese tea production areas. Populations of this pest have differentiated into four groups: the Jiangbei group, the Southwest group 1, the Southwest group 2 and the South China group. Low genetic diversity and moderate genetic differentiation of E. (M.) onukii were found in the Kagoshima population of Japan. The observed patterns may be attributable to geographic isolation, differences in feeding preference among tea germplasms and the use of pesticides in tea production areas. Additional study is needed to elucidate the relationship between selective factors (such as tea germplasm resources and geographical conditions) and genetic structure.
Materials and Methods
Sampling
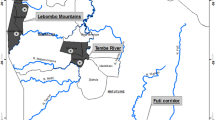
E. (M.) onukii were collected by sweep net from 19 localities in four tea production areas of China and one location in Japan, representing a total of 20 geographically delimited populations (Fig. 4 and Table 3). In order to reduce the likelihood of sample contamination from non-tea plants, specimens obtained in each tea garden were collected from five different sites (approximately five square meters per site) selected at random near the middle of each plantation. The net was swept across the top of the tea plants at each site and the collected specimens were placed in five separate vials. All specimens were preserved in absolute alcohol at −20 °C until they were identified and used for genotyping. The rest of the specimens are now deposited in the Entomological Museum, Northwest A&F University, Yangling, China (NWAFU).

Geographic distribution and Bayesian model-based cluster analysis of E. (M.) onukii. Population codes are listed in Table 3; the pie chart in each population represents the proportion of individuals from four clusters inferred by Structure analysis. SimpleMappr was used to produce a distribution map based on the geographical coordinates in Table 3. URL: http://www.simplemappr.net/#tabs=0.
Only male specimens representing each population were selected for this study because of the inability to identify females using morphological characters1. All were identified in advance in the laboratory by the first author based on morphological characters diagnostic for the species1.
DNA extraction and microsatellite genotyping
Total genomic DNA was extracted respectively from 30 male individuals per population (except for genital segments used for species identification) using the DNA Easy Blood and Tissue Kit (Qiagen, Hilden, Germany) or a modified CTAB method53. DNA concentration was measured using an ND-1000 spectrophotometer (Bio-Rad, Hercules, CA, USA). Eighteen microsatellite markers (Supplementary Table S3) out of the 21 originally characterized by Zhang et al.27 were labeled with a fluorescent dye (HEX, TAMRA or FAM). PCR protocol was performed as described by Zhang et al.27. PCR products were run by automated capillary electrophoresis on an ABI 3130xl (Applied Biosystems, Foster, CA, USA) genetic analyzer. Allele sizes were scored against a GeneScanTM 500LIZ standard using GeneMapper v4.0 (Applied Biosystems, Foster, CA, USA).
Genetic diversity analysis
FreeNA was used to evaluate the frequency of null alleles (Fnull)37. Allele frequency, observed and expected heterozygosity were calculated with Genepop v.4.2 for each marker by the allele sizes from each geographical population. Deviations from Hardy-Weinberg equilibrium (HWE) were tested with 1000 permutations across markers using Genepop v.4.254. Linkage disequilibrium (LD) for each pair of markers in the populations was assessed with Genepop v.4.2. Statistical significance (P) was corrected for multiple comparisons by applying the strict Bonferroni correction55 with the same software.
Mean observed (HO) and expected heterozygosity (HE), mean effective number of alleles per marker (Ne), mean number of alleles (Na) per marker and polymorphism information content (PIC) were calculated using GenALEx v. 6.556 and Cervus 2.057. The allelic richness (AR) was estimated using a minimum sample size of 26 diploid individuals in HP-Rare v1.058. Statistical significance of the genetic diversity indices between different populations was assessed using the independent samples t-test in SPSS Statistics 20 (IBM).
