
Abstract
The emergence of multi-drug resistant bacteria forces the therapeutic world into a position, where the development of new and alternative kind of antibiotics is highly important. Herein, we report the development of triazine-based amphiphilic small molecular antibacterial agents as mimics of lysine- and arginine-based cationic peptide antibiotics (CPAs). These compounds were screened against a panel of both Gram-positive and Gram-negative bacterial strains. Further, anti-inflammatory evaluation of these compounds led to the identification of four efficient compounds, DG-5, DG-6, DL-5, and DL-6. These compounds displayed significant potency against drug-resistant bacteria, including methicillin-resistant S. aureus (MRSA), multidrug-resistant P. aeruginosa (MDRPA), and vancomycin-resistant E. faecium (VREF). Mechanistic studies, including cytoplasmic membrane depolarization, confocal imaging and flow cytometry suggest that DG-5, DG-6, and DL-5 kill bacteria by targeting bacterial membrane, while DL-6 follows intracellular targeting mechanism. We also demonstrate that these molecules have therapeutic potential by showing the efficiency of DG-5 in preventing the lung inflammation of lipopolysaccharide (LPS)-induced acute lung injury (ALI) mouse model. More interestingly, DL-6 exhibited impressive potency on atopic dermatitis (AD)-like skin lesions in BALB/c mice model by suppressing pro-inflammatory cytokines. Collectively, these results suggest that they can serve a new class of antimicrobial, anti-inflammatory and anti-atopic agents with promising therapeutic potential.
Similar content being viewed by others

Introduction
Since the discovery of penicillin in the 1920s, a number of antibiotics have been introduced in the market to treat infectious diseases. However, in recent decades, pharmaceutical companies limited their investments for the discovery of new antibiotics because of reduced profits and short life-span of antibiotics1. Still, the frequent and excessive misuse of antibiotics leads to the inexorable emergence of drug-resistant pathogens, namely superbugs that are a serious and imminent threat to antibiotic therapy2. Moreover, the accompanying rise of antibiotic resistance is considered a serious and concerning challenge for the pharmaceutical industries, country leaders, and the public because of the social and economic burden imposed by it in treating diseases. Thus, a rapid increase in multidrug-resistant (MDR) pathogens and an apparent dearth of a new class of antibiotics necessitate the discovery of alternative and potent antimicrobial agents3,4,5. In this regard, cationic peptide antibiotics (CPAs), also known as cationic antimicrobial peptides, find importance as a new generation of antibiotics in treating drug-resistant pathogens owing to their distinctive mode of action and rapid killing rate6,7. CPAs have drawn remarkable attention because they are widely found in species ranging from bacteria to mammals and exhibit potent antimicrobial activities8,9. They exist in both α-helical and β-sheet forms, which exhibit amphiphilic conformations by presenting the cationic groups on one side and hydrophobic groups on the other side of the molecules that result in cationic/hydrophobic segregations10,11. They exhibit selectivity to prokaryotic cells over eukaryotic cells because CPAs act on the negatively charged bacterial membrane by the effective association of their positive groups in the amphiphilic structure and also by forming stable pores, thus disrupting the membrane12. Consequently, the bacterial killing is very rapid and bacterial resistance is limited; they can also permeate the outer membranes of Gram-negative bacteria, and exhibit a broad spectrum of antibacterial activity13. Despite CPAs being an alternative for treating drug-resistant bacteria, their transformation into molecules of therapeutic use is a highly daunting task owing to several disadvantages, including their large size, hemolytic activity, proteolytic instability, poor salt resistance, and cytotoxicity14. Hence, amphiphilic small molecules containing a lipophilic groups and cationic groups exhibiting controlled hemolytic activity are highly imperative for the discovery of new antibiotics and to overcome peptide-associated disadvantages, including proteolytic instability and poor cell permeability15,16. Thus, small molecules with structural features of CPAs are considered as a prominent strategy that may help in the discovery of small-molecule antibacterials. To accomplish this, we synthesized s-triazine-based cationic amphipathic small molecules comprising cationic groups including amines and guanidyl groups as mimetics of lysine and arginine-containing CPAs, and hydrophobic residues, yielding amphiphilic small molecules containing both hydrophobic and hydrophilic groups.
Over the years, synthesis of s-triazine-based heterocycles have received considerable interest in the field of medicinal chemistry, due to their remarkable pharmacological properties and easy access to array the structural diversity. s-Triazine derivatives were found to exhibit diverse biological properties such as anticancer17, antiviral18, antiprotozoal19, antimalarial20, estrogen receptor modulatory21, cyclin-dependent kinase inhibitory22, and antifungal activities23. Moreover, s-triazine derivatives were found to show antibacterial activities24,25; however, to the best of our knowledge, a detailed mechanistic study on CPA-mimicking amphipathic triazine analogues with profound anti-inflammatory and anti-atopic properties has not been reported yet. The present work stems as a part of our research program embarked on the discovery of new antibacterial agents26,27,28.
In this work, we have designed and synthesized a series of triazine-based small-molecule antibacterials containing a norspermidine (1), which is abundantly available and inexpensive, and has at least two positive charges. Moreover, both mono and bis(alkyls/alkylaryls) were anchored to achieve hydrophobicity. The resultant amphipathic structure provides insights into opportunities for finding the key factors, which are essential for displaying antibacterial activity. These small molecules were assayed for their antibacterial activity against both Gram-positive and Gram-negative bacterial strains. With an aim of identifying the most promising compound as a potential therapeutic lead, the compounds with the highest antibacterial activity and low hemolytic activity, including DL-5, DL-6, DG-5, and DG-6, were evaluated for their activity against MRSA, MDRPA, and VREF. Moreover, most of the compounds were examined for their cytotoxicity using sheep red blood cells (sRBCs) and RAW 264.7 cells. In order to understand the bacterial killing mechanism and to identify the effect of these compounds on the integrity of the bacterial membranes and intracellular region, we performed cytoplasmic membrane depolarization experiments and confocal imaging studies. We investigated the inhibition potency of DG-5, DG-6, DL-5, and DL-6 in LPS-induced inflammatory responses in vitro. In addition, the in vivo activity of DG-5 against LPS-induced inflammatory responses in a mouse model of LPS-induced ALI (acute lung injury) was evaluated. Finally, inspired by the proficient anti-inflammatory effect of DL-6, our synthesized small-molecule antibacterial compound, DL-6 has been investigated for its effect on AD-like skin lesions. Its underlying mechanisms of action was studied using BALB/c model by measuring the ear thickness in mice with AD-like skin lesions and analyzing the histopathological changes including mast cell count and cytokine expression in ear tissue.
Results and Discussion
Synthesis of triazine-based antibacterials
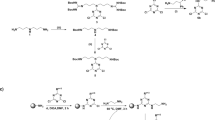
It is reported that the 2,4,6-positioned chlorine atoms in 2,4,6-trichloro-1,3,5-triazine, i.e., cyanuric chloride, are more susceptible to the nucleophilic substitution reactions, which offers the synthetic accessibility to substitute diverse nucleophiles sequentially by controlling the temperature gradient to achieve diversity in the structure25. To synthesize a series of compounds to perform the structure activity relationship study, we introduced norspermidine derivatives comprising amine or guanidyl groups and various alkyl/arylalkyl groups into s-triazine by replacing the chlorine atoms in 2,4,6-trichloro-1,3,5-triazine using the variable temperature strategy. Initially, the guanidinylation at the terminal amines of norspermidine (1) was carried out using N,N′-di-Boc-N″-trifylguanidine (2) to achieve 3, which was successfully adopted for the nucleophilic substitution reaction with 2,4,6-trichloro-1,3,5-triazine (4) in basic condition to result in the formation of 5 as delineated in Fig. 1. In order to incorporate the hydrophobic groups, various amines were treated with 5 in the presence of base at room temperature to achieve (6). Finally, the multiple Boc groups on the guanidyl groups were successfully removed using TFA and subsequent reverse phase HPLC purification resulted in the formation of MG-(1–6).

Synthesis of MG-(1–6), reagents and conditions: (i) N,N′-di-Boc-N″-trifylguanidine (2), TEA, DCM, rt, 6 h, 68%; (ii) cyanuric chloride (4), DIEA, DCM, 0 °C, 4 h, 97%; (iii) R-NH2, DIEA, DCM, rt, 4 h; (iv) TFA/DCM (3:1), 0 °C - rt, 3 h.
