
Abstract
The incidence of developing cancer should increase with the body mass, yet is not the case, a conundrum referred to as Peto’s paradox. Elephants have a lower incidence of cancer suggesting that these animals have probably evolved different ways to protect themselves against the disease. The paradox is worth revisiting with the realization that most mammals encode an endogenous APOBEC3 cytidine deaminase capable of mutating single stranded DNA. Indeed, the mutagenic activity of some APOBEC3 enzymes has been shown to introduce somatic mutations into genomic DNA. These enzymes are now recognized as causal agent responsible for the accumulation of CG- > TA transitions and DNA breaks leading to chromosomal rearrangements in human cancer genomes. Here, we identified an elephant A3Z1 gene, related to human APOBEC3A and showed that it could efficiently deaminate cytidine, 5-methylcytidine and produce DNA breaks leading to massive apoptosis, similar to other mammalian APOBEC3A enzymes where body mass varies by up to four orders of magnitude. Consequently, it could be considered that eAZ1 might contribute to cancer in elephants in a manner similar to their proposed role in humans. If so, eAZ1 might be particularly well regulated to counter Peto’s paradox.
Similar content being viewed by others

Introduction
The APOBEC3 (A3) locus is bounded by two conserved genes, chromobox 6 and 7 (CBX6 and CBX7) in most placental mammals and encodes a family of cytidine deaminases capable of converting cytidine residues to uridine in single strand DNA (ssDNA). The mutagenic activity of these enzymes is involved with the restriction of retroviruses and DNA viruses, as well as endogenous retroelements and retrotransposons through hypermutation of viral DNA in a process called editing1. The A3 repertoire is extremely variable among mammals, the locus being shaped through extensive gene duplications and functionalization in the context of a virus-host arms race. A3 enzymes are made up of three related, but distinct zinc-finger domains referred to as Z1, Z2 and Z32,3,4 presumably already present in the genome of the ancestor of placental mammals5.
The last few years has seen the identification of two human endogenous A3 cytidine deaminases, APOBEC3A (A3A) and APOBEC3B (A3B) capable of introducing multiple mutations in chromosomal DNA6,7,8,9. These findings are grounded by the analysis of many cancer genomes, revealing far more mutations and rearrangements than hitherto imagined, where CG- > TA transitions appears to be the dominant mutations10,11,12,13.
Human A3A is composed of a single Z1 domain, while A3B is composed of a double Z2Z1 domain, although only the C terminal Z1 domain being catalytically functional6. A3A and A3B enzymes are both localized in the nucleus and can edit cytidine residues to uridine in ssDNA during transcription and replication, following DNA repair, and leave TpC to TpT signature mutations that show up in cancer genomes6,8,9. Both enzymes can mutate 5-methylcytidine (5MeC) to thymidine leaving another distinct signature in cancer genomes6,14,15,16.
Although A3A and A3B are accepted as intrinsic mutators of cellular chromosomal DNA, analyzed in several cancer types8,11,17, debate still persists regarding the contribution of each enzyme in the accumulation of mutations paving the way for oncogenesis. While, it has been described that A3A and A3B could be enzymatically active in different cancers18, A3A is the more active of the two enzymes and as a consequence, only A3A can produce double stranded breaks (DSBs), at least in an experimental setting6,7,19. Editing frequencies of >0.5 can be found which is why the phenomenon is referred to as hyperediting or hypermutation9. Accumulation of substitutions localized in the A3B C-terminal domain attenuated the activity of the enzyme compared to A3A6. Interestingly, this functional attenuation was also observed for the rhesus monkey rhA3B enzyme compared to rhA3A indicating that this mutagenic dichotomy was maintained for ~38 million years6. Moreover, the deletion of most of the A3B gene results in a higher odds ratio of developing breast, ovarian or liver cancer20,21,22,23. Indeed, complete genome sequencing of ΔA3B-/- breast cancer genomes revealed a higher mutation burden24. Finally, fine analysis of signatures mutations in cancer genomes unraveled for twice as many A3A specific mutational signature (YTCA) over A3B (RTCA) suggesting a major role of A3A in cancer mutagenesis8,25.
Another difference between A3A and A3B lies in their evolutionary history. A3A is present across most placental mammals, indicating that this evolutionary experiment has been running ~150 million years26. There are some notable exceptions – an A3A gene is absent among all members of the order Rodentia, pigs, while for Felidae the gene is inactivated but identifiable3,26. By contrast A3B is unique to the order Primates and arose by gene conversion involving A3A. We have previously shown that the A3A enzymes from 8 mammalian species from rabbits to cows and horses were capable of deaminating C and 5MeC in ssDNA as well as producing DSBs, even though activities varied considerably26.