Genetic structure analysis
Bayesian analysis, Analysis of Molecular Variance (AMOVA), phylogenetic analysis using the Neighbor Joining (NJ) approach and Principal Coordinate Analysis (PCoA) were used to analyze the genetic structure of the tea green leafhopper. First, Bayesian model-based clustering analysis implemented in Structure v2.3.4 was used to estimate the number of genetically distinct clusters (K) among the Chinese populations using an admixture ancestry model and correlated allele frequencies. The range of possible clusters was set from 1 to 10, with 20 independent runs for each K. Analysis parameters included a burn-in period of 50,000 iterations followed by 1,000,000 Markov Chain Monte Carlo (MCMC) repetitions. The most likely number of genetic clusters (K) in the Chinese populations was estimated by posterior probability of the data (P (K/X)), the Dirichlet parameter Alpha (α) and the ad hoc statistic (ΔK)59,60,61,62. The output from Structure was visualized in DISTRUCT version 1.163. Second, an analysis of molecular variance (AMOVA) was performed using Arlequin 3.11 to estimate and compare the percentage of genetic variation explained by different hierarchical levels: individual (within populations, i.e., among leafhoppers in the same population), populations (i.e., among populations in the same tea production areas) and groups (i.e. Jiangbei group included XY, RZ, TA and SX populations; Southwest group 1 included CT, ZY and CY populations; Southwest group 2 included CX and PE populations; South China group included JH, HZ, HS, NC, YT, CD, YD, GL, BS and FZ populations; Japan group included JJ population). Arlequin 3.11 with 1000 permutations was also used to calculate pairwise estimates of FST and the significance (P) for each population64. The levels of differentiation between populations were measured based on FST65. Third, the genetic distances between populations were measured by DA distance in POPTREE266,67. A neighbor-joining tree (NJT) was constructed with POPTREE2 based on DA distance and 1000 replications. Finally, a principal coordinate analysis (PCoA) was carried out using GenALEX 6.502 based on Phi-st distances (GD) of populations56.
Isolation by distance
Isolation by distance was analyzed in GenALEX 6.502 to detect the relationship between genetic and geographic distances56. Geographic distance was defined as the logarithms of the geographical distance between all pairs of population locations, and genetic distance was defined by pairwise population estimates of linearized FST/(1 − FST). The correlation between the two parameter matrices was regressed using a Mantel test, with the significance estimated by P value, and the regression coefficient (R) and the mean correlation coefficient (RXY) performed over 999 random permutations.
Bottleneck analysis
Bottleneck version 1.2.0240 was used to test whether the populations experienced a recent bottleneck or expansion. The observed heterozygosity was compared with the expected heterozygosity under a two-phase mutation model (TPM) recommended for microsatellite data68 and the step-wise-mutation model (SMM) (TPM = 95% SMM; a variance among multiple steps of 12). The significance of excess heterozygosity was assessed using a Wilcoxon test (5000 iterations).
Data Availability
All data generated or analysed during current study are available within the published article (and its Supplementary Information files).
References
Qin, D. Z., Zhang, L., Xiao, Q., Dietrich, C. H. & Matsumura, M. Clarification of the identity of the tea green leafhopper based on morphological comparison between Chinese and Japanese specimens. PLoS One 10, e0139202 (2015).
Dworakowska, I. Empoascini of Japan, Korea and north-east part of China (Homoptera, Auchenorrhyncha, Cicadellidae, Typhlocybinae). Reichenbachia 20, 33–57 (1982).
Matsuda, R. A new green Empoasca leafhopper injurious to the tea shrub in Japan. Oyo-Kontyu Tokyo 8, 19–21 (1952).
Lv, W. M., Chen, X. & Luo, Q. R. Research on occurrence and control of Empoasca flavescens. Journal of Tea Science 45–55 (1964).
Kuoh, C. L. & Zhang, H. G. Research on the Cicadellidae species damaging Chinese tea (I). Journal of Tea Business 15–18 (1988).