Then, to diversify the analogs to perform the SAR studies, the DL and DG series of compounds were synthesized as shown in Fig. 2. We began our synthesis with the selective Boc protection at the primary amines of norspermidine (1) to obtain 7. Treatment of 7 with cyanuric chloride in dichloromethane (DCM) in the presence of diisopropylethylamine (DIEA) at low temperature resulted in the formation of 8. In order to introduce various hydrophobic groups at the expense of replacing two chlorine atoms by nucleophilic substitution reactions, various amines were treated with 8 in the presence of DIEA in 1,4-dioxane at reflux conditions, which resulted in the formation of various derivatives, 9(a–f). Finally, treatment of 9(a–f) with TFA in DCM yielded the desired compounds, DL-(1–6). Next, in view of introducing guanidine groups to attain the protected arginine mimic 10(a–f), the series of compounds 9(a–f) was treated with TFA in DCM, resulting in the deprotection of two Boc groups and subsequent treatment of triethylamine and N,N′-di-Boc-N″-trifylguanidine resulted in the formation of compounds, 10(a–f) with appreciable yields29. Finally, treatment with TFA in DCM followed by reverse phase HPLC purification yielded DG-(1–6).

Synthesis of DG-(1–6)/DL-(1–6), reagents and conditions: (i) Boc-ON, DIEA, THF, 0 °C - rt, 21 h, 80%; (ii) cyanuric chloride (4), DIEA, DCM, 0 °C, 3 h, 87%; (iii) R-NH2, DIEA, 1,4–dioxane, reflux, 14 h; (iv) TFA/DCM (3:1), 0 °C - rt, 3 h; (v) (a) TFA/DCM (3:1), 0 °C - rt, 3 h, (b) N,N′-di-Boc-N′-trifylguanidine (2), TEA, DCM, rt; (vi) TFA/DCM (3:1), 0 oC - rt, 3 h.
Structure-antibacterial activity study
The antibacterial activity of the MG, DG, and DL series compounds was investigated and their minimum inhibitory concentration (MIC) values determined using a panel of four organisms representative of both two Gram-negative bacteria [(E. coli (KCTC 1682) and P. aeruginosa (KCTC 1637)], and two Gram-positive bacteria [(S. epidermidis (KCTC 1917) and S. aureus (KCTC 1621)].
We began our structure activity study by synthesizing the MG series of compounds in the first phase of derivatization as shown Table 1. Initially, we introduced two hydrophobic residues including a linear alkyl (n-butyl (MG-1)) and a cyclic alkyl (cyclohexyl (MG-2)) residue by replacing a chlorine atom in the core residue of 5. Unfortunately, the MIC was not appreciable against any bacterial strain except S. aureus, where both compounds showed minimal activity. These results suggest that both compounds lacked hydrophobicity. In order to increase the hydrophobicity, ethylphenyl and butylphenyl were incorporated, which yielded MG-3 and MG-4, respectively. Interestingly, MG-3 and MG-4 showed improved results against P. aeruginosa and S. aureus. In particular, MG-4 showed better results against P. aeruginosa, S. epidermidis, and S. aureus, compared to MG-3, which demonstrated that further increase in hydrophobicity would result in a rise in antibacterial activity. To verify this hypothesis, we further enhanced the hydrophobicity by introducing naphthyl methyl (MG-5) and 3,3-diphenylpropyl (MG-6) residues. Although MG-5 did not show an appreciable antibacterial activity, MG-6 displayed better activity against almost all the strains compared to the rest of the compounds in the MG series. Collectively, the antibacterial effect of these compounds followed an order of 3,3-diphenylpropyl (MG-6) > butylphenyl (MG-4) > ethylphenyl (MG-3) > naphthylmethyl (MG-5) > cyclohexyl (MG-2) and n-butyl (MG-1) value.
Second phase derivatization
It is pertinent to note that the balance between the charge density and the hydrophobicity is one of the key determinants of the antibacterial activity30. To investigate this, initially, DG-1 was synthesized by the incorporation of two butylphenyl residues and screened against all the four bacterial strains. Interestingly, the MIC value of DG-1 was significant against all the strains; in particular, the antibacterial efficiency against S. aureus (2.5 µM) was the same as that of melittin. These results suggested that two butylphenyl residues balanced the charge produced by two guanidyl residues, compared with the mono butylphenyl substituted MG-4. This inference was further ascertained by decreasing the hydrophobicity from bis(butylphenyl) to bis(n-butyl) (DG-2), which ultimately resulted in dropping its antibacterial activity against almost all the strains compared to DG-1. Substitution of the symmetrical bis(cyclohexyl) residues (DG-3) showed an antibacterial effect nearly as equal as that of DG-1 with enhanced therapeutic index (TI) values. In general, the therapeutic potential of antibacterial agents represents their selectivity towards killing the bacterial cells without displaying significant cytotoxicity to mammalian cells. Thus, cell selectivity of the synthesized compounds is known as therapeutic index (TI) = minimum hemolytic concentration (MHC)/geometric mean (GM), where MHC denotes the antibacterial agent concentration required to cause the 10% lysis to human red blood cells, while GM is the geometric mean of MICs against four bacterial strains.
Hence, the relative safety and the proficiency of a drug candidate to differentiate between a pathogen and the host cells can be inferred from the therapeutic index (TI) values. Thus, larger TI values indicate greater cell selectivity. However, substituting DG-4 with bis(ethylphenyl) displayed little decrease in antibacterial activity compared to DG-3. From the above results, it was observed that the increase in hydrophobicity could elevate the antibacterial activity. Therefore, elevated hydrophobicity was established by synthesizing DG-5, which comprises two 3,3-diphenylpropyl groups. Predictably, DG-5 displayed potent antibacterial effect as almost similar to the control melittin, except against E. coli and S. epidermidis, where it elicited a two-fold reduced activity, while displaying significant TI value. Finally, DG-6 was synthesized and investigated for its antibacterial efficacy by incorporating bis(naphthyl) residues as hydrophobic groups. Expectedly, DG-6 also displayed an antibacterial efficiency as potent as DG-5 and the control melittin, while there was a slight drop in MIC value against P. aeruginosa (10 µM). On the whole, based on the antibacterial activities with enhanced TI values, we identified four potent compounds namely, DG-5, DG-6, DG-1, and DG-3.
Next, to investigate the effect of changing the charge from bis(guanidyl) groups to bis(amino) groups while manipulating the hydrophobic residues in a similar way as in the DG series, the DL series of compounds were derivatized as delineated in Fig. 2. First, we began with anchoring the symmetrical bis(butylphenyl) residue against positively charged two amine residues that resulted in DL-1, which displayed antibacterial effects almost as same as DG-1. However, again, manipulating the hydrophobic residues, including n-butyl (DL-2), cyclohexyl (DL-3) and ethyl phenyl (DL-4) resulted in a loss of the antibacterial effect against E. coli and S. epidermidis, while showing appreciable activity against P. aeruginosa and S. aureus compared to the control. The decreasing activity can be attributed to the imbalance of the charge to hydrophobicity ratio. To investigate this, we synthesized DL-5 by incorporating bis(3,3-diphenylpropyl). Surprisingly, DL-5 displayed excellent antibacterial activity, being as effective as the control melittin, with significant TI value. This observation led to the identification of DL-5 as the most effective compound among the MG, DG, and DL series of compounds. Further, DL-6 was also synthesized and assayed against all the bacterial strains, and was found to display as potent an antibacterial activity as DL-5; while there was a little decrease in the efficacies against P. aeruginosa and S. epidermidis, and it showed an excellent TI value. Lastly, DL-1, DL-5 and DL-6 were the most promising compounds in the DL series of compounds. Our structure activity relationship studies inferred that (i). Triazine scaffold is an excellent choice for the design of new antibacterial agents; (ii). Both guanidine and amine can be used as positive charge donor; (iii). Bulky hydrophobic residues including 3,3-diphenylpropyl and 1-naphthylmethyl are necessary to balance the charge deduced from either guanidyl or amine groups. Finally, DG-1, DG-5, DG-6, DL-1, DL-5, and DL-6 were spotted as most potent compounds based on their antimicrobial effects and TI values.