The incidence of developing cancer was hypothesized to increase with the body size, referred to as Peto’s paradox27. However, as large animals exist and do not invariably die of cancer this paradox fails to explain the presence of compensatory mechanisms that protect the genome. With this in mind, we were intrigued by a recent report showing that elephants appeared to have a lower-than-expected rate of cancer which might possibly be coupled to multiple copies of TP53 even though most were processed pseudogenes28,29. It is equally possible that the A3 enzymes of large mammals could have been attenuated by mutation. Accordingly, we decided to explore the function of the elephant A3Z1 enzyme.
Results and Discussion
Synthesis and expression of elephant APOBEC3Z1 cytidine deaminase
To explore the implication of elephant A3 enzyme in tumorigenesis, in silico data mining was performed using blast/blat analyses of the genomic for A3Z1 like sequences. We retrieved an elephant A3Z1 sequence named eA3Z1, equivalent to the p2 protein form of human A3A (hA3A), that is to say missing the first coding exon, what is also a feature of dog, horse and cow A3A genes. Furthermore, p1 and p2 forms of A3A are functionally equivalent26,30,31. There was 44% amino acid divergence between the human and elephant protein sequences (Fig. 1). The elephant sequence carried an 8 residue deletion in loop 3, which is not without precedent26 and impacts little the overall structure as can be seen from Fig. 2a. All the key amino acid residues typical of an A3A enzyme were conserved3. A phylogenetic analysis using the neighbor-joining method revealed that eA3Z1 was closely related to those from the Primate lineage (Fig. 2b). To prove that eA3Z1 is expressed in vivo and to validate the putative eA3Z1 DNA sequence inferred from elephant genome assembly, total RNA from the liver of an African Savana elephant (Loxodonta africana) that had died from an encephalomyocarditis virus infection32 was analyzed. Total RNA was extracted, cDNA generated and sequences corresponding to complete eA3Z1 transcripts were amplified by a semi-nested PCR procedure. As shown in Fig. 2c, strong eA3Z1 cDNA amplifications were obtained giving rise to two overlapping PCR products. Finally, eA3Z1 sequence (accession number: MK156802) was validated by direct sequencing (Supplementary Fig. S1) and identical to the previous BLAST/BLAT analysis search sequences from genomic data.

Comparison of APOBEC3Z1 cytidine deaminases. CLUSTALW alignment of A3A proteins. Sequence conservation is depicted in red for each residue. Asterisks represent residues involved in zinc coordination responsible for enzymatic activity. Red asterisk represents the isoleucine amino-acid specific to Z1 domain. Structural motifs structures (α helix, β sheet and loop) are indicated.

Elephant APOBEC3Z1 enzyme. (a) A3A structure with differences between human and elephant A3A presented in orange, loops 3 and 5 in green are absent in eA3Z1. (b) Phylogenetic tree of elephant A3A along with other mammalian enzymes. A3A protein sequences constructed using the Neighbor-joining method with the CLC Main Workbench 7.0.2 software. Human A3C was used as outlier. Numbers correspond to bootstrap values inferred from 1,000 replicates. Bootstrap values below the threshold of 70% are not shown. (c) Nested RT-PCR amplification of eA3Z1 transcript in liver from an African savana elephant. PCR1 corresponds to the amplification of eA3Z1 exon 2 to exon 3 and PCR2 corresponds to the amplification of exon 3 to exon 5; –:negative PCR control; M: molecular weight marker; e: exon. (d) Western blot detection of V5-tagged A3A proteins of human and elephant in HeLa and HEK-293T cells, pv (plasmid vector) and hA3AC101S were used as negative controls. β-actin probing was used as loading control. (e) Confocal microscopy analysis of V5-tagged human and elephant A3A proteins in HeLa at 48 hours post transfection. Nuclei are stained with DAPI.
Accordingly, an eA3Z1 cDNA was synthesized and cloned into pcDNA3.1D/V5-His-TOPO with a strong Kozak motif (ACCATG) for functional studies. When overexpressed in transfected HeLa or HEK-293T cells, Western blot analysis revealed a strong expression of V5-tagged eA3Z1 on a par with that for its human counterpart (hA3A) or an inactive mutant (hA3AC101S; Fig. 2d). The slightly lower molecular weight of eA3Z1 is in agreement with the calculated molecular weights (hA3A 23.0 kDa; eA3Z1 20.9 kDa). The subcellular localization was assessed in HeLa cells by confocal microscopy. Anti-V5 staining revealed that eA3Z1 exhibited the classical nuclear and cytoplasmic distribution described for human (Fig. 2e) as well as other mammalian A3A enzymes26.