Zhao, D. X., Chen, Z. M. & Cheng, J. A. Belongingness of tea leafhopper dominant species. Journal of Tea Science 20, 101–104 (2000).
Fu, J. Y. & Han, B. Y. A molecular analysis on genetic relationships among individuals of tea leafhopper. Bulletin of Science and Technology 21(549–552), 556 (2005).
Fu, J. Y., Han, B. Y. & Xiao, Q. Mitochondrial COI and 16sRNA evidence for a single species hypothesis of E. vitis, J. formosana and E. onukii in East Asia. PLoS One 9, e115259 (2014).
Backus, E. A., Serranom, M. S. & Ranger, C. M. Mechanisms of hopperburn: an overview of insect taxonomy, behavior and physiology. Annu. Rev. Entomol. 50, 125–151 (2005).
Tang, H., Tang, J. C., Li, J. L. & Wu, L. R. Overview of nuisance free control technology against tea green leafhopper Empoasca flavescens Fabricius. Guangdong Agricultural Sciences 38(92–94), 106 (2011).
Xiao, Q. Nuisanceless control techniques against tea leafhopper pest, Empoasca vitis. China. Tea 30, 25 (2008).
Yorozuya, H. Monitoring and characterization of DC electrical penetration graph waveforms of tea green leafhopper, Empoasca onukii, on tea plants. Entomol. Sci. 19, 401–409 (2016).
Van der Wal, S. Sustainability issues in the tea sector, a comparative analysis of six leading producing countries (ed. Turner, J.) 12–17 (Amsterdam, 2008).
Lakshmi, K. H., Bhuyan, M. & Budhindra, N. H. Insect pests of tea and their management. Annu. Rev. Entomol. 54, 267–284 (2009).
Dworakowska, I. Dayus takagii sp. n. and some other Empoascini (Auchenorrhyncha: Cicadellidae: Typhlocybinae). Bulletin de l’Académie Polonaise des Sciences Série des Sciences Biologiques 19, 501–509 (1971).
Tea Research Institute, Chinese Academy of Agricultural Sciences. Chinese Tea Cultivation (1st ed.) 38–48 (Shanghai, 1986).
Mu, D. et al. Behavioral responses for evaluating the attractiveness of specific tea shoot volatiles to the tea green leafhopper. Empoasca vitis. Insect Sci. 19, 229–238 (2012).
Qin, D. Z., Xiao, Q., Wang, Y. C., Qiao, L., & Zhang, L. Revision of green leafhopper species damaging tea shrub in Shaanxi and reconsideration of the species in China. Journal of Northwest A & F University (Nat. Sci. Ed.) 42, 124–134, 140 (2014).
Nakai, M. Biological control of Tortricidae in tea fields in Japan using insect viruses and parasitoids. Virologica Sinica 24, 323–332 (2009).
Fu, J. Y. & Han, B. Y. Studies on genetic relationships among populations of Empoasca vitis (Gothe) from tea gardens in seven provinces based on RAPD analysis. Acta Agriculturae Zhejiangensis 19, 11–14 (2007).
Li, L., Fu, J. Y. & Xiao, Q. Sequence analysis of the mtDNA gene and genetic differentiation in geographic populations of Empoasca vitis. J. Appl. Entomol. 50, 675–685 (2013).
Chen, S. C. et al. Genetic differentiation of Empoasca vitis (Göthe) (Hemiptera: Cicadellidae) among eleven populations based on 16S rRNA sequence. Southwest China Journal of Agricultural Sciences 28, 584–590 (2015).
Zhou, N. N. et al. Genetic variation of Empoasca vitis (Göthe) (Hemiptera: Cicadellidae) among different geographical populations based on mtDNA COI complete sequence. Acta Ecologica Sinica 34, 6879–6889 (2014).
Fu, J. Y., Li, L., Yuan, Z. J., Tang, M. J. & Xiao, Q. Molecular phylogenetic analysis of tea green leafhopper (Empoasca vitis) based on mtDNA COI sequences. Journal of Tea Science 34, 601–608 (2014).