Cell selectivity and cytotoxicity
Achieving maximum possible antibacterial effect with minimum toxicity towards the host is an attractive strategy for discovering antibacterial agents. The compounds with the most potent antibacterial activity, including DG-1, DG-5, DG-6, DL-1, DL-5, and DL-6, were evaluated for their hemolytic activity against sheep red blood cells in the range of 0–320 μM (Fig. 3a) to assess their ability in treating the bacterial cells selectively. DG-1, DG-5, DG-6, DL-1, and DL-5 showed a noticeable hemolytic activity at 320 μM, the highest concentration tested (Fig. 3a and Table 1).

(a) Hemolytic activity of the selected compounds in sheep red blood cells (s-RBCs); (b) Cytotoxic evaluation of the selected compounds in mouse macrophage RAW264.7 cells.
Interestingly, all these compounds exhibited less than 6% hemolysis until the concentration of 80 μM, at which the reference compound melittin almost induced 100% hemolysis. Even though DL-1 and DG-6 showed 50% hemolysis at a high concentration of 160 μM, the DG-1, DL-5 and DL-6 induced less than 8% hemolysis at the tested concentration, 160 μM. Astonishingly, the same lack of hemolysis was maintained even at more than 320 μM by DL-6, which was also revealed by its elevated TI values. DG-5 and DL-5 also showed appreciable cell selectivity by inducing less than 40% hemolytic even at a high concentration (more than 320 μM). These results further support the potential of these compounds for use as antibacterial therapeutics.
To further verify the cytotoxicity of DL-5, DL-6, DG-5, and DG-6 against other mammalian cells, mouse macrophage RAW264.7 cells were used. Interestingly, these compounds didn’t show noticeable cytotoxicity until 20 μM as shown in Fig. 3b, despite the fact that amphipathic moieties are often found to exhibit cytotoxicity. The low toxicity and potent antibacterial activity of these compounds suggest that these compounds may have more affinity for negatively charged bacterial membrane than for zwitterionic mammalian membranes. Together these results indicate that these compounds could potentially be model compounds for the design of antibacterial therapeutic.
Anti-inflammatory (endotoxin-neutralizing) activity
In most of the Gram-negative bacteria, the outer membranes are composed of lipopolysaccharides (LPS, endotoxin) as a major constituent. During infection, the release of LPS from bacteria is inevitable, and consequently, monocytic and phagocytic cells are induced enormously to produce pro-inflammatory cytokines such as TNF-α, IL-6, IL-b, and others. Overexpression of cytokines have been associated with the development of sepsis and septic shock, leading to multiple organs damages31,32. Hence, it is worth discovering an antibacterial agent that has dual functions such as anti-microbial and anti-inflammatory effects to treat the bacteria-induced inflammation33,34. We examined seven compounds (DG-1, DG-3, DG-4, DG-5, DG-6, DL-5, and DL-6) with enhanced antibacterial activity and TI values to test their endotoxin-neutralizing effect using NO production and TNF-α release in LPS-stimulated RAW264.7 cells, because NO and TNF-α are two major causes promoting inflammatory responses (Fig. 4). As shown in Fig. 4a, all the seven compounds tested were found to inhibit LPS-induced NO production. In particular, DG-5, DG-6, DL-5, and DL-6 were found to be more proficient in inhibiting NO production than the reference LL-37, which is well known for its characteristic LPS-neutralizing effects. Moreover, DG-6 showed the maximum inhibitory potential with a similar spectrum as the reference and control compounds.

(a) Inhibition of compounds (10 μM) against nitric oxide (NO) production in LPS-stimulated RAW264.7 cells; (b) Inhibition of compounds (20 μM) on LPS-stimulated TNF-α release from RAW264.7 cells (Inhibition of compounds (10 μM) on TNF-α production is given in Fig. S2). Asterisks dictate the significant effects of the tested compounds compared to that of LPS treated cells. One-way ANOVA with Bonferroni’s post-test (*p < 0.001 for each agonist) used for analyzing data. The data are mean ± SEM of three experiments. The results remained similar when experiments were performed using different cells. Control indicates the cell only. (c) Anti-inflammatory efficacies of the tested compounds on suppressing the mRNA expression of iNOS and TNF-α in LPS-stimulated RAW264.7 cells by RT-PCR. (Full-length blots are given in Supplementary information).
Then as shown in Fig. 4b, DG-5, DG-6, DL-5, and DL-6 significantly inhibited TNF-α and the other compounds were found to show poor efficiency. In this study, DG-6 showed the maximum TNF-α inhibition almost as similar as the reference compound, LL-37. This inference is in accordance with the results obtained for NO inhibition test, which confirms that DG-5, DG-6, DL-5, and DL-6 possess significantly potent LPS-neutralizing potency to become model compounds for the future design of antibacterials with profound anti-inflammatory activity.
Usually, mRNA expression of inducible nitric oxide synthase (iNOS) and TNF-α in LPS-stimulated RAW264.7 cells are induced by LPS and that correlates with NO and TNF-α release. Thus, the ability of the synthesized compounds on suppressing the LPS-induced mRNA expressions of iNOS and TNF-α was investigated by RT-PCR as shown in Fig. 4c. Among the seven compounds tested, DL-5, DL-6, DG-5, and DG-6 significantly reduced the expression of both iNOS and TNF-α in LPS-stimulated RAW264.7 cells. In particular, DG-5 displayed potency almost similar compared with the reference compound, LL-37. Thus, this result also correlated with the results obtained for NO inhibition and TNF-α release, additionally, confirming that DL-5, DL-6, DG-5, and DG-6 have potent anti-inflammatory activity.
Antibacterial activity of DG-5, DG-6, DL-5 and DL-6 against antibiotic-resistant bacteria
Recently, severe infection caused by escalating multidrug-resistant bacterial threats is considered as one of the most serious public health concerns35. For instance, notorious multidrug-resistant strains, including Gram-positive bacteria methicillin-resistant S. aureus, vancomycin-resistant E. faecium, and Gram-negative bacteria multidrug-resistant P. aeruginosa, have evolved as the sole reason for hospital- and community-acquired infections36. This facilitates a need for new antibiotics with great potency toward drug-resistant bacteria. Therefore, to probe the potency of our synthesized small molecules, DL-5, DL-6, DG-5, and DG-6, against drug resistance in bacteria, we screened them for their antibacterial activity against three MRSA strains (CCARM 3089, CCARM 3090, and CCARM 3095), a VREF strain (ATCC 51559), and two MDRPA strains (CCARM 2095 and CCARM 2109). Interestingly, all the compounds revealed almost considerable activities against all the strains compare to the reference compound, melittin (Table 2). Further, DL-5 and DG-5 displayed excellent efficacy against most of the bacterial strains either by showing similar or better activity with respect to the reference, and both followed similar activity trends.
In particular, even though DL-5 and DG-5 showed a small reduction in efficiency against CCARM 2095 strain compared to that of reference, they found to be as potent as melittin against CCARM 2109. This enhanced activity against Gram-negative MDRPA strains may be likely due to the interaction between positive charges in DL-5 and DG-5 and the negative charge of the bacterial membrane to kill the pathogen. Moreover, DG-6 also displayed similar or better activity against the majority of the strains except for MDRPA strains. Despite that, DL-6 showed a slight decreased in activity against all the strains, and in the case of CCARM 3090, it showed an activity as potent as reference. Collectively, these results demonstrate that symmetrical hydrophobic groups of DL-5, DL-6, DG-5, and DG-6 balance the charge produced either by two guanidyl or two amino groups. These results suggest that all our tested compounds could be used as a representative model for the design of a new class of antibacterial agents against drug-resistant bacteria.
Salt resistance
To investigate the effects of salts on antimicrobial activity, these four compounds were tested for antibacterial activity against E. coli in the presence of different salts at physiological concentrations, because the salt resistance remains a prominent limitation to transform antibacterial compounds into novel therapeutic drugs37. As shown in Table S1 (see Supplementary), even though there was a slight drop in the antibacterial activity of DL-5, DL-6, DG-5, and DG-6 compared to that in the absence of the salts, the resistance in the presence of all salts was still intact. In particular, DG-5 and DG-6 exhibited marked activity in the presence of all salts, where DG-5 activity was decreased by two-fold compared to that in the control and DG-6 showed similar or two-fold reduced activity compared to control. Similarly, DL-5 and DL-6 also showed considerable activity. These results indicate that the salt interference with the interaction between DL-5, DL-6, DG-5, and DG-6 and the bacterial membrane was not extensive.