Elephant APOBEC3Z1 editing of nuclear DNA and 5-methylcytidine
To assay catalytic activity, eA3Z1 and hA3A plasmids were transfected into HEK-293T cells and cellular lysates were used in a fluorescence resonance energy transfer assay based in vitro deamination assay where C to U conversion in a TAMRA-FAM-labeled DNA oligonucleotide allows fluorescence detection following cleavage by uracil-DNA glycosylase (UNG) activity7. Elephant eA3Z1 activity was on a par with hA3A (Fig. 3a). To explore A3 hyperediting of chromosomal DNA, the HEK-293T-UGI cell line was transfected with eA3Z1 and hA3A plasmids. The HEK-293T-UGI cell line constitutively expresses uracil N-glycosylase (UNG) inhibitor (UGI) where UNG is the crucial enzyme involved in excising uracil from DNA. As UNG is rate limiting for the detection of hyperedited chromosomal DNA, inhibition by UGI is necessary9. At 48 hours post-transfection, total DNA was extracted and TP53 DNA was amplified by 3D-PCR, a technique that selectively amplifies A3-edited ssDNA molecules33. The lowest PCR denaturation temperature (Td) allowing amplification of unedited TP53 target DNA was 87°C (Fig. 3b). For eA3Z1 and hA3A transfections, 3D-PCR products were recovered at Tds as low as 84.1°C and 84.9°C respectively which is diagnostic for A3 editing (Fig. 3b). Nonetheless, 3D-PCR products from the 85.9°C amplification (white asterisk, Fig. 3b) were cloned and sequenced as those of the last positive amplification for plasmid vector (pv) and hA3AC101S as negative controls.

Editing of TP53 with human and elephant APOBEC3A. (a) Fluorescence resonance energy transfer assay (FRET)-based in vitro deamination assay of hA3A and eA3Z1 performed on FAM-TAMRA coupled oligonucleotides using transfected HEK-293T lysates. Pv and hA3AC101S were used as negative controls. Results are expressed in Relative Fluorescence Unit per μg of protein (RFU/μg). Differences were calculated using the Unpaired t test (***p < 0.001, ns: no significance). (b) 3D-PCR recovered edited TP53 DNA down to 84.9°C and 84.1°C for hA3A and eA3Z1 respectively. The white line indicates the threshold between edited and unedited 3D-PCR products in terms of the denaturation temperature. Pv and hA3AC101S showed no editing of TP53 and were used as negative control. Asterisks refer to the samples cloned and sequenced. M, molecular weight markers. (c) CG- > TA mutation frequencies analyzed with hA3AC101S and pv at 87°C and with hA3A and eA3Z1 at 85.9°C. (d) Dinucleotide context of TP53 DNA region on minus strand DNA obtained with hA3A and eA3Z1. Chi-square test indicates dinucleotide frequencies that significantly deviate from the expected values (*p < 0.05).
Hyperedited TP53 target sequences were recovered with an average editing frequency of 10% compared to a background value of 0.6% (Fig. 3c). The monotony of editing is confirmed by the frequency of non-CG- > TA mutations which did not differ from background values. Cytidine editing was strongly associated with TpC, and to a lesser extent CpC dinucleotides (Fig. 3d) to the detriment of GpC and ApC which is typical for mammalian A3A enzymes26,34,35,36,37.
To demonstrate that no endogenous activity of hA3A present in HEK-293T-UGI cells would give rise to hypermutated sequences, hA3A or eA3Z1 transfections were performed in QT6 cell lines in presence of UGI. QT6 is a quail cell line and was chosen as there is no endogenous A3 background38. As expected, 3D-PCR product sequence analysis demonstrated the same profile of hypermutated sequences and dinucleotide contexts (data not shown).
One of the singular traits of mammalian A3A deaminases is their ability to efficiently deaminate 5MeC26. To demonstrate that eA3Z1 can deaminate 5MeC, QT6 cells were transfected with the eA3Z1 expression plasmid and subsequently transfected by 5MeC-substituted HIV env DNA fragments16. As shown in Fig. 4a, 3D-PCR products were recovered at temperatures as low as 75.7°C and 77 °C for the eA3Z1 and hA3A transfections respectively, compared to 82.1°C for the plasmid vector or hA3AC101S (Fig. 4a). The 82.1°C and 80.3°C 3D-PCR products (Fig. 4a), indicated by an asterisk were cloned, sequenced and confirmed the presence of edited 5MeC in the expected TpC dinucleotide context (Fig. 4b,c). Hyperedited 5MeC V1V2 target sequences were recovered with an average editing frequency of 5% and 6.8% respectively for hA3A and eA3Z1 compared to background value (Fig. 4c).

