
Abstract
Intratumoural heterogeneity underlies tumour escape from molecularly targeted therapy in glioblastoma. A cell-based model preserving the evolving molecular profiles of a tumour during treatment is key to understanding the recurrence mechanisms and development of strategies to overcome resistance. In this study, we established a matched pair of glioblastoma stem-like cell (GSC) cultures from patient glioblastoma samples before and after epidermal growth factor receptor (EGFR)-targeted therapy. A patient with recurrent glioblastoma (MGG70R) harboring focal, high-level EGFR amplification received the irreversible EGFR tyrosine kinase inhibitor dacomitinib. The tumour that subsequently recurred (MGG70RR) showed diploid EGFR, suggesting inhibitor-mediated elimination of EGFR-amplified tumour cells and propagation of EGFR non-amplified cell subpopulations. The MGG70R-GSC line established from MGG70R formed xenografts retaining EGFR amplification and EGFR overexpression, while MGG70RR-GSC established from MGG70RR generated tumours that lacked EGFR amplification and EGFR overexpression. MGG70R-GSC-derived intracranial xenografts were more proliferative than MGG70RR-GSC xenografts, which had upregulated mesenchymal markers, mirroring the pathological observation in the corresponding patient tumours. In vitro MGG70R-GSC was more sensitive to EGFR inhibitors than MGG70RR-GSC. Thus, these molecularly distinct GSC lines recapitulated the subpopulation alteration that occurred during glioblastoma evasion of targeted therapy, and offer a valuable model facilitating therapeutic development for recurrent glioblastoma.
Similar content being viewed by others


Introduction
Despite the standard treatment with resection, radiotherapy, and the alkylating agent temozolomide1, glioblastoma harbors a poor prognosis and remains a fatal disease for the vast majority of cases. Numerous molecularly targeted agents have been investigated in both the preclinical and clinical settings, including first-generation epidermal growth factor receptor (EGFR)-targeted agents such as gefitinib (Iressa®, AstraZeneca, London, UK), erlotinib (Tarceva®, Roche, Basel, Switzerland), and lapatinib (Tykerb®, GlaxoSmithKline, Brentford, UK)2, based on the high prevalence of aberrant EGFR activation in glioblastoma3,4. More recently, second-generation EGFR-targeted agents with irreversible inhibition and better penetration into the brain have been developed including dacomitinib (PF-00299804) (Pfizer, New York, NY)5,6. Dacomitinib is active against glioblastoma in preclinical studies7,8 and has been tested in two clinical trials (NCT01112527, NCT01520870). Of note, patient accrual in these two trials was restricted to those with EGFR gene amplification in archival tumour specimens, with an expectation of their better response to PF-002998049. The latter phase II trial (NCT01520870) reported a limited activity of the drug in recurrent glioblastoma with EGFR amplification, although a minor fraction of patients, 4 of 49 (8.2%), had durable ( > 6 months) response10.
Molecularly targeted agents have thus far been ineffective in the treatment of glioblastoma. Possible escape mechanisms include intratumoural heterogeneity11,12,13, loss of target gene expression and activation of redundant signaling pathways14,15. Elucidating these resistance mechanisms in more detail is critical for future studies of second-generation molecularly targeted agents. Our previous studies demonstrated that glioma stem-like cell (GSC)-enriched neurospheres phenotypically and genotypically recapitulate the patient tumours from which they were derived16,17. In this study, we established GSC neurospheres from patient tumour samples harvested before and after treatment with an EGFR-targeted agent, and analysed the molecular and biological characteristics that the GSC and patient tumour specimens exhibited pre- and post-treatment with this targeted drug.