Mechanistic studies
Cytoplasmic membrane depolarization
To evaluate the mode of action of our synthesized compounds on the bacterial cytoplasmic membrane depolarization, we analyzed the depolarization ability of the compounds (DL-5, DL-6, DG-5, and DG-6) on S. aureus cytoplasmic membrane. We used 3,3′-Dipropylthiadicarbocyanine Iodide (DiSC3(5)), which is a membrane potential-dependent probe. During the permeabilization and disruption of the cytoplasmic membrane, there is an increase in fluorescence intensity due to the dispersion of probe into the medium. In this study, we used melittin and LL-37 as membrane-targeting positive controls. Buforin-2, known for its intracellular targeting mechanism, served as a negative control. As shown in Fig. 5(a–c), the inclusion of the synthesized small molecules DL-5, DG-5, and DG-6 promoted a significant increase in fluorescence intensity similar to that exhibited by melittin and LL-37, indicating a rapid membrane depolarization. This result confirmed that DL-5, DG-5, and DG-6 act as membrane targeting molecules, which caused a loss of membrane potential; however, membrane depolarization was not induced by buforin-2. Although DL-5, DL-6, and DG-6 showed a similar pattern of mechanism to that exhibited by melittin, DL-6 did not induce a significant increase in fluorescence at 2x MIC (Fig. 5d). In contrast, a slight increase in fluorescence was observed at 4x MIC (Fig. S3, ESI). This inference led to the speculation that DL-6 may follow intracellular targeting mechanism similar to buforin-2.

Depolarization effects of compounds (DG-5 (a); DG-6 (b); DL-5 (c) and DL-6 (d)) and peptides on S. aureus cytoplasmic membrane. The membrane potential-sensitive fluorescent dye 3,3′-dipropylthiadicarbocyanine Iodide (DiSC3(5)) was used for determining the S. aureus membrane depolarization by measuring fluorescence of the dye release (excitation λ = 622 nm, emission λ = 670 nm). Except for melittin (2 μM), the concentration of control antimicrobial peptides and antimicrobial compounds used is 2X MIC against S. aureus (KCTC 1621) (buforin-2 (16 μM); LL-37 (5 μM); DG-5 (5 μM); DG-6 (5 μM); DL-5 (5 μM), DL-6 (5 μM)). LL-37 and melittin were used as a positive control peptides and buforin-2 was used negative control peptide. The other two triplicate images are presented in Fig. S4 (SI).
Flow cytometry and Confocal laser-scanning microscopy
To verify that DL-6 follows intracellular targeting mechanism, FITC-labeled DL-6 uptake in both Gram-negative and Gram-positive bacteria using E. coli and S. aureus respectively was confirmed by flow cytometry analysis. As shown in Fig. 6a, compared to the control, FITC-labeled DL-6 showed a substantial shift in the fluorescence intensity towards the lower right-hand side region displaying 14.89% and 93.46% in E. coli and S. aureus, respectively, which can be attributed to the differences in the cell membrane of bacteria. To further confirm the hypothesis, we performed FITC-labeled DL-6 (Fig. S1) uptake in E. coli and S. aureus using confocal laser-scanning microscopy (Fig. 6b). As observed in the merged portion (FITC/DAPI) of the images in both bacteria, there is a substantial increase in the fluorescence. This result indicated that FITC-labeled DL-6 translocates inside the bacterial cell by accumulating at the cytoplasm of the cell, which suggest that DL-6 exhibits intracellular targeting mechanism via cell penetration. However, still, we observed a few left out FITC-DL-6 in E. coli because of the low permeability barrier of inner and outer membranes of bacteria (Fig. S5). This evidence further confirms that DL-6 can translocate inside of both E. coli and S. aureus bacteria.

(a) Flow cytometry analysis of FITC-labeled DL-6 uptake in bacteria; Quantification of positive bacterial cells with FITC labels. Each point represents +/−SEM of six repeated experiments p < 0.001. (b) Confocal fluorescence microscopy images of localization of FITC-labeled DL-6 in E. coli and S. aureus.
Protease stability
The complexity rendered by the proteolytic instability of peptides in the presence of enzymes remains one of the major challenges in the advancement of peptide-based antibacterial for therapy, including cationic peptide antibiotics. It is known that lysine and arginine are highly prone to trypsin-mediated hydrolysis. Because the synthesized antibacterials (DL-5, DL-6, DG-5, DG-6) emerge as mimics of lysine and arginine, the proteolytic stability of these small molecules was investigated by tryptic degradation (see Supplementary Fig. S6). Incubation of these compounds with trypsin for 24 hours did not alter their antibacterial activity against E. coli. However, antibacterial activity of the reference melittin was lost entirely because of the proteolytic degradation by trypsin.
Atopic dermatitis (AD)
AD can be classified as a complex and chronic inflammatory skin disease, which is characterized by a type 2 helper T cell (Th2) immune response and sensitization of immunoglobulin (Ig)E38. The pathogenesis of AD involves various factors including skin barrier dysfunction, susceptibility genes, and immunologic and environmental factors39. The most common environmental allergens for humans is house dust mites, including Dermatophagoides pteronyssinus and Dermatophagoides farina (DFE)40. In general, the imbalance between Th1/Th2 cells remains the sole reason for the induction of AD, where Th2 and Th1 cells are responsible for promoting the acute and chronic phases of AD, respectively41,42.
We found that most of our synthesized compounds possessed antibacterial activity along with considerable anti-inflammatory effects in LPS-induced inflammation. Hence, considering the proficient anti-inflammatory effects of our compounds, we anticipated that they could be effective in treating AD. Therefore, initially, we screened all the compounds for their ability to suppress the TNF-α expression (see Supplementary Fig. S7) in HaCaT cells using conventional RT-PCR. The results inferred that DL-6 effectively suppressed TNF-α expression, even at 15 μM. Further, DL-6 was evaluated for its cytotoxicity at different concentrations in HaCaT cells. The results revealed that DL-6 did not show considerable toxicity at an elevated concentration (50 μM); more than 75% cell viability was observed (see Supplementary Fig. S8). Hence, the preliminary prerequisite for treating AD was successfully satisfied by DL-6 in terms of both cytotoxicity and TNF-α suppressing effect. Therefore, DL-6 was used for further studies on AD-like skin lesion in a BALB/c mouse model that was developed by alternate painting with dermatophagoides farinae extract (DFE) and 2,4-dinitrochlorobenzene (DNCB) for four weeks on both the earlobes of mouse (Fig. 7a). The repetitive application of DFE and DNCB induced swelling with a significant increase in the thickness of the ears.

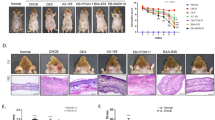
Histopathological analysis of DL-6 on the ear thickness and mast cell infiltration. (a) The ear thickness measurement chart and day 28 mice photographs; (b) Measurement of the thickness of epidermis and dermis using hematoxylin and eosin-stained microphotographs; (c) Measurement of mast cells infiltration using toluidine blue staining. The data denote mean ± SD of six determinations of each group when n = 8. Asterisks indicate the significant difference from the AD value at p < 0.05. Original magnification was 200×. CON denotes the control. The pictures are representative images of each group (n = 8).
However, treatment of DL-6 with DFE and DNCB daily for 28 days exhibited remarkable effect on AD, which was evidenced by the suppression of the inflammation-induced thickness of the earlobes (Fig. 7a ear photographs). Microscopic images of DFE and DNCB-induced epidermal and dermal thickness were analyzed after treating with DL-6. As shown in Fig. 7b, treatment with DL-6 significantly reduced the thickness of both epidermis and dermis. In particular, epidermal thickness was reduced by approximately 30% and dermal thickness by more than 50%, which proves the efficiency of DL-6 over AD-lesion. The infiltration of mast cells in the ear tissue was also investigated by the toluidine blue staining of skin sections. Interestingly, as shown in Fig. 7c, DL-6 decreased the infiltration of mast cells, which further proves the efficacy of DL-6 against AD-like lesions.
It is reported that the Th2 cytokine, IL-4 is not only overexpressed in the acute phase of AD lesions but also, in many cases, IL-10 expressions also is predominant in AD lesions43. Also, there are reports pertaining to the increased levels of IL-17 in cutaneous lesions of AD patients in the acute lesions44. However, in the chronic phase of AD, Th1 producing IFN-γ and keratinocytes inducing proinflammatory cytokines TNF-α and IL-6 were abundant. We investigated the inhibitory effect of DL-6 on inflammatory cytokine expressions in earlobes of mice with AD-like skin lesions using real-time PCR. As shown in Fig. 8, repeated oral application of DFE and DNCB increased the expressions of IFN-γ, TNF-α, IL-6, IL-4, IL-10, and IL-17. The treatment of DL-6 significantly reduced the upregulated expression of cytokines in AD-like skin, irrespective of all Th1, Th2, and Th17.

