
Abstract
Switches in heterogamety are known to occur in both animals and plants. Although plant sex determination systems probably often evolved more recently than those in several well-studied animals, including mammals, and have had less time for switches to occur, we previously detected a switch in heterogamety in the plant genus Silene: section Otites has both female and male heterogamety, whereas S. latifolia and its close relatives, in a different section of the genus, Melandrium (subgenus Behenantha), all have male heterogamety. Here we analyse the evolution of sex chromosomes in section Otites, which is estimated to have evolved only about 0.55 MYA. Our study confirms female heterogamety in S. otites and newly reveals female heterogamety in S. borysthenica. Sequence analyses and genetic mapping show that the sex-linked regions of these two species are the same, but the region in S. colpophylla, a close relative with male heterogamety, is different. The sex chromosome pairs of S. colpophylla and S. otites each correspond to an autosome of the other species, and both differ from the XY pair in S. latifolia. Silene section Otites species are suitable for detailed studies of the events involved in such changes, and our phylogenetic analysis suggests a possible change from female to male heterogamety within this section. Our analyses suggest a possibility that has so far not been considered, change in heterogamety through hybridization, in which a male-determining chromosome from one species is introgressed into another one, and over-rides its previous sex-determining system.
Similar content being viewed by others

Introduction
Genetic sex-determining systems in plants with separate sexes (dioecious plants) are thought mostly to have evolved via gynodioecy, involving a male sterility mutation in a hermaphroditic or monoecious ancestor as a first step1, or perhaps sometimes gradually from a monoecious ancestor, in the “paradioecy pathway”2,3,4. The gynodioecy route is predicted to generate male heterogamety more often than female heterogamety, because male sterility mutations are mostly recessive, consistent with females being the homozygous sex1. It has therefore been suggested that species with female heterogamety evolved this state secondarily, from ancestral systems with male heterogamety. Although changes in heterogamety are known in several animal and plant taxa, empirical evidence on the directions of the changes is scanty. However, some species with female heterogamety appear to have evolved without a previous male heterogametic system. For example, in the genus Fragaria (Rosaceae), subdioecious and dioecious species have female heterogamety, with dominant male sterility alleles in F. virginiana5,6 and F. chiloensis7. The analysis of the genetic determination of male sterility in a gynodioecious species, F. vesca subsp. bracteata, revealed the presence of a dominant nuclear male sterility gene (it is epistatically dominant to a dominant fertility restorer that is also present)8. In two other plant genera where multiple species have been studied, Cotula9 (Asteraceae), and Pistacia10,11 (Anacardiaceae), only female heterogamety has been found, again suggesting that there might have been no previous history of male heterogamety. In two further genera with female heterogamety, only single species have been tested (Distichlis12 in the Poaceae, and Potentilla13 in the Rosaceae).
Several animal genera with both male and female heterogamety are known14, but there is firm evidence in only two plant genera, Populus15, in the family Salicaceae, and Silene16,17, in the family Caryophyllaceae. In fish, many examples of differences in the chromosome carrying the sex-determining locus in related species have been documented, including in medaka and related species18,19 and in cichlids20,21,22, sometimes changing the heterogamety. Similar events have also occurred in Silene. Turnovers of sex-determining regions, with no change in heterogamety, have been described in Fragaria species, involving inferred repeated translocation of small regions between homeologous chromosomes in octoploid species23. In the Salicaceae, almost all species are dioecious, and the available data on fossils suggest that dioecy could be as old as 45 My24,25, so multiple changes could have occurred during this evolutionary history. The current sex chromosomes in Populus trichocarpa are estimated to have evolved only ~6–7 MYA ago, based on divergence between X- and Y-linked sequences26, supporting the view that multiple switches in sex determination occurred or even a possibility of independent evolution of dioecy in this lineage of Salicaceae, rather than dioecy being ancestral to the family27,28,29,30. Despite the different localization of sex-determining regions in Salix and Populus (chromosome 15 and 19), recent work in S. purpurea suggests a possible common origin of some sex-linked loci in Salix and Populus31. The sex-linked region in S. purpurea is estimated to have evolved recently31, so it is not possible to test whether female heterogamety was ancestral to Salicaceae.
The genus Silene (family Caryophyllaceae) has a diversity of sexual systems32, including naturally occurring cytoplasmic male sterility (gynodioecy), which have been studied in detail in S. vulgaris and S. nutans33,34,35. The evolution of dioecy in the genus Silene is therefore thought to have involved the gynodioecy-based two-locus pathway. The chromosome number in almost all species of the genus Silene is 2n = 2436 (although polyploids have been described in several species of the genus Silene and are most common among North American species37). In contrast to the genus Fragaria, all dioecious species of the genus Silene so far studied are diploid in natural populations38,39. Dioecy in the Caryophyllaceae probably evolved within the past 10 million years17,40,41,42,43. Multiple changes in sex determination are therefore less likely than in the Salicaceae, and changes in state can reliably be inferred. In the genus Silene, at least three independent origins of genetic sex determination (dioecy and/or subdioecy) have been reported17, as well as a switch from XY to ZW, or vice versa, in section Otites17. In the present study, we identified sex-linked genes in three section Otites species (Fig. 1), S. borysthenica (group Cyri), and S. colpophylla and S. otites (group Otites), allowing us to test (i) whether male heterogamety was ancestral in section Otites (which also includes S. pseudotites), and (ii) whether the chromosomes carrying the sex-determining loci of different dioecious species with the same heterogamety evolved from the same or different autosome pairs, or are likely to represent turnovers in sex determination, and to detect the lineages that underwent switches in heterogamety, potentially helping to test between different hypotheses for the causes of these changes.

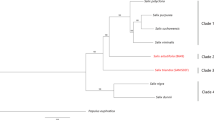
Results of StarBeast phylogenetic analysis of 662 single-copy orthogroup sequences. Species with female heterogamety are indicated by red boxes, and male heterogamety by blue boxes. Female heterogamety in Silene borysthenica, and S. otites, and male heterogamety in S. colpophylla, were confirmed or inferred from the molecular data presented here, and the male heterogamety in S. pseudotites and female heterogamety in S. wolgensis from results of interspecific crosses. The values indicated at the nodes are posterior probabilities. Branch colours indicate the most probable scenario for the evolution of the sex-determining system in section Otites. Branches and nodes where female heterogamety is inferred are coloured red, while the branches with male heterogamety are in blue. The branch to S. baschkirorum is coloured green as its heterogametic state is not known. Silene sibirica is not dioecious.
The causes of turnovers in sex determination and switches in heterogamety have been classified into three main categories (reviewed by Beukeboom & Perrin14). (1) Selectively neutral changes. Recent modelling of finite populations44,45 has shown that genetic drift favors the spread and fixation of dominant masculinising or feminising alleles, which can lead to changed sex determination in a population in which a new genotype involved in sex determination has no fitness advantage (for example a dominant female-determining mutation on an X chromosome in an XY system); dominant alleles are therefore predicted to replace ones with lesser dominance. (2) Changes caused by fitness differences between sex phenotypes can be due to a new sex-determining mutation arising closely linked to a gene with a polymorphism for sexually antagonistic alleles (reviewed by van Doorn46), or be advantageous due to genetic degeneration of an ancestral non-recombining sex linked region (the mutational load hypothesis of Blaser et al.47). In this type of model, the combined effects of sexually antagonistic mutations and mutational load can potentially lead to repeated turnovers in the locus responsible for sex determination, possibly involving a limited set of chromosomes that carry genes affecting gender development (the “hot-potato model“ of Blaser et al.48). In these models, the ancestral heterogamety is preserved, as is observed in anurans, though Glandirana rugosa (syn. Rana rugosa) is an exception49,50,51 and this is supported by the data from other vertebrates reviewed by Blaser et al.48. (3) Finally, sex ratio selection can play a role if a population’s optimal sex ratio in the population is non-1:1, or if the sex ratio has been distorted by a genomic conflict (reviewed by Beukeboom & Perrin14).
As described more fully in the Discussion section, our results allow us to exclude some of these models for Silene section Otites.