Results
Phenotypic and genotypic comparison of paired patient tumour samples
Using FFPE samples of the original tumour, recurrent tumour, re-recurrent tumour and autopsy of this glioblastoma case (Fig. 1), we first characterized histopathological phenotypes of the tumours. Immunohistochemical analysis showed that MGG70R (pre-dacomitinib tumour) had diffuse and intense immunopositivity for EGFR and its activated form phospho-EGFR, a very similar staining pattern to that observed in the original tumour MGG70 (Fig. 2, Supplementary Fig. S1). In contrast to these tumours (MGG70 and MGG70R), the expression of EGFR and phospho-EGFR was substantially decreased in the post-dacomitinib tumour MGG70RR (Fig. 2). MIB-1 (Ki-67) staining revealed that MGG70RR exhibited a significantly lower proliferative rate compared to MGG70 and MGG70R (P = 0.03) (Fig. 2). Genetically, FISH analysis showed focal EGFR amplification in the newly diagnosed tumour MGG70, which was retained at a higher level in the recurrent MGG70R specimen (Fig. 3), suggesting that the treatment with radiotherapy and temozolomide did not preferentially target cell populations harboring amplified EGFR. In contrast, all tumour cells had diploid EGFR signals in the post-dacomitinib MGG70RR (Fig. 3). In the brain obtained at autopsy, there were scattered foci with relatively strong immunostaining of EGFR/phospho-EGFR (Fig. 2), but FISH analysis did not detect any cells with EGFR amplification (Fig. 3). Of note, gene amplification of other receptor tyrosine kinases (RTKs) such as platelet-derived growth factor receptor (PDGFR) and MET was not noted in any of the tumour samples (Supplementary Fig. S2). Thus, in this glioblastoma patient, prominent phenotypic and genotypic changes, most notably the elimination of EGFR-amplified tumour cells, occurred after targeted treatment with an EGFR tyrosine kinase inhibitor.

MRI and schema of clinical course of the patient GBM and glioma stem-like cell establishment. (A) The newly diagnosed tumour was located in the right posterior temporal lobe associated with perifocal edema. This was subtotally resected, followed by radiotherapy and chemotherapy with temozolomide (TMZ). (B) The tumour recurred after one year (MGG70R), which was resected. The patient was then treated with an EGFR inhibitor, dacomitinib. (C) After two months with two cycles of dacomitinib, the tumour recurred again (MGG70RR), which was surgically removed. After subsequent treatment with Bevacizumab and lomustine, the patient passed away and autopsy was performed. 70R-GSC and 70RR-GSC were established from the surgical specimens of MGG70R and MGG70RR, respectively.

Histopathological analysis reveals drastic changes in EGFR and MIB1 status in re-recurrent GBM after EGFR inhibitor therapy. Microscopic pictures of H&E staining (top row) and IHC for EGFR (2nd row), phospho-EGFR (Tyr1068, 3rd row) and MIB-1 (Ki-67, the bottom row) for, from left to right column, original tumour MGG70 (70), recurrent tumour MGG70R (70R), re-recurrent tumour MGG70RR (70RR), and autopsy material MGG70A (70A). Original magnification, x200. Note negative staining of EGFR and phospho-EGFR in tumour-associated blood vessels (arrows). pEGFR positivity (% area) and MIB-1 labeling indices of each tumour are plotted on the right.

EGFR inhibition resulted in elimination of EGFR-amplified populations. FISH images of the patient tumours, with EGFR probe in green and centromere 7 (CEN7) control probe in red. From left to right, panel represents the original tumour MGG70 (70), the first recurrent tumour before dacomitinib treatment MGG70R (70R), the re-recurrent tumour after dacomitinib treatment MGG70RR (70RR) and the autopsy material MGG70A (70A). Clumped amplification of EGFR is noted in 70 and 70 R, but not in 70RR and 70 A.
Phenotypic and genotypic characterization of GSC-derived xenografts and comparison to patient tumour specimens
We successfully established neurosphere cultures from pre- and post-dacomitinib patient tumour samples (MGG70R and MGG70RR) (Figs 1, 4A). MGG70R-GSC and MGG70RR-GSC had comparable abilities to generate a sphere from a single cell. However, cell proliferation assays demonstrated that MGG70R-GSC proliferated at a faster rate than MGG70RR-GSC (Fig. 4A). Both MGG70R-GSC and MGG70RR-GSC were able to generate orthotopic xenografts in SCID mice (Fig. 4B). Although MGG70R-GSC as well as MGG70RR-GSC-derived intracranial tumours became lethal within a similar time frame (~2 months), the size of the brains harboring MGG70R-GSC xenografts were considerably larger than that of the brains with MGG70RR-GSC xenografts (Supplementary Fig. S3). H&E stain showed that the MGG70R-GSC tumour was much larger than MGG70RR-GSC tumour, causing a striking enlargement of the implanted hemisphere while both xenografts displayed a semi-invasive phenotype with signs of moderate invasiveness (Fig. 4B). In concordance with the aforementioned changes observed in the patient tumours, the post-dacomitinib MGG70RR-GSC xenografts exhibited reduced levels of EGFR/phospho-EGFR expression as well as reduced MIB-1 labeling index, i.e., the proliferation rate (P = 0.007), compared to the pre-dacomitinib MGG70R-GSC xenografts (Figs 4C,D). EGFR amplification was observed in the MGG70R-GSC-derived xenografts but not in the MGG70RR-GSC-derived xenografts, indicating that the GSC-generated xenografts preserved the EGFR status observed in the respective patient tumour specimens (Fig. 4E). These results show that GSC neurospheres established from pre- and post-dacomitinib treatment recapitulated the phenotypic and genotypic profiles of the corresponding tumour samples in the same patient.