Anti-atopic efficacies of DL-6 on the expression of various pathogenic factors in mice ear. After day 28, mice ears were excised, and total RNA was isolated, and performed the quantitative real-time polymerase chain reaction. The data indicate mean ± SD of triplicate experiments of each group, where n = 8. CON represents the control and asterisks indicate the significant differences from the AD value at p < 0.05.
It is pertinent to note that TNF-α expression was reduced by approximately 50%. Surprisingly, DL-6 inhibited the expression of IL-4 and IL-6 by more than 75%, while that of IL-10 and IL-17 by approximately 25%. These results collectively infer the potency of DL-6 in both acute and chronic inflammation in AD-lesions, which effectively suppressed cytokine expressions. As bacterial colonization is involved in the skin infection of AD patients, DL-6 provides an opportunity for the development of AD inhibitors with potent antibacterial properties.
In vivo anti-inflammatory activity of DG-5
Inspired by the inhibitory potency of DG-5 over in vitro LPS-induced mRNA expressions of iNOS and TNF-α, we tested its efficacy in LPS-induced ALI mouse model to evaluate its therapeutic potential in vivo. To examine the effect of DG-5, we prepared LPS-induced ALI mouse models and compared the inflammatory efficiency by counting the inflammatory cells in bronchoalveolar lavage fluid (BALF) as shown in Fig. 9a. In the LPS-induced ALI mouse model, the numbers of total and differential inflammatory cells in BALF were remarkably high compared to those in the normal controls (NC). Oral administration of DG-5 in two doses (5 mg and 10 mg) resulted in the appreciable suppression of inflammation in LPS-induced ALI mouse model based on the reduction in BALF cell counts (Fig. 9b).

Anti-inflammatory effects of DG-5 in LPS-induced mice BALF. (a) Timeline of LPS and DG-5 treatment; (b) Total cells in mice BALF were isolated and quantified using Diff-Quick-stained reagent. #p < 0.05, ##p < 0.01, and ###p < 0.001, compared with normal control (NC); *p < 0.05, **p < 0.01, and ***p < 0.001, compared with model group (two-way ANOVA); (c) Microscopic images of DG-5 (5 mg and 10 mg doses) and dexamethasone (Dex) treatments in LPS induced lung inflammation cells.
Although oral administration of 5 mg dose showed reduced effects in suppression of inflammation, interestingly, treatment with 10 mg dose of DG-5 displayed significant effects in suppression of inflammation as Dex. This observation was further ascertained by the microscopic images of DG-5-treated LPS-induced lung inflammation cells (Fig. 9c). Compared to the 5 mg dose of DG-5, 10 mg of DG-5 displayed superior effect in the eradication of lung inflammation cells. These results strongly support that DG-5 is capable of suppressing acute lung inflammation in addition to showing strong antibacterial activity.
Conclusion
Though triazine derived antibacterials are reported24,25,45, the present work advances in terms of highly symmetrical design, enhanced antibacterial and anti-inflammatory effects, and enhanced potency against drug-resistant pathogens. Our findings from this work demonstrate that substituted triazines are suitable for designing potent antibacterial agents due to their symmetric structures holding amphipathicity as in CPAs and easy access to the synthesis of various derivatives, which led to the identification of four lead compounds DG-5, DG-6, DL-5, and DL-6. These compounds have not only antibacterial and anti-inflammatory effects but also show significant potency against drug-resistant bacteria. Mechanistic evidence revealed that DG-5, DG-6, and DL-5 were found to be membrane targeting and in contrast, DL-6 followed an intracellular targeting mechanism. Along with the potent in vitro anti-inflammatory effects, DG-5 and DL-6 also showed their remarkable potency in in vivo ALI mouse model and atopic BALB/c mouse model, respectively. Thus, these compounds have the potential for future use as a therapeutic model. Together with their facile synthesis, these compounds could be a new class of drug-resistant, protease stable small molecular antibacterial agents with potent anti-inflammatory and anti-atopic dermatitis. In addition, further studies on atopic dermatitis are under progress using other in vivo models.
Materials and Methods
Chemistry
As described in our previous article46, all reagents and starting materials were purchased from commercial chemical suppliers (Sigma-Aldrich, TCI and Across Organics) and used as received. All the anhydrous organic solvents of purity greater than 99.9% were purchased from Aldrich and used directly. Thin layer chromatography (TLC) was performed on Merck aluminum sheets with silica gel 60 F254 and was visualized by ultraviolet light and staining with KMnO4 and ninhydrin. For the purification of compounds, column chromatography was performed on Merck silica gel 60 (70–230 mesh or 230–400 mesh). NMR spectra including, 1H and 13C NMR spectra were recorded on a Bruker DRX-400 and DRX-500 spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) measured and coupling constants (J) are given in hertz (Hz). MALDI-TOF mass were recorded using Shimadzu mass spectrometer. Reverse-phase HPLC analysis (RP-HPLC) was carried out at 230 nm on an Agilent HPLC system equipped with a C18 analytical column (250 × 22 mm, 10 microns). Two different linear gradients of 0.05% aq. trifluoroacetic acid (TFA) (eluent A) and 0.05% TFA in CH3CN (eluent B) were used with a flow rate of 5.0 mL/min at 25 °C.
Bacterial strains and Growth Conditions
As described in our previous articles26, two Gram-positive bacterial strains (Staphylococcus aureus [KCTC 1621] and Staphylococcus epidermidis [KCTC 1917]), and two Gram-negative bacterial strains (Pseudomonas aeruginosa [KCTC 1637] and Escherichia coli [KCTC 1682]) were received from the Korean Collection for Type Cultures (KCTC) of the Korea Research Institute of Bioscience and Biotechnology (KRIBB). Culture Collection of Antibiotic-Resistant Microbes (CCARM) (Seoul Women’s University in Korea) supplied the methicillin-resistant Staphylococcus aureus strains (MRSA; CCARM 3089, CCARM 3090, and CCARM 3095) and multidrug-resistant Pseudomonas aeruginosa strains (MDRPA; CCARM 2095, and CCARM 2109) were obtained from the. American Type Culture Collection (Manassas, VA, USA) supplied the vancomycin-resistant Enterococcus faecium (VREF; ATCC 51559). All bacterial strains were made as glycerol stocks and stored at −80°, cultured on Luria Broth agar plates, and stored at 4 °C. Before the experiments, the single bacterial colony was chosen from the agar plates and cultured in LB media for overnight at 37 °C. Then, subcultures were prepared and incubated at 37 °C for 3 h or until the OD600 reached 0.5, which corresponds to 2 × 108 CFU/mL.
Antimicrobial activity
As described in our previous articles47, MICs were determined by the standard broth microdilution method as previously described. Briefly, 96-well polystyrene plates were inoculated with two-fold serial dilutions of the small molecule compounds (starting at 128 μM) and a log-phase culture of the target bacterial strain (2 × 106 CFU/mL), which were both diluted in 1% peptone. The plates were subsequently incubated for 18–24 h at 37 °C and 200 rpm in a shaking incubator. The MIC is defined as the lowest concentration that inhibits bacterial growth as determined visually or spectrophotometrically by OD600. In order to assess the effects of different salts on the antimicrobial activity of the synthesized small molecules, MICs were also determined in the presence of salts. E. coli (KCTC 1682) at a concentration of 2 × 106 CFU/mL were treated with small molecule compounds in solutions containing physiological concentrations of various salts (150 mM NaCl, 4.5 mM KCl, 6 µM NH4Cl, 1 mM MgCl2, 2.5 mM CaCl2, 4 µM FeCl3), added to 1% peptone. With the exception of the added salt, MICs were determined using the assay described above.
Hemolytic Activity
As similar to our previous articles47, sheep red blood cells (sRBCs) in PBS were washed repeatedly three times or until the supernatant was clear (centrifuged at 2000 rpm for 5 min). Two-fold serial dilutions of the synthesized antibacterial agents in phosphate buffered saline (PBS) (concentrations varying from 1 to 320 μM) were added to 96-well polystyrene plates incubated with sRBCs to a final concentration of 4% (v/v) for 1 h at 37 °C. Samples were centrifuged at the speed of 800 × g for 10 min, cell supernatants were transferred to clear 96-well plates, and their absorbance was measured at 405 nm. The OD value of PBS was considered to be 0% hemolysis, while 100% hemolysis was established using 0.1% Triton X-100.