Results
Illumina RNAseq sequencing, 454 RNAseq sequencing and genetic analyses
The overall results of the different approaches employed in this study suggest that sex-determining regions are found on three different chromosomes in the genus Silene: two different S. latifolia autosomes carry homologues of the S. otites and S. borysthenica (W) and S. colpophylla (Y) sex-determining regions, and neither is homologous with the S. latifolia XY pair (Fig. 2A). We next describe the evidence that supports these conclusions.

Comparison of the genetic maps of S. otites and S. colpophylla. The figure shows that the S. colpophylla homologs of the sequences that are sex-linked in S. otites are not sex-linked in S. colpophylla (A) and vice versa (B). The completely sex-linked sequences (no recombinants found in the mapping population) that also show complete linkage disequilibrium in natural populations of S. otites are marked by a red line, and by a violet line in S. colpophylla.
S. otites
Candidate fully or partially sex-linked genes in S. otites were identified and genetically mapped (Fig. 2A) using several approaches (see Materials and Methods and Supporting information Table S1A). Initial 454 sequencing yielded 36 sequences suitable for genetic mapping in our S. otites family of 113 plants (see Material and Methods), which identified seven partially sex-linked sequences. These markers are denoted by names beginning with oti, followed by a number. Orthology with these genes identified the S. otites sex chromosome as the homolog of S. latifolia linkage group LG6, in the numbering of Bergero et al.52, or LG3 in the numbering of Papadopulos et al.42, which we adopt in what follows. Ten further sequences that were previously mapped to this S. latifolia linkage group52 were then identified and genetically mapped in our S. otites family; these markers’ names begin with E (following Bergero et al.52). To search for more sex-linked genes, we did Illumina RNAseq using the parents of the same S. otites family and 6 progeny of each sex. The results were analysed using the SNP based workflow in the LINKYX pipeline53 (see Methods), which identified 318 sequences with potentially completely Z-linked and/or W-linked SNPs; these markers are denoted by names beginning with oti, followed by WS (standing for W-linked SNP) and a number. 25 of the sequences identified by the LINKYX pipeline were genetically mapped using the SNP variants, of which 16 were identified as pseudoautosomal and 9 were not localized for technical reasons (unexpected segregation patterns due to multiple copies amplified in PCR). In a third approach, we tested sequences that show female-specific transcription in leaves and flower buds (using LINKYX’s workflow for detecting sex-specific transcripts); this yielded a set of 83 sequences detected only in females in both leaf and flower bud tissues (denoted by names beginning with otiWT, followed by a number (where T indicates that the sequence was detected by analysing transcripts). Further testing using 29 sequences chosen from the set of 83 sequences was done using PCR on genomic DNA in mapping population. In the cases where sex-linked presence/absence PCR polymorphism was not found, we have searched for sex-linked SNP polymorphisms in the corresponding sequences. This study identified one pseudoautosomal sequence (based on mapping an SNP in the mapping population) and four completely sex-linked sequences (based on PCR presence/absence polymorphisms in the mapping population, while the other 24 sequences did not show any sex linkage.
Overall, these three approaches yielded a genetic map with a total of 4 fully sex-linked and 34 pseudoautosomal genes in S. otites. Fig. 2A shows the genetic map based on all 38 markers (Supporting information Table S1A). The sex-determining locus maps at 138.0 cM, and only the four markers just mentioned showed complete sex linkage; almost the whole of the chromosome’s total map length of 302.7 cM consists of two genetically large pseudo-autosomal regions flanking a fully sex-linked region. All four candidate fully sex-linked polymorphisms showed complete agreement with the sex phenotype in two natural population samples; PCR amplifications of these sequences yielded products only from female genomic DNA samples, and not from males, confirming that they are fully sex-linked (Supporting information Table S2).
To estimate the physical size of the fully sex-linked region, BACs carrying the four fully sex-linked sequences were isolated and sequenced. The W-linked BAC clones proved to have low gene density, consistent with this genome region being non-recombining, and this sequencing revealed only one new sex-linked gene (Supporting information Table S3). The sum of the lengths of the two smallest BACs sequences, plus the smallest distances of the mapped genes from the BAC ends in two another BACs, yields a size of at least 180 kb for the region, yielding a mean of one gene per 36 kb, a very low density.
S. colpophylla
In S. colpophylla, the LINKYX (its workflow based on the study of SNPs in RNAseq data) identified 88 sequences with SNPs suggesting either X- and/or Y-linkage. SNPs in 48 of the 88 putatively sex-linked sequences just described were mapped in our sibship of 69 plants. This mapping confirmed full sex-linkage for 16 sequences, and partial sex-linkage for 28 (pseudoautosomal) genes, while 4 were not localized from technical reasons (Supporting information Table S1B). A further 1,671 sequences showed male-limited transcription in flower buds in the same sample of plants (as identified by workflow for detection of sex specific transcripts in LINKYX), but only 22 of these showed male-specific expression in leaves, and only two of them showed male-specific PCR amplification from genomic DNA (one is a sequence with no known homologues, and the other shows homology to F-box proteins). The complete sex linkage of these two sequences was confirmed by mapping of the presence/absence PCR polymorphism in the mapping population. In contrast, only 19 sequences showed female-specific transcription in flower buds, and only one also did so in leaves, and no sequence showed female-specific PCR amplification from genomic DNA. These results support the previous evidence that this species has male heterogamety38. In total, we have identified 18 fully sex-linked and 28 pseudoautosomal sequences.
Homologs could be identified between S. otites or S. colpophylla for 14 genes showing complete or partial, sex-linkage in one species or the other, and in all cases genes that are sex-linked in one species are autosomal in the other (Fig. 2). The conclusion that the two species have non-homologous sex chromosomes is further supported by screening recently mapped scaffolds from S. latifolia42 with homologs of sex-linked sequences from S. otites and S. colpophylla. 29 of the 38 S. otites sex-linked sequences detected in these scaffolds map to the linkage group identified by our genetic mapping, LG3 (see above), while nine map to five other LGs (Supporting information Fig. S1). 27 homologs of the 29 S. colpophylla sex-linked sequences detected in these scaffolds map to LG6, and two to LG1 (Supporting information Fig. S1). The discrepancies may reflect incorrect assignments of some S. latifolia sequences to these scaffolds, or the truly homologous scaffolds were not yet sequenced in some cases and the other ones contained a sequence with sufficient homology to tested sequence as well. Errors in our genetic mapping are much less likely.
Figure 2B shows the genetic map of the S. colpophylla chromosome carrying the sex-determining locus. The genetic results are consistent with male heterogamety, and our 46 markers place the estimated location of a male-determining locus at 15.8 cM, out of a total map length of 39.4 cM for this chromosome. The fully sex-linked region includes a higher proportion of the sex-linked genes identified than in S. otites.
The 18 completely sex-linked markers (with no recombinants with the sex-determining locus in our family of 69 progeny) correspond to at a least 16 different genes (in two cases the mapped markers could correspond to different parts of the same gene). However, as markers that were expected to be closely linked to the sex-determining region in S. colpophylla were preferentially selected for mapping, no reliable comparison can be made of the sizes of the non-recombining region with those in the other species studied here.
S. borysthenica
For S. borysthenica, no information on heterogamety was previously available. We again tested sex linkage by LINKYX analysis of SNPs in RNAseq data, using the parents and 12 progeny plants in a full sib family from this species. This approach directly yielded 99 sequences showing probable Z- and/or W-linkage as confirmed in IGV browser. In addition, we tested 318 sequences that showed Z- or W-linked SNP(s) in S. otites, of which 62 also showed (in IGV browser) complete sex-linkage in this small sample of S. borysthenica family; conversely, 47 of the 99 sequences with sex-linkage ascertained directly in S. borysthenica also showed sex-linkage in our RNAseq sequenced samples of S. otites family. We therefore conclude that the same chromosome carries the sex-determining gene in both these species. 19S. borysthenica putatively sex-linked sequences showed homology with mapped S. latifolia scaffolds. Consistent with our identification of the chromosome carrying the S. otites sex-determining locus, 13 of these (7 newly identified in S. borysthenica and 6 that are sex-linked both in S. borysthenica and S. otites) were detected in LG3 scaffolds; all except one of the scaffolds carrying homologs of the S. otites and S. borysthenica sex-linked genes are located in the upper arm of S. latifolia LG3 (Supporting information Figs S1 and S2). Finally, we tested (using presence/absence PCR polymorphism) sex linkage of the S. borysthenica gene borW44973, which is the homolog of otiW43556, one of the four S. otites genes showing complete W linkage and female specific expression; we used a larger mapping population of 32 plants from the same sibship as the used for RNAseqs. As expected under a ZW system, all females but no males had the allele that was assigned as W-linked in our smaller sample of this S. borysthenica sibship.