Paired GSC orthotopic xenografts recapitulate the molecular and phenotypic characteristics of patient tumours. (A) Cell counting assay showing different growth kinetics of MGG70R-GSCs (70R-GSC) and MGG70RR-GSCs (70RR-GSC) in culture. Shown right are representative microscopic pictures of a neurosphere generated from a single GSC. (B) H&E staining of coronal sections of the brains bearing GSC-derived orthotopic tumours. Scale bar, 5 mm. (C) H&E staining and IHC for EGFR, phospho-EGFR, and Ki-67 (MIB-1) in orthotopic GSC xenografts. Top row, MGG70R-GSC tumours. Bottom row, MGG70RR-GSC tumours. (D) MIB-1 labeling indices and pEGFR immunopositivity (% area) of MGG70R-GSC and MGG70RR-GSC tumours. (E) FISH images of the GSC xenografts, with EGFR probe in green and CEN7 control probe in red. Left, MGG70R (pre-dacomitinib) GSC tumour. Right, MGG70RR (post-dacomitinib) GSC tumour.
Phenotypic and molecular characterization of GSC neurospheres in vitro
We further molecularly characterized patient-derived GSCs in vitro. Semi-quantitative RT-PCR and western blotting showed that MGG70R-GSC overexpressed wild-type EGFR at both the transcript and protein levels and had a higher amount of phospho-EGFR in comparison to MGG70RR-GSC (Fig. 5A,B). However, western analysis of the PI3K pathway downstream of EGFR revealed that the status of Akt phosphorylation (at Thr308 and Ser473) was comparable or slightly more pronounced in MGG70RR-GSC (Fig. 5B). Similarly, activation of Erk1/2, a MAPK pathway signal transducer, was present in the paired GSC lines, but was higher in MGG70RR-GSC (Fig. 5B).

Pre-EGFR inhibitor GSCs overexpress EGFR and phospho-EGFR and are sensitive to EGFR inhibition. (A) RT-PCR showing EGFR expression in the paired GSCs, MGG70R-GSC (70R-GSC) and MGG70RR-GSC (70RR-GSC). GAPDH was used as control. (B) Western blotting showing EGFR, phospho-EGFR (Tyr1068), phospho-Akt (Thr308), phospho-Akt (Ser473), Akt, and phospho-Erk1/2 in 70R and 70RR GSCs. Actin served as control. (C) GSC sensitivities to molecular targeted agents. Cell viability assay was performed on the paired GSCs after 3-day exposure to the EGFR inhibitor lapatinib (upper), the PI3K inhibitor PF-05212384 (middle) and the MEK inhibitor PD98509 (bottom).
Sensitivities of the paired GSC neurospheres to different pathway inhibitors in vitro
We sought to determine the sensitivities of the paired GSC neurospheres to small molecule inhibitors that target different oncogenic pathways. Based on the results of western blotting and immunohistochemistry, we selected an EGFR inhibitor (lapatinib), a PI3K inhibitor (PF-05212384), and a MEK inhibitor (PD98509). In vitro cell viability assays revealed that, as expected, the paired GSC neurospheres showed differential sensitivities to lapatinib; MGG70RR-GSC was significantly more resistant compared to MGG70R-GSC (Fig. 5C). However, both GSC lines displayed a comparable sensitivity to PF-05212384 with an EC50 value below 1 μM (Fig. 5C). Both GSC lines were resistant to PD98509 (Fig. 5C).