Zhang, Y. J. et al. Sequence analysis of mtDNA gene and genetic differentiation in geographic populations of Empoasca onukii in southestwest (Guizhou Province) of China International. Int. J. Agric. Biol. 20, 404–414 (2018).
Li, Q., Chen, X. X. & Han, B. Y. Bioinformatic analysis of microsatellites in the tea green leafhopper, Empoasca vitis (Hemiptera: Cicadellidae). Acta Entomologica Sinica 54, 421–426 (2016).
Zhang, L., Dietrich, C. H. & Qin, D. Z. Microsatellite markers from tea green leafhopper Empoasca (Matsumurasca) onukii: a powerful tool for studying genetic structure in tea plantations. BMC Genet. 17, 112–121 (2016).
Papura, D. et al. Ten polymorphic microsatellite markers for Scaphoideus titanus, the vector of flavescence dorée phytoplasma. Mol. Ecol. Notes 6, 1114–1116 (2006).
Papura, D. et al. Isolation and characterization of polymorphic microsatellite loci in the green leafhopper Empoasca vitis Goethe (Homoptera). Mol. Ecol. Resour. 9, 827–829 (2009).
Papura, D. et al. Microsatellite and mitochondrial data provide evidence for a single major introduction for the Neartic leafhopper Scaphoideus titanusin Europe. PLoS One 7, e36882 (2012).
Shabani, M. et al. Population genetic structure and ecological niche modelling of the leafhopper Hishimonus phycitis. J. Pest Sci. 86, 173–183 (2012).
Shabani, M. et al. Isolation and characterization of novel microsatellite markers from the leafhopper Hishimonus phycitis distant (Hemiptera: Cicadellidae). Conserv. Genet. Resour. 3, 493–495 (2011).
Martinez, A. J., Ritter, S. G., Doremus, M. R., Russell, J. A. & Oliver, K. M. Aphid-encoded variability in susceptibility to a parasitoid. BMC Evol. Biol. 14, 127–136 (2014).
Park, M., Kim, K. S. & Lee, J. H. Genetic structure of Lycorma delicatula (Hemiptera: Fulgoridae) populations in Korea: implication for invasion processes in heterogeneous landscapes. B. Entomol. Res. 103, 414–424 (2013).
Tang, X. T., Tao, H. H. & Du, Y. Z. Microsatellite-based analysis of the genetic structure and diversity of Aleurocanthus spiniferus (Hemiptera: Aleyrodidae) from tea plants in China. Gene 560, 107–113 (2015).
Zhu, J. Y., Ze, S. Z., Cao, L. J. & Yang, B. Development of microsatellite markers for the plant bug, Pachypeltis micranthus (Hemiptera: Miridae). Appl. Entomol. Zool. 51, 327–331 (2016).
Chapuis, M. P. & Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631 (2007).
Dakin, E. E. & Avise, J. C. Microsatellite null alleles in parentage analysis. Heredity 93, 504–509 (2004).
Botstein, D., White, R. L., Skolnick, M. & Davis, R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331 (1980).
Cornuet, J. M. & Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014 (1996).
Chen, Z. Guizhou tea tree species resources and tea industrial economic development analysis. Seed 28, 74–76 (2009).
Tadakazu, T. et al. One speculation on the origin and dispersion of tea plant in China — One speculation based on the chemotaxonomy by using the contest-ration of terpene-alcohols found in the aroma composition. Journal of Tea Science 12, 81–86 (1992).
Kaundun, S. S., Zhyvoloup, A. & Park, Y. G. Evaluation of the genetic diversity among elite tea (Camellia sinensis var. sinensis) accessions using RAPD markers. Euphytica 115, 7–16 (2000).
Wachira, F. N., Tanaka, J. & Takeda, Y. Genetic variation and differentiation in tea (Camellia sinensis) germplasm revealed by RAPD and AFLP variation. J. Hortic. Sci. Biotech. 76, 557–563 (2001).