Phylogenetic analyses: Origin of female heterogamety within section Otites
Our Bayesian phylogenetic analysis yielded estimated phylogenetic relationships between members of the section Otites (see Fig. 1). S. sibirica appears to be a very close relative of section Otites (the estimated time of the most recent common ancestor is about 0.72 MYA). Our phylogenetic analysis estimates that the dioecious section Otites evolved very recently, with an age of 0.55 MYA. Silene sibirica is not dioecious, but is gynodioecious, having female and hermaphrodite sex morphs. Gynomonoecious individuals with hermaphrodite and female flowers are also occasionally found (Supporting information Fig. S3).
Our BayesTraits analysis infers (see Methods) female heterogamety for both the most recent common ancestor of the section Otites, and of the Otites group within this section (Supporting information Table S4a,b). MultiState analysis in BayesTraits V3.0 evaluates this scenario as 5 times more probable than the next most probable scenario, constituting a significant difference according to the scale proposed by Kass and Raftery54, which considers twofold or greater differences in ln(marginal likelihood) values to be significant. Given the inferred tree, ancestral female heterogamety is consistent with the fact that both the extant species with female heterogamety, S. otites and S. borysthenica (see Fig. 2) have sex-determining loci located on a chromosome homologous with the same S. latifolia LG (LG3); see Supporting information Fig. S1).
In the case of the two Otites group species S. colpophylla into S. pseudotites, however, their shared male heterogamety could reflect gene flow from S. colpophylla into S. pseudotites (or its ancestor). This possibility is evaluated in the Discussion section. We, therefore, also allowed S. pseudotites to have either male or female heterogamety. We again calculated the probabilities of all nine possible following scenarios ((i) both section and group Otites ZW, (ii) section Otites ZW, group Otites XY, (iii) section Otites ZW, group Otites non-dioecious, (iv) section Otites XY, group Otites ZW, and (v) both section and group Otites XY), (vi) section Otites XY, group Otites non-dioecious), (vii) section Otites N, group Otites ZW), (viii) section Otites non-dioecious, group Otites XY) and (ix) both section and group Otites non-dioecious. The conclusion that the most recent common ancestor of both the section Otites and group Otites had female heterogamety is then 17 times more probable than the next alternative. As S. colpophylla males are heterogametic, this implies that there was a change in heterogamety, as well as a change in the chromosome carrying the sex-linked genes, during the evolution of this species. The chronogram therefore suggests that male heterogamety evolved on the S. colpophylla branch within group Otites about 0.35 MYA (Fig. 1).
Discussion
Our results confirm the male heterogamety in S. colpophylla and female heterogamety in Silene otites previously inferred using AFLP17,38 and markers from 454 sequencing17, and demonstrate the presence of a large region with suppressed XY recombination in S. colpophylla (corresponding to a large homologous region spanning at least 18 cM in S. otites), and our new data reveal that the chromosome carrying the sex-determining genes in S. colpophylla evolved from different autosome pair from that carrying the S. otites sex-determining genes. The overall results (Fig. 1, Supporting information Table S4a,b) suggest that the ancestral state in both section Otites and group Otites was female heterogamety, and that the original sex-determining region evolved on the homolog of autosomal linkage group 3 of S. latifolia.
What are possible scenarios for changes in heterogamety in section Otites? The scenario most strongly supported by our phylogenetic analyses suggests that the most recent common ancestors of the whole section Otites and of the group Otites had female heterogamety (Supporting information Table S4). Combined with the results of our genetic analyses, we can reject a simple scenario involving single origin of dioecy and a single change in heterogamety without any further event such as introgression of a sex chromosome from another species. Four alternatives are possible. The first two scenarios involve male heterogamety as the ancestral state in section Otites, while the other two involve ancestral female heterogamety, as the analysis of ancestral states suggests. We next consider the arguments against the first three scenarios, and the evidence that the fourth one is more plausible.
First, even if male heterogamety was ancestral in section Otites and female heterogamety evolved independently, it is not parsimonious to propose that the same autosome evolved independently to become a sex chromosome. In the second possibility, male heterogamety is again ancestral in section Otites but it changed to female heterogamety in section Cyri and a W chromosome introgressed into S. otites, over-riding its previous sex-determining system, and resulting in female heterogamety in this species. Again, the analysis of ancestral states does not support this. Moreover, introgression is unlikely, given the geographic and phylogenetic distance between species of groups Otites and Cyri. Species of the Cyri group have mostly eastern distributions (in Ukraine and Russia) or are endemic (S. velebitica in Croatia), whereas S. otites occupies western and central European localities55. Contact in the past cannot, however, be excluded. The third possibility is that, as the analysis of ancestral states suggests, female heterogamety was ancestral in section Otites and male heterogamety evolved independently. Again, we argue against this because the hypothesis that the sex chromosomes evolved from the same autosome is not parsimonious.
The final possibility again involves ancestral female heterogamety, as supported by the analysis of ancestral states, and that this changed to male heterogamety in S. colpophylla and that the Y chromosome from this lineage was introgressed into S. pseudotites. Although this scenario is also not parsimonious, introgression between these species is likely. There is evidence that hybrids occur naturally between S. colpophylla and S. pseudotites, and between S. otites and S. pseudotites, in central eastern France, and hybrids between S. colpophylla and S. otites are phenotypically similar to S. pseudotites55. Our molecular data, however, suggest that our S. pseudotites sample from Trieste is not such a hybrid, and suggest that it could be a separate species, sister to S. otites (we estimate the age of the most recent common ancestor to be > 0.2 MYA). Introgression from S. colpophylla cannot have occurred recently in Italian populations of S. pseudotites as S. colpophylla does not occur in Italy, but it could have occurred in the past. The hybridization hypothesis is further supported by evidence that the sex chromosomes of S. pseudotites correspond to the S. latifolia LG6 and male heterogamety in S. pseudotites was recently confirmed by molecular methods (unpublished results of the group of Prof. Pascal Touzet, University of Lille 1, France, Prof. Touzet personal communication). If so, these two species probably share a common sex-determining locus, as required under this hypothesis. This scenario predicts that S. colpophylla’s sex chromosomes should be younger than those of the ZW species. This is not incompatible with our finding that the S. colpophylla Y chromosome includes a large non-recombining region, as a single large inversion could create such a region56, and a large amount of evolutionary time is not required to lead to a large non-recombining region, assuming that the selective situation favouring an inversion has evolved, perhaps involving a sexually antagonistic polymorphism in a partially sex-linked region57 (see next section). This inversion hypothesis is empirically testable, because it predicts that S. colpophylla should have evolutionary strata58, but no data are yet available.
An origin of S. colpophylla’s XY sex-determining system from an ancestral ZW sex-determining system in section Otites is also consistent with the phenotypes of interspecific hybrids between S. otites and S. pseudotites16,59. Hybrids possessing both the W-chromosome and Y-chromosome have male phenotypes, i.e. the Y-linked sex determiner is dominant and epistatic to the previously existing factor. In the future, the study of synonymous site divergence between sequences of Y- versus X-linked genes, and W- versus Z-linked ones should enable us to further characterize the XY systems (in S. colpophylla, S. pseudotites) and the ZW systems (in S. borysthenica, S. wolgensis and S. otites). The possibility of change in heterogamety through hybridization is an interesting one that has so far not been considered.
Why might species in section Otites have changed heterogamety? All three categories of hypotheses outlined in the introduction require the new sex-determining gene to determine sex even in the presence of the ancestral gene (i.e. to be epistatic to the original sex-determining master gene). Although our results do not identify the mechanism responsible for the switch in heterogamety in section Otites, the male determiner of S. pseudotites is indeed epistatic to the female determiner of S. otites, as WY hybrid plants show the male phenotype16, as expected if female heterogamety was the primary sex-determining system, and the male-determining Y-linked regions of S. pseudotites and S. colpophylla represent derived states. As the age of the sex chromosomes in this section is very small, and our results suggest that the non-recombining region of the S. otites W-chromosome carries few genes, models of type 2 (see Introduction) involving genetic degeneration of this region of the W-chromosome appear improbable.