Evaluation of mesenchymal markers and Met expression in the paired tumour specimens
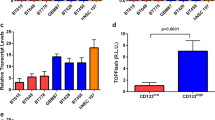
Since epithelial to mesenchymal transition (EMT) is implicated in cancer progression and resistance18,19, we explored whether a similar process is involved in glioblastoma resistance to dacomitinib. We observed that the expression of the mesenchymal markers CD44 and YKL-40 was upregulated at the time of recurrence on dacomitinib (MGG70RR) and that this change was phenocopied in the GSC-derived xenografts (Fig. 6). Intratumoural microvascular density was also elevated in MGG70RR-GSC-derived intracranial tumours (Supplementary Fig. S4). Furthermore, since Met overexpression and MET amplification has been implicated in acquired resistance to EGFR inhibition20,21, we examined Met expression in MGG70R and MGG70RR patient tumour specimens and the respective GSC tumours. We found that Met was not expressed in any of these tumours (Supplementary Fig. S5A), suggesting that Met was not involved in the resistance of MGG70RR-GSC to EGFR inhibitors. Accordingly, both MGG70R-GSC and MGG70RR-GSC were resistant to the Met inhibitor crizotinib (Supplementary Fig. S5B).

MGG70RR tumour and its GSC xenografts overexpress mesenchymal markers. Immunohistochemistry (IHC) for CD44 and YKL-40 in the patient tumours, MGG70R (70R) and MGG70RR (70RR), and the corresponding orthotopic GSC xenografts. Upper two rows, patient tumours. Lower two rows, xenografts. For CD44 IHC in MGG70RR-GSC tumours, a tumour-brain border area was depicted to demonstrate tumour-selective staining. Note negative staining of CD44 in tumour-associated blood vessels (arrow). Image quantification of IHC is shown on the right: upper plots, patient tumors; lower plots, xenografts.
Discussion
Here we present a case of recurrent glioblastoma in which EGFR gene amplification present in the pre-treatment tumour subsequently disappeared in the tumour that progressed on targeted treatment with the EGFR tyrosine kinase inhibitor dacomitinib. We successfully established GSC neurospheres from the above longitudinal tumours and demonstrated that they recapitulated the molecular changes noted in the patient tumour specimens.
To the best of our knowledge, this is the first report in glioblastoma that documents loss of receptor tyrosine kinase gene amplification in patient’s tumour after direct molecularly targeted inhibition with a small molecule agent, an observation reported in other cancers22,23. EGFRvIII, a mutant form of EGFR, was eliminated on immunohistochemistry in the majority of glioblastomas after treatment with the anti-EGFRvIII peptide vaccine rindopepimut24. In contrast, we show that EGFR amplification present in the primary tumour was retained in the recurrent tumour after treatment with radiotherapy and temozolomide, in line with a recent report showing relative stability of EGFR amplification and EGFRvIII mutation within paired primary and recurrent glioblastomas25,26.
The pre-dacomitinib MGG70R contained focal clusters with EGFR-amplified cells, suggesting intratumoural heterogeneity, a hallmark of glioblastoma11,12,13,27. Loss of EGFR amplification in the post-dacomitinib tumour indicates differential efficacy of EGFR-targeting within a single tumour depending upon EGFR amplification status28; it appears that EGFR-amplified cells were selectively eliminated by irreversible EGFR inhibition with dacomitinib and the non-amplified cells that survived seeded the re-recurrent tumour. Darwinian selection of preexisting drug-resistant clones, or so-called “subpopulation switch” has been recognized in cancer29. The current case highlights intratumoural heterogeneity as a key mediator of tumour evasion30,31 and recurrence after molecularly targeted therapy.
The unique cell models based on the paired GSC-enriched neurosphere cultures that we established before and after targeted treatment allowed us to characterize their molecular signatures and in vivo phenotypes. FISH analyses on the paired GSC xenografts demonstrated amplified and non-amplified EGFR in pre- and post-dacomitinib tumours, respectively, which reflected the differential EGFR expression levels on immunohistochemistry. In addition, the cell proliferation marker, MIB-1 in the GSC xenografts corresponded to the patient tumours. Each of these GSC models thus mirrors the biological behavior of the patient tumour specimen from which the GSCs were derived, representing the respective stage of the tumour during the clinical course of tumour evolution. This is in line with previous work from our group and others demonstrating that the GSC-enriched neurosphere models faithfully recapitulate the molecular and phenotypic characteristics of original patient tumours16,17,32,33,34.