Cytotoxicity against Mammalian Cells
As described in our previous article47 MTT assays were performed to determine the cytotoxicity of the synthesized small molecule compounds against RAW 264.7 macrophage cells. The cells were seeded into 96-well plates at 2 × 104 cells per well and incubated for 24 h at 37 °C in 5% CO2 to ensure full confluency. Cells were treated with serial dilutions of small molecule compounds in DMEM ranging from 0.313 to 20 μM (final) and further incubated for 48 h. Cells without the added small molecule antibacterial agents served as controls. After incubation, the cell media were replaced with 100 μL of fresh media containing MTT solution (5 mg/mL), and cells were incubated for another 4 h at 37 °C. Next, the media were replaced with 100 μL of DMSO and metabolic activities were detected spectrophotometrically by measuring the absorbance at 550 nm as previously described. Cell proliferation values were expressed as percentages and calculated as follows: (Abs550 nm of treated sample)/(Abs550 nm of control) × 100.
Measurement of NO production from LPS-stimulated RAW264.7 cells
As described in our previous articles26,47, NO production from LPS-stimulated RAW264.7 cells was determined. Briefly, RAW264.7 cells were plated at a density of 5 × 105 cells/mL in 96-well culture plates and stimulated with LPS (20 ng/mL) in the presence or absence of synthesized small molecule compounds for 24 h. Isolated supernatant fractions were mixed with an equal volume of Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride and 2% phosphoric acid) and incubated at room temperature for 10 min. Nitrite production was quantified by measuring absorbance at 540 nm, and concentrations were determined using a standard curve generated with NaNO2.
Measurement of release of TNF-α from LPS-stimulated RAW264.7 cells
As described in our previous articles26,47, RAW264.7 cells were seeded in 96-well plates (5 × 104 cells/well) and incubated overnight. The synthesized small molecule compounds were added and the cultures were incubated at 37 °C for 1 h. Subsequently, 20 ng/mL of LPS was added and the cells were incubated for another 6 h at 37 °C. Cell supernatants were collected and stored at −20 °C until needed for analysis of cytokine levels. Levels of TNF-α in the samples were measured by the commercial enzyme-linked immunosorbent assay kit (R&D Systems, Minnespolis, MN, USA) according to the manufacturer’s instructions.
Reverse-transcription polymerase chain reaction (RT-PCR)
As described in our previous article47, RAW264.7 cells were plated at a concentration of 5 × 105 cells/well in six-well plates and incubated overnight. Each peptide was added to the wells. The final concentration of the small molecule compounds was 10 μM (for NO) or 20 μM (for TNF-α). Cells to be used for quantification of TNF-α and iNOS were treated for 3 h and 6 h, respectively, with or without (negative control) 20 ng/mL LPS, in the presence or absence of peptide, in DMEM supplemented with 10% bovine serum. The cells were detached from the wells and washed once with PBS. Total RNA was extracted by Trizol Reagent (Ambion, USA). Then, 1 μg of total RNA was converted to cDNA using a TakaRa PrimescriptTM Reverse Transcriptase (TakaRa, Seoul, Korea). The primers used were purchased from Bioneer (Seoul, Korea). The cDNA products were amplified by AccuPower® PCR PreMix (Bioneer, Daejeon, Korea). For iNOS (forward, 5′-CTGCAGCACTTGGATCAGGAACCTG-3′; reverse, 5′-GGGAGTAGCCTGTGTGCACCTGGAA-3′), TNF-α (forward, 5′-CCTGTAGCCCACGTCGTAGC-3′; reverse, 5′-TTGACCTCAGCGCTGAGTTG-3′), or GAPDH (forward, 5′-GACATCAAGAAGGTGGTGAA-3′; reverse, 5′-TGTCATACCAGGAAATGAGC-3′). The amplification procedure consisted of an initial denaturation step of 5 min at 94 °C, followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 55 °C for 1.5 min, and extension at 72 °C for 1 min, and afterwards by a final extension step of 5 min at 72 °C.
Protease stability by radial diffusion assay
As described in our previous article26, E. coli (KCTC 1682) was grown overnight for 18 h at 37 °C in 10 mL of LB broth, and then 10 mL of this culture was inoculated into 10 mL of fresh LB and incubated for an additional 3 h at 37 °C to obtain mid-logarithmic-phase organisms. A bacterial suspension (2 × 106 CFU/mL in LB) was mixed with 0.7% agarose. This mixture was poured into a 10-cm petri dish, and it dispersed rapidly. Five microliters of an aqueous peptide stock solution (10 mg/mL) were added to 25 mL of trypsin solution in PBS (0.2 mg/mL) and incubated at 37 °C for 4 h. The reaction was stopped by freezing with liquid nitrogen, after which 10 mL aliquots were placed in each circle paper (<6 mm in diameter), put on the agarose plates, and then incubated at 37 °C overnight. The diameters of the bacterial clearance zones surrounding the circle paper were measured for the quantitation of inhibitory activities.
Cytoplasmic membrane depolarization assay
As described in our previous articles26,47, the effect of synthesized small molecule antibacterial agents on cytoplasmic membrane depolarization was determined using the membrane potential-sensitive dye, DiSC3(5), as previously described48,49. Briefly, S. aureus (KCTC 1621) grown at 37 °C with agitation to the mid-log phase (OD600 = 0.4) was harvested by centrifugation. Cells were washed twice with washing buffer (20 mM glucose and 5 mM HEPES, pH 7.4) and resuspended to an OD600 of 0.05 in the same buffer. The cell suspension was incubated with 20 nM DiSC3(5) until a stable fluorescence value was achieved, implying the full incorporation of the dye into the bacterial membrane. Membrane depolarization was monitored based on the changes in the intensity of the fluorescence emitted from the DiSC3(5) (excitation λ = 622 nm, emission λ = 670 nm) after peptide addition. The membrane potential was fully abolished by adding 0.1% Triton X100.
Statistical analysis
As described in our previous article47, all results are presented as averages of values derived from three independent experiments, with triplicates used in each experiment. Error bars represent the mean ± standard deviation of the mean. The statistical significance of differences between samples and respective controls (no added antimicrobial agent) were determined by one-way analysis of variance (ANOVA) with Bonferroni’s post-test method using Sigma plot v12.0 (Systat Software Inc., San Jose, CA, USA). Differences with P < 0.001 were considered statistically significant.
Confocal fluorescence microscopy
Bacteria was collected to the extent of 107 CFU, then it was resuspended in phosphate buffer (PBS) containing FITC-labeled DL-6 at 10 μg/mL and incubated for 1 hour at 37 °C. Subsequently, the cells were washed, fixed, DAPI stained and immobilized on poly-L-lysine coated glass slides. Before mounting, ProLong Gold antifade reagent (Invitrogen) was added to the slides. Finally, localization of FITC-labeled DL-6 was observed using LSM710 (Carl Zeiss, Germany) confocal microscopy equipped with a 100X oil immersion lens.
Flow cytometry analysis
FITC-labeled DL-6 uptake in bacteria was confirmed by flow cytometry analysis. Bacteria was resuspended in PBS containing FITC-labeled DL-6 at a 10 μg/mL and incubated for 1 hour at 37 °C. Subsequently, cells were washed and analyzed using a flow cytometer (CytoFlex, Beckman Coulter, Miami, USA). The laser excitation wavelength remained at 488 nm and emission band-pass wavelengths were at 525/40 nm. The results are described as the median of the distribution of FITC intensity obtained by analyzing 10,000 bacteria.
In vivo study
Animals
As reported our previously reported article50, six-week-old male C57BL/6J mice (20–22 g) were obtained from the KRIBB Laboratory Animal Resource Center (Ochang, Korea) and housed in environmentally controlled pathogen-free conditions throughout the experiments. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Korea Research Institute of Bioscience and Biotechnology and performed in compliance with the National Institute of Health Guidelines for the care and use of laboratory animals and the Korean National Animal Welfare Law.