A further argument against a mutational load model to explain the changes in heterogamety in section Otites (in either direction), is that changes in the chromosome on which the sex-determining locus is located are not predicted by this type of model. The extent of genetic degeneration is important because, after such a change, the pre-existing sex-determining chromosome can then become homozygous. If this chromosome carries a degenerated Y- or W-linked region, the change may be prevented due to low fitness or complete inviability of homozygotes of such chromosomes. Changed heterogamety in Silene section Otites is consistent with our observations suggesting that degeneration is likely to be minor in the ZW species. In such conditions, changed heterogamety is, however, possible under the turnover version of category 2 models (see Introduction) that involves a sexually antagonistic polymorphism60. Overall, we conclude that the change to a XY sex-determining system in section Otites could have involved either sexually antagonistic selection or neutral processes which allowed spread of a new epistatic sex-determining factor. The same two possibilities could also explain sex determination switches in fish such as Cichlidae21, which also involve changes in the linkage group carrying the sex-determining region.
A difference in the ages, and the extent of genetic degeneration, can potentially explain the difference between the changes in section Otites species, versus maintenance of male heterogamety in species in section Melandrium (subgenus Behenantha), which have retained XY sex chromosomes with no major changes apart from some translocations, including a reciprocal Y-autosome translocation in S. diclinis61 and expansion of the pseudoautosomal region in S. latifolia52. Species in section Melandrium have extensive non-recombining Y-linked regions, carrying large numbers of genes, including anther development genes, some of them essential for male fertility62,63,64,65. Given its numerous genes, making degeneration expected after sufficient evolutionary time, and the estimated 10 MY time since a fully Y-linked region first evolved, the lethality of YY genotypes (reviewed by Westergaard62) is predicted, and there is direct evidence that the non-recombining region of the S. latifolia Y-chromosome has undergone some degeneration42,66,67,68. Further important differences between sex-determining systems in the section Melandrium and that in S. otites are revealed by the genetic properties of so called “androhermaphrodites” (plants with male and hermaphrodite flowers). Such plants are occasionally found in natural populations of S. dioica69 and North American populations of S. latifolia70. They can also be induced by treatment with hypomethylating agents71,72. As expected, given that they are modified males, their progeny include male, females and androhermaphrodites. Phenotypic “androhermaphrodites” were found at an estimated frequency of 0.2% in two natural populations of S. otites (based on data of Correns73). Their progeny included only males and androhermaphrodites, but no females16,73, suggesting, in combination with our current data, that their genotype was ZZ, not ZW. An alternative possibility is that these androhermaphrodites do carry W-chromosomes, but that these are abnormal, having compromised female-promoting function, and compromised ability to suppress male functions; in other words, W-chromosomes in some natural populations may be similar to Z-chromosomes, in terms of their sex-determining functions. If so, the female-specific part of the S. otites W-chromosome cannot be essential for gynoecium formation, i.e. at least some Z-chromosomes carry all genetic information necessary for gynoecium formation.
Scenario for the evolution of ZW sex determination in section Otites
In speculating about the possible de novo evolution of ZW sex determination in section Otites species (or in the genus Fragaria), a two-locus gynodioecy model is most parsimonious, as the closest relatives (S. sibirica in section Otites) are gynodioecious. If female heterogamety was the first sex-determining system to evolve, male function must have been suppressed by a dominant male-suppressing mutation (rather than a recessive loss-of-function male-sterility mutation as proposed for the evolution of male heterogamety1. Such an effect could certainly arise by a dominant negative mutation directly creating a dominant female-determining chromosome (the mutation would define a proto-W-chromosome, see Fig. 3A). This mechanism is similar to the action of the W-linked homologue of the Xenopus laevis gene that counteracts DMRT1 expression by repressing its transcriptional targets74. Alternatively, it might involve a loss-of-function proto-W linked mutation of a haplo-insufficient gene important in anther development, such that proto-Z/proto-W plants, with only a single copy of the functional allele cannot develop normally functioning anthers (Fig. 3B). This mechanism resembles the presumed action of the DMRT1 gene in the gonadal sex determination in birds (reviewed by Kopp75).

Possible routes to female heterogamety from gynodioecy in the section Otites. Possible types of anther suppression mutations on the proto-W-chromosomes (A,B) and possible mutations causing gynoecium suppression (C,D). (A) A dominant negative W-linked anther suppressor causes male sterility by repressing the transcriptional targets of a Z-linked transcription factor. A W-linked inactive variant of the transcription factor competes with the active variant for its binding site in S. otites. (B) Anther suppression caused by a haploinsufficient loss of function mutation on the proto-W chromosome (C) A recessive loss-of-function mutation on the proto-Z chromosome. The presence of androhermaphrodites in S. otites is difficult to explain under this model. (D) Recruitment of dosage-dependent Z-linked gynoecium-suppressing alleles. The W chromosome carries no gynoecium-suppressing alleles. This hypothesis can explain the existence of ZZ androhermaphrodites. Anthers or gynoecium with compromised functions are indicated by red or blue crosses over their primordia.
To create males, a second mutation is required. A recessive loss-of-function female-sterility mutation (Fig. 3C) cannot readily account for the S. otites androhermaphrodites, which (as explained above) appear to be ZZ individuals. Therefore, one must invoke a dosage-dependent proto-Z linked allele causing gynoecium suppression when homozygous, but not when present in a single copy in ZW plants (Fig. 3D). As in the two-locus model of the origin of sex determination with male heterogamety (reviewed by Charlesworth76), such an allele is more likely to invade the population if it occurs in a locus that is closely linked (in repulsion) to the male-suppressing locus. This mechanism would again be similar to the proposed action of the DMRT1 gene in birds. In birds, however, DMRT1’s female-suppressing effect is indirect, through its role in the formation of male gonad from an originally bipotential tissue (reviewed by Kopp75) and so one gene can perform both “male promoting” and “female suppressing” functions.
An interesting aspect of the scenarios outlined here, which assume dosage-dependence, is that the W-chromosome need not contain any sex-determining genes. This would make the ZW sex-determining system prone to become epistatically recessive to the male-determining Y in XY systems. For example, a Y-chromosome might arise through a duplication of a Z-linked gene that promotes male functions in ZZ individuals. Under the model of Veller et al.44, no special driving force (such as a sexually antagonistic allele) is necessary for a change in sex determination, and the spread of a new dominant male-determiner can occur by drift, potentially explaining the change(s) in section Otites within a short time period.
Further progress in understanding of sex determination in the section Otites can be achieved by the study of the genes from the non-recombining regions of the species of section Otites in this work. In S. otites, four genes isolated from the non-recombining region represent good candidates for the putative sex-determining genes. OtiWT_51286 shows homology to leucine rich repeat receptor kinase proteins. This large protein family shows various functions, one of its members take part in meristem maintenance (CLV1)77. OtiWT_52979 is pentatricopeptide (PPR) repeat-containing protein. This large family of modular RNA-binding proteins is important in regulation of gene expression primarily in organelles but also in the nucleus. Some of them act as fertility restorers78. OtiWT_43556 shows homology to zinc-finger motif containing protein CRABS CLAW that is among others involved in the control of the flower meristem termination79. OtiWT_51403 codes for putative tetrapeptide repeat homeobox like protein so it is probably a transcription factor80 and its role in sex determination cannot be excluded. In further studies of these sequences, it should be kept in mind that the W-linked alleles can act as dominant negatives or they can be just inactive. There are also several putative sex-determining genes among the genes from non-recombining region of S. colpophylla. The role in the control of male fertility can show colpYS_12469 that is putative LYR protein (leucine/tyrosine/arginine motif containing proteins interacting with mitochondrial protein complexes)81. The role in the control of the carpel development (probable gynoecium suppression role in males) is expected in colpYS_7668 and colpYS_25642 showing similarity to CLV182. These two sequences can represent distant part of the same gene. In the case of S. colpophylla, the sex-determining genes are epistatically dominant to the previous sex-determining system. We therefore expect that the products of Y-linked alleles are biologically active and the future studies of sex determination should be easier in this species.