Since MGG70RR GSCs must have been derived from subpopulations that had survived dacomitinib, we expected that these GSCs would be resistant to EGFR inhibition. Indeed, the post-dacomitinib GSCs exhibited an elevated resistance to EGFR tyrosine kinase inhibitors compared to the pre-dacomitinib GSCs. This finding provides experimental support to our clinical observation that non-EGFR-amplified glioblastoma cells were preferentially resistant to dacomitinib. Interestingly, small molecule inhibitors of the major EGFR downstream signaling pathway, the PI3K-Akt axis, resulted in comparable viability reduction of the pre- and post-dacomitinib GSCs. Phosphorylation status of both Akt and Erk was comparable between the GSCs despite clearly reduced EGFR phosphorylation in post-dacomitinib GSCs. Thus, the loss of EGFR amplification rendered GSCs less dependent on the EGFR signaling for survival/proliferation, however did not alter the dependence on the hub pathway of PI3K-Akt. Compensatory activation of other ErbB family35 or alternative receptor tyrosine kinases such as Met20,21,36 through gain of gene amplification, and downstream signaling by acquired genetic alterations such as PTEN loss37,38 could contribute to the acquired resistance to EGFR tyrosine kinase inhibitors. Lack of MET amplification, Met and phosphorylated Met levels, and insensitivity to the Met inhibitor crizotinib in pre and post-dacomitinib GSCs support the notion that Met does not play a significant role in the emergent tumour resistance in the current case. Alternatively, EMT could contribute to the acquired resistance to EGFR inhibitors as implicated in lung cancer18. Indeed, we did observe increased expression of representative mesenchymal markers in the post-dacomitinib tumours, both in the patient and in the xenografts, as seen with other recurrent GSC xenografts39. Further study is necessary to determine whether an EMT-like phenomenon observed in relapsed glioblastoma is associated with resistance to targeted therapy.
Here we show that paired GSC neurospheres serve as a unique preclinical model that allows investigations of the genetic and biological evolution of tumours, and treatment evasion mechanisms in glioblastoma. However, whether the findings derived from the particular case are applicable beyond the single patient is currently uncertain, which represents the limitation of the current work. Future studies are necessary to clarify whether GSC cultures may provide a robust tool enabling selection of patients potentially receiving benefit from a certain molecularly targeted agent40,41,42. Resection of relapsed disease could bring opportunities for not only assessing pharmacodynamic treatment effect and molecular profiling, but also for isolation of GSCs to screen drugs potentially efficacious for recurrent disease.
Conclusion
GSC neurospheres established from longitudinal patient glioblastoma samples before and after targeted treatment with the EGFR inhibitor dacomitinib recapitulated the molecular changes observed in the patient tumours. Paired GSCs before and after therapy serve as a powerful tool for understanding of tumour evolution through the clinical course of molecularly targeted therapy as well as for drug screening for the recurrent disease.
Materials and Methods
All methods were performed in accordance with the relevant guidelines and regulations. The Institutional Review Board (IRB) at MGH approved the portion of this study involving a human participant. Clinical information and tumour specimens were collected under IRB-approved protocols and informed consent had been previously obtained from the patient. The animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at MGH.
Clinical course of an EGFR-amplified glioblastoma case and establishment of paired glioma stem-like cells
A 62-year-old, right-handed man presented with personality changes, fatigue, and a left hemiparesis and was found to have a heterogeneously enhancing mass in the right temporal lobe associated with surrounding edema (Fig. 1A). The tumour was subtotally resected and the pathological diagnosis was glioblastoma (patient tumour designated as MGG70). He received postoperative standard therapy with focal, fractionated radiotherapy combined with concurrent and adjuvant temozolomide. The tumour recurred after one year (Fig. 1B), which was resected (designated MGG70R). A sample of fresh surgical specimen was used to generate GSC culture, designated MGG70R-GSC. As the recurrent tumour harbored focal, high-level EGFR amplification, he was enrolled in a phase II trial of an oral, irreversible EGFR tyrosine kinase inhibitor, dacomitinib (NCT01112527). However, the glioblastoma progressed on MRI scans after two months of treatment with dacomitinib (Fig. 1C). At the time of this second recurrence the tumour was resected (designated MGG70RR). Another GSC culture, designated MGG70RR-GSC, was generated from this surgical specimen Despite further treatment with bevacizumab and lomustine, he expired 7 months later. An autopsy was obtained (tumour designated MGG70A).