Yao, M. Z., Chen, L. & Liang, R. L. Genetic diversity among tea cultivars from China, Japan and Kenya revealed by ISSR markers and its implication for parental selection in tea breeding programmes. Plant Breeding 127, 166–172 (2008).
Chu, D. et al. Evidence for rapid spatiotemporal changes in genetic structure of an alien whitefly during initial invasion. Sci. Rep. 4, 4396 (2014).
Fenton, B., Margaritopoulos, J., Malloch, G. & Foster, S. Micro-evolutionary change in relation to insecticide resistance in the peach–potato aphid. Myzus persicae. Ecol. Entomol. 35, 131–146 (2010).
Franck, P., Reyes, P. F. M., Olivares, J. & Sauphanor, B. Genetic architecture in codling moth populations: comparison between microsatellite and insecticide resistance markers. Mol. Ecol. 16, 3554–3564 (2007).
Zhang, J. D., Tu, X. L., Mao, Y. X., Pan, G. X. & Cheng, F. Y. Recent advances on Empoasca (Matsumurasca) onukii Matsuda in tea plantation in China. Hubei Agricultural Sciences 56(2204–2208), 2211 (2017).
Zhang, Y. J., Chen, W. L., Yang, L. & Chen, X. S. Pesticides in chemical control and resistance development of Empoasca onukii. Agrochemicals 56, 239–245 (2017).
Coates, B. S. et al. Comparative performance of single nucleotide polymorphism and microsatellite markers for population genetic analysis. Journal of Heredity 100, 556–564 (2009).
Morin, P. A., Luikart, G. & Wayne, R. K. SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 19, 208–216 (2004).
Marzachì, C., Veratti, F. & Bosco, D. Direct PCR detection of phytoplasmas in experimentally infected insects. Ann. Appl. Biol. 133, 45–54 (1998).
Rousset, F. Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106 (2008).
Rice, W. R. Analyzing tables of statistical tests. Evolution 43, 223–225 (1989).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research─an update. Bioinformatics 28, 2537–2539 (2012).
Marshall, T. C., Slate, J., Kruuk, L. E. B. & Pemberton, J. M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 7, 639–655 (1998).
Kalinowski, S. T. HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 5, 187–189 (2005).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Pritchard, J. K. Wen, X. Q. & Falush, D. Documentation for STRUCTURE software: Version 2. 3. (Chicago, USA, University of Chicago, 2010).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. & von Holdt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Rosenberg, N. A. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138 (2004).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Takezaki, N., Nei, M. & Tamura, K. POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with windows interface. Mol. Biol. Evol. 27, 747–752 (2010).
Nei, M., Tajima, F. & Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 19, 153–170 (1983).
Piry, S., Luikart, G. & Cornuet, J. M. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 90, 502–503 (1999).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31270689) and the Natural Science Foundation of Shaanxi Province (No. 2018JM3009). We thank Prof. John Richard Schrock (Emporia State University, USA) and anonymous reviewers for reviewing this manuscript and providing valuable comments.
Author information
Authors and Affiliations
Contributions
D.Z.Q. and L.Z. conceived and designed the experiments. F.P.W., L.Q. and M.M. helped to collect the specimens used in this study. L.Z. and F.P.W. performed the experiments. L.Z., F.P.W. and D.Z.Q. participated in data analysis. D.Z.Q., L.Z. and C.H.D. drafted the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Wang, F., Qiao, L. et al. Population structure and genetic differentiation of tea green leafhopper, Empoasca (Matsumurasca) onukii, in China based on microsatellite markers. Sci Rep 9, 1202 (2019). https://doi.org/10.1038/s41598-018-37881-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37881-0
This article is cited by
-
Conservation concerns associated with low genetic diversity for K’gari–Fraser Island dingoes
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