Material and Methods
Plant material and sex systems of the species studied
Information about all the species in our phylogenetic and/or genetic analyses is summarized in the Supporting information Table S5. The male heterogamety in S. pseudotites and female heterogamety in S. wolgensis were inferred from results of interspecific crosses16,59, and the S. pseudotites conclusion was recently confirmed by results obtained by the group of Prof. Pascal Touzet, University of Lille 1, Lille, France). The ZW system in S. otites was previously known17, and is confirmed by the present study. S. otites was used as the second parent in the interspecific crosses whose results are summarized above16,59. In all dioecious species studied here, the male and female phenotypes of individual plants are easily distinguishable from the flowers without dissection.
Candidate fully or partially sex-linked genes were identified using several different approaches (see the main text and Supporting information Table S1A,B). In all three species studied, sex linkage was tested in F1 full sibships produced by controlled crosses; the family sizes were 113 (58 females and 55 males) in S. otites, 32 (15 females and 17 males) in S. borysthenica, and 69 (27 females and 42 males) in S. colpophylla. In S. otites, tests for sequences completely linked to the W-chromosome (showing presence/absence polymorphism in PCR reactions with primers targeting sequences for which preliminary data suggested sex linkage) were done by PCR amplifications in plants sampled from two natural populations from the Czech Republic, located in Rohatec (Hodonin district) (sampled 15 females and 17 males) and Brno-Kralovo Pole (Brno city) (sampled 12 females and 18 males). Presence of a product indicates linkage disequilibrium with a female-determining allele.
Illumina RNAseq sequencing, Roche 454 RNAseq sequencing and genetic analyses
Initial Roche 454 RNAseq of S. otites, using cDNA from the seed parent of the F1 sibship used by Slancarova et al.17 to identify the first sex-linked genes from this species, was performed by GATC Biotech (Constance, Germany). The assembly was performed using mira3 assembler83 using default setting. To minimise amplification of paralogs, which would complicate tests for sex linkage, single-copy sequences were chosen from genes in this dataset that showed homology to single-copy nuclear genes in a plant species set that includes species in several genera, Arabidopsis, Populus, Vitis and Oryza84. Tests for sex linkage, including partial sex linkage, used the families described above, and identified several such genes, identifying pseudoautosomal regions (see below). The linkage group carrying the S. latifolia homologs of partially or completely sex-linked genes identified in this species was determined using the S. latifolia genetic map52, and proved to be LG6 (in the numbering of Bergero et al.52). The S. otites homologs of other genes from this chromosome were then tested for sex linkage in S. otites and mapped using the same S. otites sibship.
The main approach used for the identification of sex-linked sequences, and to obtain the sequence data for phylogenetic analysis was Illumina RNAseq (Illumina HiSeq. 2000 PE, 2 × 50 bp and Illumina NextSeq. 500 PE, 2 × 75 bp), performed at the EMBL Genomics Core Facility, Heidelberg (Germany). Sequencing libraries were prepared according to the Illumina protocol using oligo-dT attached magnetic beads. The sequences are deposited in GenBank (https://www.ncbi.nlm.nih.gov/genbank/; see accession numbers in Supporting information Table S6. In S. otites, S. borysthenica and S. colpophylla, the complete list of the sequenced samples including the batch ID, raw read counts, paired read counts after trimming, and mapped and unmapped read counts is summarised in Supporting information Table S7. The evaluation of the quality of Illumina RNA seqs in particular samples has been also performed, to check for possible batch effects see Supporting information Figs S4–S5. Rlog function was used for raw read counts normalization, as implemented in DESeq. 2R package. Adapters and low quality sequences were trimmed using Trimmomatic85, and errors corrected using Rcorrector86. An RNAseq dataset was assembled separately for each of the species using Trinity (with default setting)87. The longest ORFs were identified using Transdecoder and annotated using Trinotate88. 1082 orthogroups were used in phylogenetic analyses (see below), and were also annotated using the Blast2go pipeline89.
In S. otites, Illumina RNAseq sequences were obtained separately from both parent plants of the family used for genetic mapping (flower buds and leaves, separately), and from six progeny of each sex (flower buds only). For S. colpophylla, both flower buds and leaves were sampled from the seed parent of our family, but only leaves were available from the pollen donor. However, leaf and flower bud samples of a “full brother” of this plant were also sequenced, as well as from six progeny of each sex. In S. borysthenica, sequences were obtained from flower buds of both parents and a sample of their progeny (five females and seven males).
The Illumina RNAseq data were used for preliminary screening for candidate sex-linked genes in S. colpophylla, S. borysthenica and S. otites. Sequences with sex-linked inheritance patterns were first identified using the LINKYX pipeline53. This uses reference sequences generated using Trinity87 and maps reads to this reference using BWA90 (with standard settings). Read duplicates are then removed using Picard’s MarkDuplicates function (http://broadinstitute.github.io/picard/). Subsequently, variants are called. SAMtools91 with mpileup set to -uf, and bcftools view used with the following parameters: “-p 0.85 -cgv -”. To identify sequences with sex -linked SNPs, the reference is assembled from reads of the homogametic parent (XX mother or ZZ father), using rules for filtering SNPs as described53 (see also Supplementary Methods S1–S3).
The LINKYX pipeline was also used to identify genes with sex-specific expression in S. otites, and S. colpophylla. To identify sequences with sex specific expression, separate reference sequences were built using all individuals of each sex. The process is otherwise similar to the detection of sex-specific SNPs. If the contigs are male-specifically expressed represent sequences that are present only in males, the female reads will not map to them. The quantification rules used for the filtering to detect such sequences were as previously described53 (see also Supplementary Methods S4).
To detect sequences with SNPs with X-linked inheritance patterns in S. colpophylla, the pipeline was used without modifications. Modifications were made to reflect female heterogamety, to detect SNPs with Z- or W-linked inheritance patterns in S. otites, and Y-linked inheritance patterns in species with male heterogamety. In S. borysthenica, for which no previous genetic information was available, we tested for both male and female heterogamety. The latest version of the pipeline, including all the modifications used here, is available at GitHub repository: https://github.com/biomonika/linkyx. To validate the conclusions from LINKYX, reads from individual plants were mapped to the candidate sex-linked sequences using Bowtie 292 and the output was visually checked in the IGV browser93. The examples of the output from this control are shown in Supporting information Fig. S6.
The variants used for genetic mapping included DFLP (DNA fragment length polymorphism)94, intron size variants (ISVS, see Bergero et al.40), and single nucleotide polymorphisms (SNPs). SNPs in our mapping populations were genotyped as CAPS or dCAPS markers95, or by a high-resolution melting technique96,97. Genetic maps were constructed using JoinMap version 4.198, using the algorithm for a full-sib family in an outbreeder (called CP as “cross pollinators”). The linkage groups were inferred based on LOD scores for independent assortment higher than 4. We used regression mapping and Haldane’s mapping function (the Kosambi mapping function is not supported for CP mapping populations in JoinMap 4.1). Genetic map figures were produced using MapChart99. Homologies between the sex chromosomes of S. otites, S. borysthenica or S. colpophylla and S. latifolia chromosomes were further studied using blastn100,101 searches of S. latifolia scaffolds carrying homologs of genes showing sex linkage in section Otites species (with the following parameter values: minimal homology = 75%, minimal length of homologous region = 160 bp, and maximal E-value = 1.e-35). For scaffolds that have been genetically mapped in S. latifolia42, we recorded the locations of those carrying the homologs of sex-linked genes from section Otites species.