Mouse model of LPS-induced ALI (Acute Lung Injury)
Mice (n = 3) were grouped based on the experimental design. (i) control group (NC), which received saline; (ii) LPS-induced ALI group (model), which received LPS; (iii) LPS-received low-dose DG-5 group, which received 5 mg/kg DG-5 and LPS; (iv) LPS-received high-dose DG-5 group, which received 10 mg/kg DG-5 and LPS; (v) LPS-received dexamethasone group (Dex), which received 1 mg/kg dexamethasone and LPS. Experimental mice were subjected to an intranasal instillation of 10 μg of LPS (Sigma-Aldrich, St. Louis, MO) or PBS. DG-5 and dexamethasone were administered to mice by oral gavage (1 h before and 24 h after LPS treatment). The dose of DG-5 chosen in the present study was based on the results from in vitro assays.
Bronchoalveolar lavage fluid (BALF)
BALF collection was performed as previously described50. The trachea was cannulated, and the BALF was obtained by injecting ice-cold PBS (0.5 mL) and withdrawing as much fluid as possible using a tracheal cannula with a 20G blunt needle. The procedure was repeated once for each mouse, and the fluid was transferred to a test tube on ice. For total and differential cell counting, BALF was centrifuged on cytospin slides for 5 min at 1500 rpm. The slides were dried, and the inflammatory cells were fixed and stained using Diff-Quik® staining reagent (IMEB Inc, Deerfield, IL) according to the manufacturer’s instructions.
Atopic dermatitis
Cell culture
As described in our previous article51, in this AD study, Human keratinocytes HaCaT cells were broadly used. The HaCaT cells were cultured in a 5% CO2 atmosphere at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics (10 μg/100 mL penicillin/streptomycin). To perform PCR and western blotting, HaCaT cells were pre-incubated with 10 ng/mL of both TNF-α and IFN-γ for the stimulation. After 6 h of stimulation, cells were harvested.
Cytotoxicity Assay
As described in52, cell viability was determined by 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) assay in 96-well plates. Cells were incubated with various concentrations of DL-6 for 48 h followed by MTT for 4 h, and then 100 μL isopropanol (in 0.04 N-hydrochloric acid) was added to dissolve the formazan crystals. The absorbance was read at 570 nm using a spectrophotometer (Tecan, Switzerland). Cell viability was calculated as the relative absorbance compared to DMSO vehicle control absorbance52.
Animals
As described in our previous article53, eight-week-old female BALB/c mice were purchased from Samtako and housed under specific pathogen-free conditions. All experiments were approved by the Institutional Animal Care and Use Committee of Konkuk University (KU17089).
Induction of AD lesions in the ear
As previously described in our article53, AD was induced in the mice by repeated local exposure of Dermatophagoides farinae extract (DFE; house dust mite extract) and 2,4-dinitrochlorobenzene (DNCB) to the ears. For the induction of AD, the mice were divided into four groups (control, DL-6-only, AD-only, and AD + DL-6), and the surfaces of both earlobes were stripped five times with surgical tape (Nichiban, Tokyo, Japan). After stripping, 20 µL of 1% DNCB was painted onto each ear, followed by 20 µL of DFE (10 mg/mL) 4 days later. Animals received DL-6 (20 μL of 20 mg/mL, topical application) throughout the 4 weeks of AD induction. The ear thickness was measured 24 h after DNCB and DFE application with a dial thickness gauge (Kori Seiki MFG, Co., Japan).
Histological observations
As described in our previous article53, excised ears were fixed in 4% paraformaldehyde for 16 h and embedded in paraffin. Thin (6 µm) sections were stained with hematoxylin and eosin (H&E). The thickness of the epidermis and dermis was measured under a microscope. For measurement of mast cell infiltration, skin sections were stained with toluidine blue and the number of mast cells was counted in five randomly chosen fields of view.
Real-time polymerase chain reaction (PCR)
The conditions for PCR were similar to those previously described53. The total RNA was isolated from cells using TRIzol according to the manufacturer’s protocol. The first-strand complementary DNA (cDNA) was synthesized using Superscript II reverse transcriptase (Invitrogen). Quantitative real-time PCR was carried out using a Thermal Cycler Dice TP850 (Takarabio Inc., Shiga, Japan) according to the manufacturer’s protocol. Total RNA was isolated from the ear tissues of each group. Procedure in Briefly, 2 μL of cDNA (100 ng), 1 µL of sense and antisense primer solution (0.4 µM), 12.5 µL of SYBR Premix Ex Taq (Takarabio Inc.), and 9.5 µL of dH2O were mixed to obtain a final 25 µL reaction mixture in each reaction tube. The primers used for PCR were: mouse tumor necrosis factor-alpha (TNF-α, 5′-AAGCCTGTAGCCCACGTCGTA-3′ and 5′-GGCACCACTAGTTGGTTGTCTTTG-3′; mouse IFN-γ, 5′-TCAAGTGGCATAGATGTGGAAGAA-3′ and 5′-TGGCTCTGCAGGATTTTCATG-3′; mouse IL-4, 5′-ACAGGAGAAGGGACGCCAT-3′ and 5′-GAAGCCGTACAGACGAGCTCA-3′; mouse IL-6, 5′-CCGGAGAGGAGACTTCACAG-3′ and 5′-GGAAATTGGGGTAGGAAGGA-3′; mouse IL-10, 5′-TCAGCTGTGTCTGGGCCACT-3′ and 5′-TTATGAGTAGGGACAGGAAG-3′; mouse IL-17, 5′-CCTACCAGACCAAGGTCAAC-3′ and 5′-AGGGGGTAATAAAGGGATTG-3′; mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5′-GCACAGTCAAGGCCGAGAAT-3′ and 5′-GCCTTCTCCATGGTGGTGAA-3′. The amplification conditions were 10 s at 95 °C, 40 cycles of 5 s at 95 °C and 30 s at 60 °C, 15 s at 95 °C, 30 s at 60 °C, and 15 s at 95 °C. The mRNA levels of the target genes, relative to GAPDH, were normalized using the following formula: relative mRNA expression = 2−(ΔCt of target gene−ΔCt of GAPDH), where Ct is the threshold cycle value. In each sample, the expression of the analyzed gene was normalized to that of GAPDH and presented as the relative mRNA level.
Data Availability
All data generated or analysed during this study are included in this article and its supplementary information files.
References
Ventola, C. L. The Antibiotic Resistance Crisis. Pharm. Ther. 40, 277–283 (2015).
Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 415, 89–395 (2002).
Brown, E. D. & Wright, G. D. Antibacterial drug discovery in the resistance era. Nature 529, 336–343.
O’Neill, J. Tackling Drug-resistant Infections Globally: Final Report and Recommendations. The Review on Antimicrobial Resistance, https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (2016).
Hou, C. D. et al. Insights into an evolutionary strategy leading to antibiotic resistance. Sci. Rep. 7, 40357 (2017).
Mangoni, M. L. Host-defense peptides: from biology to therapeutic strategies. Cell. Mol. Life Sci. 68, 2157–2159 (2011).
Yeung, A. T. Y., Gellatly, S. L. & Hancock, R. E. W. Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 68, 2161–2176 (2011).
Hancock, R. E. W. & Chapple, D. S. Peptide antibiotics. Antimicrob. Agents Chemother. 43, 1317–1323 (1999).
Lehrer, R. I. & Ganz, T. Antimicrobial peptides in mammalian and insect host defence. Curr. Opin. Immunol. 11, 23–27 (1999).
Cheng, Y. et al. Facial amphiphiles. J. Am. Chem. Soc. 114, 7319–7320 (1992).
Liu, D. & DeGrado, W. F. De novo design, synthesis, and characterization of antimicrobial β–peptides. J. Am. Chem. Soc. 123, 7553–7559 (2001).
Scorciapino, M. A. & Rinaldi, A. C. Antimicrobial Peptidomimetics: Reinterpreting Nature to Deliver Innovative Therapeutics. Front immunol. 3, 171 (2012).
Savage, P. B. Multidrug-resistant bacteria: overcoming antibiotic permeability barriers of gram-negative bacteria. Ann. Med. 33, 167–171 (2001).
Peters, B. M., Shirtliff, M. E. & Jabra-Rizk, M. A. Antimicrobial peptides: primeval molecules or future drugs? PLOS Pathog. 6, e1001067 (2010).
Ding, B. et al. Savage, Correlation of the antibacterial activities of cationic peptide antibiotics and cationic steroid antibiotics. J. Med. Chem. 45, 663–669 (2002).