Tumour samples, MGG70R and 70RR, were sterilely obtained at surgery and were used to establish GSC-enriched neurospheres as described previously43. After enzymatic dissociation, cells were grown in Neurobasal medium (Invitrogen) supplemented with L-glutamine (3 mM; Mediatech), B27 supplement (Invitrogen), N2 supplement (Invitrogen), heparin (5 mg/mL; Sigma), EGF (20 ng/mL; R and D systems), and FGF2 (20 ng/mL; Peprotec). Neurospheres were passaged using TrypLE (Gibco).
Pathology and immunohistochemistry
Hematoxylin and eosin (H&E) staining and immunohistochemistry were performed on formalin-fixed, paraffin-embedded (FFPE) tissues from MGG70, MGG70R, MGG70RR, and MGG70A, as described16,43. Primary antibodies used were MIB-1 (Dako, 1:150), EGFR, phosphorylated EGFR (phospho-EGFR), CD44, Mesenchymal-epithelial transition (Met) (all Cell Signaling, 1:500), and YKL40 (Quidel, 1:300). For antigen retrieval, microwave treatment in citrate buffer (for MIB-1), EDTA (1 mM, pH = 8, for EGFR and phosphorylated EGFR) and saponin (0.1%, for CD44 and YKL-40) were used. MIB-1 labeling index was calculated by counting MIB-1 positive cells and total cells (more than 1000 cells) in 4 randomly chosen high power fields within the different tumour specimens. Immuno-positivity of P-EGFR, CD44 and YKL40 was quantified by using Image J (NIH) in an unbiased manner and expressed as % positive area with SD.
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) for EGFR, PDGFRA and MET was performed using BAC probes CTD-2113A18 (7p EGFR locus), RP11-114O6 (7q MET locus), RP11-819D11 (4q PDGFRA locus) and centromere 7 copy number control as described11,44. Cells were counted in at least 3 different high-power fields, and gene amplification was defined when gene/control probe copy number ratio was 2.0 or greater.
Western blotting
Cells were lysed in RIPA buffer and lysates were analyzed via western blot as previously described16. The primary antibodies used were against EGFR, phospho-EGFR, Akt, phosphorylated-Akt (Ser473 & Thr308), extracellular signal-regulated kinase (Erk) 1/2, and phosphorylated-Erk 1/2 (all Cell Signaling, 1:1,000), and Actin (Sigma, 1: 5,000).
Reverse transcription – polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells using Trizol (Invitrogen). First strand cDNA was synthesized from 2 μg of RNA using Superscript 3 (Invitrogen). PCR was performed using Hotstar Taq polymerase (Qiagen) and the DNA samples were run on a 1% agarose gel containing ethidium bromide for visualization. The PCR primer sequences were: GAPDH (forward, CAATGACCCCTTCATTGACC; reverse, GACAAGCTTCCCGTTCTCAG), EGFR (forward, ATGCGACCCTCCGGGACG; reverse, ATTCCGTTACACACTTTGCGGC), and EGFR from exon 4 to 6 (forward, CATGTCGATGGACTTCCAGA; reverse, GGGCACAGATGATTTTGGTC).
Cell proliferation assay
Dissociated cells were seeded at 20,000 cells per well to 24-well plates. Neurospheres were collected and dissociated after 2, 4, 6, and 8 days of culture, and viable cells were counted using trypan blue. The experiment was performed in triplicate.
Mouse intracranial orthotopic xenograft models
GSC neurospheres were dissociated and 50,000 cells in 3 µL were stereotactically injected into the right striatum of 6–8 week-old female SCID mice as described previously16,43. Mice were sacrificed and brains removed when animals became symptomatic.
Cell viability assay
Dissociated cells were seeded at 5,000 cells per well to 96-well plates; serially diluted drugs were added and cells were cultured for 3 days. Cell viability was measured by MTS cell viability assay with CellTiter 96 Aqueous One Solution (Promega, Madison, WI) to determine effective concentration 50 (EC50) values. Experiments were performed in triplicate. Targeted agents tested include the EGFR and HER2 inhibitor lapatinib (Tykerb®, GlaxoSmithKline, Brentford, UK), the phosphatidylinositol-3 kinase (PI3K) and mammalian target of rapamycin inhibitor PF-05212384 (gedatolisib) (Pfizer), the mitogen-activated protein kinase (MAPK)/Erk kinase (MEK) inhibitor PD98509 (Cell Signaling), and the mesenchymal-epithelial transition (Met) and anaplastic lymphoma kinase inhibitor crizotinib (Xalkori®, Pfizer, New York, NY).