Phylogenetic analyses, species tree estimation and analysis of ancestral states
Phylogenetic analyses were based on sequences of single-copy genes with orthologues that were found (based on Illumina RNAseqs and transcriptome assemblies) in 10 species of the subgenus Silene of the genus Silene (we refer to these sequences as orthogroups). We identified 1,082 such orthogroups using Orthofinder102, which is based on an MCL algorithm103. For the phylogenetic analysis, we mapped the reads from each species to the S. otites reference sequence using Bowtie 292. We also applied a phasing procedure (to avoid biases in estimates of divergence times, as Lischer et al.104, have shown that unphased data can produce errors). For individuals without pedigree information, we performed read-based phasing in WhatsHap software105, and for samples from S. borysthenica, S. colpophylla and S. otites with pedigree information, we did read-based pedigree phasing106, using pedigree phasing mode in WhatsHap105,106. Estimated haplotypes were extracted from VCF files using a custom python script and the BCFtools-call method of Samtools v1.3.191. When several alternative phases were found (40 orthogroups), the most parsimonious (the one requiring the fewest substitutions in the data) were chosen. Three out of 40 orthogroups were discarded due to phasing ambiguity (more than one equally parsimonious phase). Low coverage sites with a read depth below 2 were masked using the BEDtools107 maskfasta method. Multiple sequence alignments were performed using the FFT-NS-i option108 of MAFFT v7.13109.
In order to conduct a fully parameterized multiple coalescent analysis, only S. sibirica, the closest non-dioecious species, was used as the outgroup. To avoid possible paralogous sequences, all alignments with more than two alleles in the single individual representing any of the taxa were removed before further analyses. All remaining alignments were manually checked for phasing ambiguities, and branch lengths appearing excessively long were excluded after visual inspection. This affected fourteen orthogroup alignments (2% of the dataset after removal of alignments with more than two alleles). In eight of these, the long branches appeared likely to be due to paralogy, as the aberrant allele formed a separate branch from the other alleles, but the species topology was the same in both branches (”mirror image topologies”). The final phased data set consisted of 662 alignments.
The data were analyzed using the Starbeast2 version 0.13 plugin110 and BEAST2111 version 2.4.7. We used a strict clock and a Jukes-Cantor substitution model for all alignments. A lognormal distribution with parameters (4.6, 2) was used for the speciation rate, and, for the population size, a lognormal distribution (−7, 2) was used as prior. The Markov Chain Monte Carlo was run for 1,000,000,000 iterations. For the time estimates, we used calibration based on estimated divergence per synonymous site dS (substitutions per synonymous site) and the divergence time estimates for several angiosperm lineages (Brassicaceae, Malvaceae, Euphorbiaceae, Fabaceae, Cucurbitaceae, Rosaceae, Solanaceae, and Poaceae), so as not to depend on a single fossil record or phylogenetic tree position, which yielded a mean substitution rate of 5.35 × 10−9 synonymous substitutions/site/year112. Mean dS values between the S. sibirica outgroup species and each section Otites species were estimated for each gene using the HyPhy package113. The mean dS is 0.0077; using the above angiosperm substitution rate per year, and assuming one generation per year for the Silene species, the estimated age of the most recent common ancestor of the S. sibirica and the section Otites species is about 720 thousand years (SE = 13 thousand years).
Ancestral state analysis was performed in BayesTraits V3.0 using the MultiState method114. The analysis took into account all currently available information about male and female heterogamety in the section Otites, including previously published phenotypic data16,59. All nine possible combinations of ancestral states of the most recent common ancestor of the whole section Otites and of the group Otites (see Supporting information Table S4a,b) were tested ((i) section Otites ZW, group Otites ZW, (ii) section Otites ZW, group Otites XY, (iii) section Otites ZW, group Otites N, (iv) section Otites XY, group Otites ZW, and (v) section Otites XY, group Otites XY), (vi) section Otites XY, group Otites N), (vii) section Otites N, group Otites ZW), (viii) section Otites N, group Otites XY) and (ix) section Otites N, group Otites N; female heterogamety-ZW, male heterogamety –XY, non-dioecious-N). As the input we used the set of the species trees obtained using the Starbeast2 plugin110 and BEAST2111. The chains were run for 109 states, using a gamma hyper-prior (mean from uniform: 0–10, variance from uniform: 0–10). Again, the process was checked using Tracer115. The marginal likelihoods for the Bayes factors were obtained using the stepping-stones method following the BayesTraits V3.0 tutorial. The significance of differences was evaluated according to Kass and Raftery54.
BAC library construction, screening and the subsequent analyses
A partial genomic BAC library was constructed for Silene otites (sample NN1 from the Nantes (France) population) using high molecular weight DNA prepared from flow-sorted nuclei. The protocol was modified from Cegan et al.116, for the details see Supporting information Methods S5. Library screening was performed by radioactive hybridization with α32P using the Prime-It II Random Primer Labelling Kit (Stratagene) following Cegan et al.117. Probes were prepared by PCR amplification of selected markers. Positively hybridizing BAC clones were selected, and the presence of a gene of interest was confirmed by PCR and sequencing. BAC DNA was isolated from the positive BACs and sequenced using an Illumina MiSeq (2 × 300PE) machine at the Centre of Plant Structural and Functional Genomics (Olomouc, Czech Republic). The reads were assembled de novo using the Edena assembler118.
Data Availability
The Illumina RNAseq raw data and Roche 454 RNAseq raw data have been deposited in GenBank (https://www.ncbi.nlm.nih.gov/genbank/; see accession numbers in Supporting information Table S6).
References
Charlesworth, B. & Charlesworth, D. A model for the evolution of dioecy and gynodioecy. Am. Nat. 112, 975 (1978).
Lloyd, D. G. The distribution of gender in four angiosperm species illustrating two evolutionary pathways to dioecy. Evolution 34, 123–134 (1980).
Webb, B. C. J. In Gender and Sexual Dimorphism in Flowering Plants (eds Geber, M. A., Dawson, T. E. & Delph, L. F.) 61–95 (Springer, 1999).
Renner, S. S. & Won, H. Repeated evolution of dioecy from monoecy in Siparunaceae (Laurales). Syst. Biol. 50, 700–712 (2001).
Spigler, R. B., Lewers, K. S., Main, D. S. & Ashman, T.-L. Genetic mapping of sex determination in a wild strawberry, Fragaria virginiana, reveals earliest form of sex chromosome. Heredity 101, 507–517 (2008).
Spigler, R. B., Lewers, K. S., Johnson, A. L. & Ashman, T.-L. Comparative mapping reveals autosomal origin of sex chromosome in octoploid Fragaria virginiana. J. Hered. 101, Suppl, S107–117 (2010).
Goldberg, M. T., Spigler, R. B. & Ashman, T.-L. Comparative genetic mapping points to different sex chromosomes in sibling species of wild strawberry (Fragaria). Genetics 186, https://doi.org/10.1534/genetics.110.122911 (2010).
Ashman, T.-L. et al. Multilocus sex determination revealed in two populations of gynodioecious wild strawberry. Fragaria vesca subsp. bracteata. G3(5), 2759–2773 (2015).
Lloyd, D. G. Breeding systems in Cotula III. dioecious populations. New Phytologist 74, 109–123 (1975).
Hormaza, J. I., Dollo, L. & Polito, V. S. Identification of a RAPD marker linked to sex determination in Pistacia vera using bulked segregant analysis. Theor. Appl. Genet. 89, 9–13 (1994).
Esfandiyari, B., Davarynejad, G. H., Shahriari, F., Kiani, M. & Mathe, A. Data to the sex determination in Pistacia species using molecular markers. Euphytica 185, 227–231 (2012).
Eppley, S. M., O’Quinn, R. & Brown, A. L. New sequence-tagged site molecular markers for identification of sex in Distichlis spicata. Mol. Ecol. Resour. 9, 1373–1374 (2009).
Grewal, M. S. & Ellis, J. R. Sex determination in Potentilla fruticosa. Heredity 29, 359 (1972).
Beukeboom, L. W. & Perrin, N. The evolution of sex determination. (Oxford University Press, 2014), https://doi.org/10.1093/acprof:oso/9780199657148.001.0001.
Yin, T. et al. Genome structure and emerging evidence of an incipient sex chromosome in. Populus. Genome Res. 18, 422–430 (2008).
Sansome, F. W. Sex determination in Silene otites and related species. Journal of Genetics 35, 387–396 (1938).
Slancarova, V. et al. Evolution of sex determination systems with heterogametic males and females in Silene. Evolution 67, 3669–77 (2013).
Myosho, T. et al. Tracing the emergence of a novel sex-determining gene in medaka. Oryzias luzonensis. Genetics 191, 163–70 (2012).
Myosho, T., Takehana, Y., Hamaguchi, S. & Sakaizumi, M. Turnover of sex chromosomes in Celebensis group medaka fishes. G3 5, 2685–91 (2015).