Seydlova, G. et al. Lipophosphonoxins II: Design, Synthesis, and Properties of Novel Broad Spectrum Antibacterial Agents. J. Med. Chem. 60, 6098–6118 (2017).
Menicagli, R. et al. In vitro cytotoxic activities of 2-alkyl-4,6-diheteroalkyl-1,3,5-triazines: new molecules in anticancer research. J. Med. Chem. 47, 4649–4652 (2004).
Chen, X. et al. Design, synthesis, anti-HIV evaluation and molecular modeling of piperidine-linked amino-triazine derivatives as potent non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 20, 3856–3864 (2012).
Baliani, A. et al. Design and synthesis of a series of melamine-based nitroheterocycles with activity against trypanosomatid parasites. J. Med. Chem. 48, 5570–5579 (2005).
Gahtori, P. et al. Antimalarial evaluation and docking studies of hybrid phenylthiazolyl-1,3,5-triazine derivatives: a novel and potential antifolate lead for Pf-DHFR-TS inhibition. Exp. Parasitol. 130, 292–299 (2012).
Henke, B. R. et al. A new series of estrogen receptor modulators that display selectivity for estrogen receptor β. J. Med. Chem. 45, 5492–5505 (2002).
Kuo, G. H. et al. Synthesis and identification of [1,3,5] triazine-pyridine biheteroaryl as a novel series of potent cyclin-dependent kinase inhibitors. J. Med. Chem. 48, 4535–4546 (2005).
Singh, U. P., Bhat, H. R. & Gahtori, P. Antifungal activity, SAR and physicochemical correlation of some thiazole-1,3,5-triazine derivatives. J. Mycol. Med. 22, 134–141 (2012).
Zhou, C. et al. Synthesis and biological evaluation of novel 1,3,5-triazine derivatives as antimicrobial agents. Bioorg. Med. Chem. Lett. 18, 1308–1311 (2008).
Srinivas, K. et al. Synthesis and antibacterial activity of various substituted s-triazines. Eur. J. Med. Chem. 41, 1240–1246 (2006).
Ahn, M. et al. Pyrazole derived ultra-short antimicrobial peptidomimetics with potent anti-biofilm activity. Eur. J. Med. Chem. 125, 551–564 (2017).
Ahn, M. et al. Poly-lysine peptidomimetics having potent antimicrobial activity without hemolytic activity. Amino Acids 46, 2259–2269 (2014).
Murugan, R. N. et al. Non hemolytic short peptidomimetics as a new class of potent and broad-spectrum antimicrobial agents. Bioorg. Med. Chem. Lett. 23, 4633–4636 (2013).
Fillon, Y. A., Anderson, J. P. & Chmielewski, J. Cell penetrating agents based on a polyproline helix scaffold. J. Am. Chem. Soc. 127, 11798–11803 (2005).
Lee, E. et al. Functional Roles of Aromatic Residues and Helices of Papiliocin in its Antimicrobial and Anti-inflammatory Activities. Sci. Rep. 5, 12048 (2015).
Rietschel, E. T. et al. Bacterial endotoxin: chemical constitution, biological recognition, host response, and immunological detoxification. Curr. Top. Microbiol. Immunol. 216, 39–81 (1996).
Dinarello, C. A. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest 112, 321S–329S (1997).
Rosenfeld, Y., Papo, N. & Shai, Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J. Biol. Chem. 281, 1636–1643 (2006).
Pulido, D., Nogues, M. V., Boix, E. & Torrent, M. Lipopolysaccharide neutralization by antimicrobial peptides: a gambit in the innate host defense strategy. J. Innate Immun. 4, 327–336 (2012).
Mohamed, F. M., Abdelkhalek, A. & Seleem, M. N. Evaluation of short synthetic antimicrobial peptides for treatment of drug-resistant and intracellular Staphylococcus aureus. Sci. Rep. 6, 29707 (2016).
WHO. Antimicrobial resistance: global report on surveillance. World Health Organization, http://www.who.int/drugresistance/documents/surveillancereport/en/ (2014).
Takahashi, D., Shukla, S. K., Prakash, O. & Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 92, 1236–1241 (2010).
Leung, D. Y. M. et al. New insights into atopic dermatitis. J. Clin. Invest. 113, 651–657 (2004).
Dai, X. et al. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J. Allergy Clin. Immunol. 127, 806–814 (2011).
Matsuoka, H. et al. A mouse model of the atopic eczema/dermatitis syndrome by repeated application of a crude extract of house-dust mite Dermatophagoides farina. Allergy 58, 139–145 (2003).
Gittler, J. K. et al. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 130, 1344–1354 (2012).
Purushothaman, B., Arumugam, P., Kulsi, G. & Song, J. M. Design, synthesis, and biological evaluation of novel catecholopyrimidine based PDE4 inhibitor for the treatment of atopic dermatitis. Eur. J. Med. Chem. 145, 673–690 (2018).
Ohmen, J. D. et al. Overexpression of IL-10 in atopic dermatitis. Contrasting cytokine patterns with delayed-type hypersensitivity reactions. J. Immunol. 154, 1956–1963 (1995).
Cesare, A. D., Meglio, P. D. & Nestle, F. O. A role for Th17 cells in the Immunopathogenesis of Atopic Dermatitis? J. Investig. Dermatol. 128, 2569–2571 (2008).
Bahar, A. A. et al. Controlling persister and biofilm cells of Gram-negative bacteria with a new 1,3,5-triazine derivative. Pharmaceuticals 8, 696–710 (2015).
Gunasekaran, P. et al. Pyrrole-Based Macrocyclic Small-Molecule Inhibitors That Target Oocyte Maturation. Chem Med Chem. 12, 580–589 (2017).
Rajasekaran, G., Kim, E. Y. & Shin, S. Y. LL-37-derived membrane-active FK-13 analogs possessing cell selectivity, anti-biofilm activity and synergy with chloramphenicol and anti-inflammatory activity. Biochim. Biophys. Acta. 1859, 722–733 (2017).
Wu, M. & Hancock, R. E. Interaction of the cyclic antimicrobial cationic peptide bactenecin with the outer and cytoplasmic membrane. J. Biol. Chem. 274, 29–35 (1999).
Friedrich, C. L., Rozek, A., Patrzykat, A. & Hancock, R. E. Structure and mechanism of action of an indolicidin peptide derivative with improved activity against gram-positive bacteria. J. Biol. Chem. 276, 24015–24022 (2001).
Choi, J. et al. Picroside II Attenuates Airway Inflammation by Downregulating the Transcription Factor GATA3 and Th2-Related Cytokines in a Mouse Model of HDM-Induced Allergic Asthma. PLoS One 11, e0167098 (2016).
Choi, E. J., Park, Z. Y. & Kim, E. K. Chemical Composition and Inhibitory Effect of Lentinula edodes Ethanolic Extract on Experimentally Induced Atopic Dermatitis in Vitro and in Vivo. Molecules 21, 993 (2016).
Kim, S. H., Johnson, V. J. & Sharma, R. P. Mercury inhibits nitric oxide production but activates proinflammatory cytokine expression in murine macrophage: differential modulation of NF-kappa B and p38 MAPK signaling pathways. Nitric Oxide. 7, 67–74 (2002).
Choi, E. J. et al. Heat-Killed Enterococcus faecalis EF-2001 Ameliorates Atopic Dermatitis in a Murine Model. Nutrients 8, 146 (2016).
Acknowledgements
This work was supported by National Research Council of Science & Technology (NST) grant CAP-17-03-KRIBB, (J.K.B.), the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A01057988 to S.Y.S.) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2018R1A2B6003250 to S.Y.S.).
Author information
Authors and Affiliations
Contributions
P.G. and G.R. conceptualized and performed experiments, E.H.H., Y.H.C., Y.J.C., Y.J.Y., J.E.L., H.N.K., K.L., J.S.K., H.J.L. and E.J.C. performed the experiments, E.K.K., S.Y.S. and J.K.B. directed the research. All the authors discussed the results and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gunasekaran, P., Rajasekaran, G., Han, E.H. et al. Cationic Amphipathic Triazines with Potent Anti-bacterial, Anti-inflammatory and Anti-atopic Dermatitis Properties. Sci Rep 9, 1292 (2019). https://doi.org/10.1038/s41598-018-37785-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37785-z
This article is cited by
-
Amphiphilic Triazine Polymer Derivatives as Antibacterial And Anti-atopic Agents in Mice Model
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