Statistical analyses
All statistical analyses were performed using JMP9.0 (SAS, Cary, NC). Two-tailed t-tests (unpaired) with Welch’s correction were employed to compare continuous variables between two groups. P values less than 0.05 were considered significant.
References
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine 352, 987–996 (2005).
Tanaka, S., Louis, D. N., Curry, W. T., Batchelor, T. T. & Dietrich, J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol 10, 14–26, https://doi.org/10.1038/nrclinonc.2012.204 (2013).
Bigner, S. H. et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res 50, 8017–8022 (1990).
Sugawa, N., Ekstrand, A. J., James, C. D. & Collins, V. P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA 87, 8602–8606 (1990).
Gonzales, A. J. et al. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Mol Cancer Ther 7, 1880–1889, https://doi.org/10.1158/1535-7163.MCT-07-2232 (2008).
Janne, P. A. et al. Dacomitinib as first-line treatment in patients with clinically or molecularly selected advanced non-small-cell lung cancer: a multicentre, open-label, phase 2 trial. Lancet Oncol 15, 1433–1441, https://doi.org/10.1016/S1470-2045(14)70461-9 (2014).
Zahonero, C. et al. Preclinical Test of Dacomitinib, an Irreversible EGFR Inhibitor, Confirms Its Effectiveness for Glioblastoma. Mol Cancer Ther 14, 1548–1558, https://doi.org/10.1158/1535-7163.MCT-14-0736 (2015).
Zhu, Y. & Shah, K. Multiple lesions in receptor tyrosine kinase pathway determine glioblastoma response to pan-ERBB inhibitor PF-00299804 and PI3K/mTOR dual inhibitor PF-05212384. Cancer biology & therapy 15, 815–822, https://doi.org/10.4161/cbt.28585 (2014).
Haas-Kogan, D. A. et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst 97, 880–887, https://doi.org/10.1093/jnci/dji161 (2005).
Sepulveda-Sanchez, J. M. et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol 19, 1522–1531, https://doi.org/10.1093/neuonc/nox105 (2017).
Snuderl, M. et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20, 810–817, https://doi.org/10.1016/j.ccr.2011.11.005 (2011).
Szerlip, N. J. et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA 109, 3041–3046, https://doi.org/10.1073/pnas.1114033109 (2012).
Sottoriva, A. et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 110, 4009–4014, https://doi.org/10.1073/pnas.1219747110 (2013).
Network, C. G. A. R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068, https://doi.org/10.1038/nature07385 (2008).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477, https://doi.org/10.1016/j.cell.2013.09.034 (2013).
Wakimoto, H. et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. https://doi.org/10.1093/neuonc/nor195 (2011).
Wakimoto, H. et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res 20, 2898–2909, https://doi.org/10.1158/1078-0432.CCR-13-3052 (2014).
Sequist, L. V. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3, 75ra26, https://doi.org/10.1126/scitranslmed.3002003 (2011).
Suda, K. et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer 6, 1152–1161, https://doi.org/10.1097/JTO.0b013e318216ee52 (2011).
Jun, H. J. et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 31, 3039–3050, https://doi.org/10.1038/onc.2011.474 (2012).
Engelman, J. A. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043, https://doi.org/10.1126/science.1141478 (2007).
Hurley, J. et al. Docetaxel, cisplatin, and trastuzumab as primary systemic therapy for human epidermal growth factor receptor 2-positive locally advanced breast cancer. J Clin Oncol 24, 1831–1838, https://doi.org/10.1200/JCO.2005.02.8886 (2006).
Mittendorf, E. A. et al. Loss of HER2 amplification following trastuzumab-based neoadjuvant systemic therapy and survival outcomes. Clin Cancer Res 15, 7381–7388, https://doi.org/10.1158/1078-0432.CCR-09-1735 (2009).
Schuster, J. et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol 17, 854–861, https://doi.org/10.1093/neuonc/nou348 (2015).
van den Bent, M. J. et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol 17, 935–941, https://doi.org/10.1093/neuonc/nov013 (2015).
Felsberg, J. et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin Cancer Res 23, 6846–6855, https://doi.org/10.1158/1078-0432.CCR-17-0890 (2017).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401, https://doi.org/10.1126/science.1254257 (2014).