Böhne, A., Wilson, C. A., Postlethwait, J. H. & Salzburger, W. Variations on a theme: Genomics of sex determination in the cichlid fish Astatotilapia burtoni. BMC Genomics 17, 883 (2016).
Roberts, R. B., Ser, J. R. & Kocher, T. D. Sexual conflict resolved by invasion of a novel sex determiner in lake Malawi cichlid fishes. Science 326, 998–1001 (2009).
Gammerdinger, W. J. et al. Novel Sex Chromosomes in 3 Cichlid Fishes from Lake Tanganyika. J. Hered. 109, 489–500 (2018).
Tennessen, J. A. et al. Repeated translocation of a gene cassette drives sex-chromosome turnover in strawberries. PLoS Biol. 16, e2006062 (2018).
Boucher, L. D., Manchester, S. R. & Judd, W. S. An extinct genus of Salicaceae based on twigs with attached flowers, fruits, and foliage from the Eocene Green River Formation of Utah and Colorado, USA. Am. J. Bot. 90, 1389–99 (2003).
Manchester, S. R., Judd, W. S. & Handley, B. Foliage and fruits of early poplars (Salicaceae: Populus) from the Eocene of Utah, Colorado, and Wyoming. Int. J. Plant Sci. 167, 897–908 (2006).
Geraldes, A. et al. Recent Y chromosome divergence despite ancient origin of dioecy in poplars (Populus). Mol. Ecol. 24, 3243–56 (2015).
Hou, J. et al. Different autosomes evolved into sex chromosomes in the sister genera of Salix and Populus. Sci. Rep. 5, 9076 (2015).
Pucholt, P., Rönnberg-Wästljung, A.-C. & Berlin, S. Single locus sex determination and female heterogamety in the basket willow (Salix viminalis L.). Heredity 114, 575–83 (2015).
Pucholt, P., Wright, A. E., Conze, L. L., Mank, J. E. & Berlin, S. Recent sex chromosome divergence despite ancient dioecy in the willow Salix viminalis. Mol. Biol. Evol. 34, 1991–2001, https://doi.org/10.1093/molbev/msx144 (2017).
Tuskan, G. A. et al. The obscure events contributing to the evolution of an incipient sex chromosome in Populus: a retrospective working hypothesis. Tree Genet. Genomes 8, 559–571 (2012).
Zhou, R. et al. Characterization of a large sex determination region in Salix purpurea L. (Salicaceae). Mol. Genet. Genomics 293, 1437–1452, https://doi.org/10.1007/s00438-018-1473-y (2018).
Casimiro-Soriguer, I., Buide, M. L. & Narbona, E. Diversity of sexual systems within different lineages of the genus Silene. AoB Plants 7 (2015).
Charlesworth, D. & Laporte, V. The male-sterility polymorphism of Silene vulgaris: analysis of genetic dat from two populations and comparison with Thymus vulgaris. Genetics 150, 1267–82 (1998).
Stone, J. D., Kolouskova, P., Sloan, D. B. & Storchova, H. Non-coding RNA may be associated with cytoplasmic male sterility in Silene vulgaris. J. Exp. Bot. 68, 1599–1612 (2017).
Garraud, C., Brachi, B., Dufay, M., Touzet, P. & Shykoff, J. A. Genetic determination of male sterility in gynodioecious Silene nutans. Heredity 106, 757–764 (2011).
Bari, E. A. Cytological studies in the genus Silene L. New Phytol. 72, 833–838 (1973).
Kruckeberg, A. R. Chromosome numbers in Silene (Caryophylaceae): I. Madroño 12, 238–246 (1954).
Mrackova, M. et al. Independent origin of sex chromosomes in two species of the genus Silene. Genetics 179, 1129–1133 (2008).
Blackburn, K. B. Sex chromosomes in plants. Nature 112, 687–688 (1923).
Bergero, R., Forrest, A., Kamau, E. & Charlesworth, D. Evolutionary strata on the X chromosomes of the dioecious plant Silene latifolia: evidence from new sex-linked genes. Genetics 175, 1945–1954 (2007).
Rautenberg, A., Hathaway, L., Oxelman, B. & Prentice, H. C. Geographic and phylogenetic patterns in Silene section Melandrium (Caryophyllaceae) as inferred from chloroplast and nuclear DNA sequences. Mol. Phylogenet. Evol. 57, 978–91, https://doi.org/10.1016/j.ympev.2010.08.003 (2010).
Papadopulos, A. S. T., Chester, M., Ridout, K. & Filatov, D. A. Rapid Y degeneration and dosage compensation in plant sex chromosomes. Proc. Natl. Acad. Sci. USA 112, 13021–6 (2015).
Krasovec, M., Chester, M., Ridout, K. & Filatov, D. A. The mutation rate and the age of the sex chromosomes in Silene latifolia. Curr. Biol. 28, 1832–1838.e4 (2018).
Veller, C., Muralidhar, P., Constable, G. W. A. & Nowak, M. A. Drift-induced selection between male and female heterogamety. Genetics 207, 711–727 (2017).
Saunders, P. A., Neuenschwander, S. & Perrin, N. Sex chromosome turnovers and genetic drift: a simulation study. J. Evol. Biol. 31, 1413–1419 (2018).
van Doorn, G. S. Evolutionary transitions between sex-determining mechanisms: a review of theory. Sex Dev. 8, 7–19 (2014).
Blaser, O., Grossen, C., Neuenschwander, S. & Perrin, N. Sex-chromosome turnovers induced by deleterious mutation load. Evolution 67, 635–645 (2013).
Blaser, O., Neuenschwander, S. & Perrin, N. Sex-chromosome turnovers: the hot-potato model. Am. Nat. 183, 140–146 (2014).
Brelsford, A. et al. Homologous sex chromosomes in three deeply divergent anuran species. Evolution 67, 2434–2440 (2013).
Ogata, M. et al. Change of the heterogametic sex from male to female in the frog. Genetics 164, 613–620 (2003).
Ogata, M., Lambert, M., Ezaz, T. & Miura, I. Reconstruction of female heterogamety from admixture of XX-XY and ZZ-ZW sex-chromosome systems within a frog species. Mol. Ecol. 27, 4078–4089 (2018).
Bergero, R., Qiu, S., Forrest, A., Borthwick, H. & Charlesworth, D. Expansion of the pseudo-autosomal region and ongoing recombination suppression in the Silene latifolia sex chromosomes. Genetics 194, 673–86 (2013).
Michalovova, M., Kubat, Z., Hobza, R., Vyskot, B. & Kejnovsky, E. Fully automated pipeline for detection of sex linked genes using RNA-Seq data. BMC Bioinformatics 16, 78 (2015).
Kass, R. E. & Raftery, A. E. Bayes factors. J. Am. Stat. Assoc. 90, 773–795 (1995).
Wrigley, F. Taxonomy and chorology of Silene section Otites (Caryophyllaceae). Ann. Bot. Fenn. 23, 69–81 (1986).
Suarez-Villota, E. Y., Pansonato-Alves, J. C., Foresti, F. & Gallardo, M. H. Homomorphic sex chromosomes and the intriguing Y chromosome of Ctenomys rodent species (Rodentia, Ctenomyidae). Cytogenet. Genome Res. 143, 232–240 (2014).
Kirkpatrick, M. The evolution of genome structure by natural and sexual selection. J. Hered. 108, 3–11 (2017).
Lahn, B. T. & Page, D. C. Four evolutionary strata on the human X chromosome. Science 286, 964–967 (1999).
Newton, W. C. F. Genetical experiments with Silene otites and related species. J. Genet. 24, 109–120 (1931).
van Doorn, G. S. & Kirkpatrick, M. Transitions between male and female heterogamety caused by sex-antagonistic selection. Genetics 186, 629–45 (2010).
Howell, E. C., Armstrong, S. J. & Filatov, D. A. Evolution of neo-sex chromosomes in Silene diclinis. Genetics 182, 1109–1115 (2009).
Westergaard, M. The mechanism of sex determination in dioecious flowering plants. Adv. Genet. 9, 217–281 (1958).
Zluvova, J. et al. The inter-specific hybrid Silene latifolia x S. viscosa reveals early events of sex chromosome evolution. Evol. Dev. 7, 327–336 (2005).
Zluvova, J. et al. Early events in the evolution of the Silene latifolia Y chromosome: male specialization and recombination arrest. Genetics 177, 375–386 (2007).
Kazama, Y. et al. A new physical mapping approach refines the sex-determining gene positions on the Silene latifolia Y-chromosome. Sci. Rep. 6, 18917 (2016).
Marais, G. A. B. et al. Evidence for degeneration of the Y chromosome in the dioecious plant Silene latifolia. Curr. Biol. 18, 545–549 (2008).
Muyle, A. et al. Rapid de novo evolution of X chromosome dosage compensation in Silene latifolia, a plant with young sex chromosomes. PLoS Biol. 10, e1001308 (2012).
Bergero, R., Qiu, S. & Charlesworth, D. Gene loss from a plant sex chromosome system. Curr. Biol. 25, 1234–1240 (2015).
Shull, G. H. Reversible sex mutants in Lychnis dioica. Bot. Gaz. 52, 329–368 (1911).
Miller, P. M. & Kesseli, R. V. A sex-chromosome mutation in Silene latifolia. Sex. Plant Reprod. 24, 211–217 (2011).
Janousek, B., Siroký, J. & Vyskot, B. Epigenetic control of sexual phenotype in a dioecious plant. Melandrium album. Mol. Gen. Genet. 250, 483–490 (1996).
Janoušek, B., Grant, S. R. & Vyskot, B. Non-transmissibility of the Y chromosome through the female line in androhermaphrodite plants of Melandrium album. Heredity 80, 576–583 (1998).
Correns, C. B. Vererbung and Verteilung des geschlechtes bei den hoheren Pflanzen. Handb. Vererbungsw. 2, 1–138 (1928).
Yoshimoto, S. et al. A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proc. Natl. Acad. Sci. 105, 2469–2474 (2008).
Kopp, A. Dmrt genes in the development and evolution of sexual dimorphism. Trends in Genetics 28, 175–184 (2012).
Charlesworth, D. Plant contributions to our understanding of sex chromosome evolution. New Phytol. 208, 52–65 (2015).
Torii, K. U. Leucine-rich repeat receptor kinases in plants: structure, function, and signal transduction pathways. Int. Rev. Cytol. 234, 1–46 (2004).
Manna, S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie 113, 93–9 (2015).
Gross, T., Broholm, S. & Becker, A. CRABS CLAW acts as a bifunctional transcription factor in flower development. Front. Plant Sci. 9, 835 (2018).
Mukherjee, K., Brocchieri, L. & Bürglin, T. R. A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol. Biol. Evol. 26, 2775–2794 (2009).
Angerer, H. Eukaryotic LYR Proteins Interact with Mitochondrial Protein Complexes. Biology (Basel) 4, 133–150 (2015).
Hazak, O. & Hardtke, C. S. CLAVATA 1-type receptors in plant development. J. Exp. Bot. 67, 4827–4833 (2016).
Chevreux, B. et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14, 1147–1159 (2004).
Duarte, J. M. et al. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 10, 61 (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–20 (2014).
Song, L. et al. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. Gigascience 4, 48 (2015).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–512 (2013).
Hebert, F. O. Trinotate_pipeline: Annotation Pipeline – Trinotate, https://doi.org/10.5281/zenodo.50974 (2016).
Conesa, A. & Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 619832 (2008).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–9 (2009).
Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics Chapter 11, Unit11.7 (2010).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013).
Hongtrakul, V., Slabaugh, M. B. & Knapp, S. J. DFLP, SSCP, and SSR markers for Δ9-stearoyl-acyl carrier protein desaturases strongly expressed in developing seeds of sunflower: intron lengths are polymorphic among elite inbred lines. Mol. Breed. 4, 195–203 (1998).
Neff, M. M., Neff, J. D., Chory, J. & Pepper, A. E. dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J. 14, 387–92 (1998).
Reed, G. H. & Wittwer, C. T. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin. Chem. 50, 1748–54 (2004).
Simko, I. High-resolution DNA melting analysis in plant research. Trends Plant Sci. 21, 528–537 (2016).
van Ooijen, J. W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 93, 343–349 (2011).
Voorrips, R. E. MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78 (2002).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Emms, D. M. et al. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–84 (2002).
Lischer, H. E. L., Excoffier, L. & Heckel, G. Ignoring heterozygous sites biases phylogenomic estimates of divergence times: implications for the evolutionary history of microtus voles. Mol. Biol. Evol. 31, 817–831 (2014).
Patterson, M. et al. WhatsHap: weighted haplotype assembly for future-generation sequencing reads. J. Comput. Biol. 22, 498–509 (2015).
Garg, S., Martin, M. & Marschall, T. Read-based phasing of related individuals. Bioinformatics 32, i234–i242 (2016).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–80 (2013).
Ogilvie, H. A., Bouckaert, R. R. & Drummond, A. J. StarBEAST2 brings faster species tree inference and accurate estimates of substitution rates. Mol. Biol. Evol. 34, 2101–2114 (2017).
Bouckaert, R. et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10, e1003537 (2014).
De La Torre, A. R., Li, Z., Van de Peer, Y. & Ingvarsson, P. K. Contrasting rates of molecular evolution and patterns of selection among gymnosperms and flowering plants. Mol. Biol. Evol. 34, 1363–1377 (2017).
Pond, S. L. K., Frost, S. D. W. & Muse, S. V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–9 (2005).
Pagel, M., Meade, A. & Barker, D. Bayesian estimation of ancestral character states on phylogenies. Syst. Biol. 53, 673–684 (2004).
Rambaut, A., Suchard, M. A., Xie, D. & Drummond, A. J. Tracer v1.5. (2013). Available at: http://beast.bio.ed.ac.uk/Tracer.
Cegan, R. et al. Structure and evolution of Apetala3, a sex-linked gene in Silene latifolia. BMC Plant Biol. 10, 180 (2010).
Cegan, R. et al. Genomic diversity in two related plant species with and without sex chromosomes - Silene latifolia and S. vulgaris. PLoS One 7, e31898 (2012).
Hernandez, D., Francois, P., Farinelli, L., Osteras, M. & Schrenzel, J. De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 18, 802–809 (2008).
Acknowledgements
This project was supported by grants from the Czech Science Foundation (GACR) 13-06264S to BJ, and 18-06147S and 16-08698S to RH, and from the Swedish Research Council (2015-03836) to BO. Computational resources were provided by the CESNET LM2015042 and the CERIT Scientific Cloud LM2015085, provided under the programme “Projects of Large Research, Development, and Innovations Infrastructures”. We thank The Genomics Core Facility (EMBL Heidelberg, http://www.genecore.embl.de) for RNA sequencing. We thank Dr. Jiri Danihelka (Masaryk University, Brno) and the Botanical Garden of Trieste for the seed material. We also thank to Monika Cechova (Pennsylvania State University), Pascal Touzet (University of Lille1, Lille) and Gabriel Marais (University Lyon 1, Lyon) for discussions.
Author information
Authors and Affiliations
Contributions
B.J., V.B., R.G., J.S., R.H. and R.B. conceived and designed the experiments. V.B., R.G., J.S., B.J. and R.B. performed the experiments. B.J., R.C., P.C., B.O., J.Z., V.B., R.G. and V.K. analysed the data. B.J. contributed reagents/materials/analysis tools. B.V., V.B., R.G. and R.H. discussed the paper and B.J., J.Z., D.C. and B.O. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Balounova, V., Gogela, R., Cegan, R. et al. Evolution of sex determination and heterogamety changes in section Otites of the genus Silene. Sci Rep 9, 1045 (2019). https://doi.org/10.1038/s41598-018-37412-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37412-x
This article is cited by
-
The male-heterogametic sex determination system on chromosome 15 of Salix triandra and Salix arbutifolia reveals ancestral male heterogamety and subsequent turnover events in the genus Salix
Heredity (2023)
-
Evolution of a ZW sex chromosome system in willows
Nature Communications (2023)
-
Plant sex chromosomes defy evolutionary models of expanding recombination suppression and genetic degeneration
Nature Plants (2021)
-
A willow sex chromosome reveals convergent evolution of complex palindromic repeats
Genome Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