Eller, J. L., Longo, S. L., Hicklin, D. J. & Canute, G. W. Activity of anti-epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery 51, 1005–1013, discussion 1013–1004 (2002).
Inukai, M. et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res 66, 7854–7858, https://doi.org/10.1158/0008-5472.CAN-06-1951 (2006).
Kim, J. et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 28, 318–328, https://doi.org/10.1016/j.ccell.2015.07.013 (2015).
Lee, J. K. et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat Genet 49, 594–599, https://doi.org/10.1038/ng.3806 (2017).
Lee, J. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9, 391–403, https://doi.org/10.1016/j.ccr.2006.03.030 (2006).
Davis, B. et al. Comparative genomic and genetic analysis of glioblastoma-derived brain tumor-initiating cells and their parent tumors. Neuro Oncol 18, 350–360, https://doi.org/10.1093/neuonc/nov143 (2016).
Rosenberg, S. et al. Multi-omics analysis of primary glioblastoma cell lines shows recapitulation of pivotal molecular features of parental tumors. Neuro Oncol 19, 219–228, https://doi.org/10.1093/neuonc/now160 (2017).
Clark, P. A. et al. Activation of multiple ERBB family receptors mediates glioblastoma cancer stem-like cell resistance to EGFR-targeted inhibition. Neoplasia 14, 420–428 (2012).
Stommel, J. M. et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318, 287–290, https://doi.org/10.1126/science.1142946 (2007).
Mellinghoff, I. K. et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. The New England journal of medicine 353, 2012–2024, https://doi.org/10.1056/NEJMoa051918 (2005).
Bianco, R. et al. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene 22, 2812–2822, https://doi.org/10.1038/sj.onc.1206388 (2003).
Esaki, S. et al. Blockade of transforming growth factor-beta signaling enhances oncolytic herpes simplex virus efficacy in patient-derived recurrent glioblastoma models. Int J Cancer 141, 2348–2358, https://doi.org/10.1002/ijc.30929 (2017).
Pollard, S. M. et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell stem cell 4, 568–580, https://doi.org/10.1016/j.stem.2009.03.014 (2009).
Daher, A. & de Groot, J. Rapid identification and validation of novel targeted approaches for Glioblastoma: A combined ex vivo-in vivo pharmaco-omic model. Exp Neurol. https://doi.org/10.1016/j.expneurol.2017.09.006 (2017).
Saito, N. et al. A high Notch pathway activation predicts response to gamma secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 32, 301–312, https://doi.org/10.1002/stem.1528 (2014).
Wakimoto, H. et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res 69, 3472–3481, https://doi.org/10.1158/0008-5472.CAN-08-3886 (2009).
Chi, A. S. et al. Prospective, high-throughput molecular profiling of human gliomas. J Neurooncol 110, 89–98, https://doi.org/10.1007/s11060-012-0938-9 (2012).
Acknowledgements
We acknowledge excellent technical assistance from Clarice Chang (Department of Pathology, Massachusetts General Hospital). This work was supported by NIH R01NS032677 (RLM), ABTA Discovery grant (HW), R21CA183686 (AJI) and Burroughs Wellcome Fund (DPC). Pfizer, Inc, provided funding support for the clinical trial that the patient described in the manuscript was treated on.
Author information
Authors and Affiliations
Contributions
S.T. and H.W. designed the research. S.T., S.L., J.K., M.L.O. and C.C. performed experiments. A.J.I. and H.W. supervised experiments. A.J.I., K.S., R.L.M., S.D.R., D.P.C., A.S.C. and H.W. interpreted and analyzed data. S.T., T.T.B., S.D.R., A.S.C., and H.W. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
TTB received consulting honoraria from Merck, Genomicare, NXDC, UpToDate, Inc, and Champions Biotechnology. AJI received equity from ArcherDx. The other authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tanaka, S., Luk, S., Kiyokawa, J. et al. Genetically distinct glioma stem-like cell xenografts established from paired glioblastoma samples harvested before and after molecularly targeted therapy. Sci Rep 9, 139 (2019). https://doi.org/10.1038/s41598-018-37437-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37437-2
This article is cited by
-
Exosome-transmitted circCABIN1 promotes temozolomide resistance in glioblastoma via sustaining ErbB downstream signaling
Journal of Nanobiotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