Elephant APOBEC3Z1 deamination of 5-methylcytidine. (a) 3D-PCR recovered edited 5MeC substituted HIV env DNA down to 77°C and 75.7°C for hA3A and eA3Z1 respectively. The white line indicates the threshold between edited and unedited 3D-PCR products in terms of the denaturation temperature. Pv and hA3AC101S showed no editing of 5MeC substituted HIV env DNA and were used as negative control. Asterisks refer to the samples cloned and sequenced. M, molecular weight markers. (b) Dinucleotide context for human and elephant A3A deamination of 5MeC. Chi-square test indicates dinucleotide frequencies that significantly deviate from the expected values (*p < 0.05). (c) CG- > TA mutation frequencies analyzed with hA3AC101S and pv at 82.1°C and with hA3A and eA3Z1 at 80.3°C.
Elephant APOBEC3Z1 induces double strand DNA breaks and apoptosis
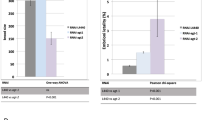
Human A3A editing of chromosomal DNA results in the formation of DSBs and can be readily scored by analysis of histone variant H2AX phosphorylation at serine 139 (γH2AX), a well-known marker for DSBs and the DNA damage response39. HeLa and QT6 cell lines were transfected with eA3Z1 and hA3A constructs ±UGI using plasmid vector as negative control. As can be seen in Fig. 5a, eA3Z1 and hA3A generated DSBs in HeLa cells ∼35 and ∼20-fold over background. While DSBs were more pronounced in QT6 with ∼70 and ∼40-fold higher over plasmid control with eA3Z1 being the more active of the two enzymes (Fig. 5b). In the presence of UGI, a decrease in A3-induced DSBs was noted for hA3A and eA3Z1 transfected cells indicating that UNG plays an important role in the formation of DSBs upon DNA editing. As A3-induced DSBs lead to apoptosis, we measured cytochrome c release (Fig. 5c). Transfection of equal amounts of plasmid DNA resulted in greater cytochrome c release for eA3Z1 using Annexin V and propidium iodide as markers for apoptosis (Fig. 5c,d). These data demonstrate that eA3Z1 is a strong editor of ssDNA and can induce DSB leading to apoptosis.

APOBEC3Z1 proteins can induce DSBs and apoptosis. (a,b) FACS analysis of γH2AX-positive HeLa and QT6 cells at 48 hours post transfection with pv, hA3A, eA3Z1 in presence or absence of UGI. Percentage of γH2AX positive cells are gated on V5 positive cells except for mock and UGI transfections. Error bars represent s.d. from three independent transfections. Differences were calculated using the Unpaired t test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (c) FACS analysis of cytochrome c release in transfected cells with pv, hA3AC101S, hA3A, eA3Z1 and etoposide used as positive control. Differences were calculated using the Unpaired t test (*p < 0.05, **p < 0.01, ns: no significance). (d) FACS analysis of early apoptosis and late apoptosis/necrosis in HeLa cells transfected with pv, hA3AC101S, hA3A, eA3Z1 and etoposide used as positive control. Early apoptosis (Annexin V-positive, PI-negative cells, white) and late apoptosis/necrosis (Annexin V-positive, PI double-positive cells, grey) were analyzed at 48 hours post-transfection. Error bars represent s.d. from three independent transfections. Top error bars correlate with Annexin staining, whereas lower error bars represent s.d. for PI staining among Annexin + cells. Differences were calculated using the Unpaired t test (**p < 0.01, ***p < 0.001, ns: no significance). (e) FACS analysis of γH2AX-positive HeLa cells gated on the V5-tagged A3A from different animal species. Differences were calculated using the Unpaired t test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Taken together, these data demonstrate that eA3Z1 clearly exhibit an enzymatic activity similar to human and other mammalian A3A cytidine deaminases since eA3Z1 edits both C and 5MeC residues in ssDNA and can make DSBs leading to apoptosis. A side-by-side comparison of eA3Z1 to 8 mammalian A3As is shown in Fig. 5e. All constructs are well expressed by Western blotting and immunofluorescence with the signal exception of the tamarin construct26. While eA3Z1 is well ranked among the series, it is less efficient than the cow and horse constructs which, although they have large body mass and longevity (wild cattle 18–25 + years, up to 900 kg; horses 30–40 years, up to 600 kg), are not comparable to the elephant (median 56 years, up to 70 years, up to 7000 kg). Obviously, there are many variables that can alter A3 function: mi- and lncRNAs and transcription factor sites in the promoter as well as the role of negative interactors like TRIB334,40 which is part of the broad CtIP-Rb-BRCA1-ATM protein network that involves cell cycle control, cell survival, DNA repair, and genome stability.
The eA3Z1 described here is clearly comparable to those of many large mammals, being able to damage chromosomal DNA and might therefore contribute to oncogenesis. If so, perhaps the A3s of elephants must be tightly regulated to lower the incidence of cancer.
Methods
Plasmids and samples
Elephant A3Z1 cDNAs were synthetized (GeneCust) and subsequently cloned into pcDNA3.1/V5-His-TOPO vector (Invitrogen). All constructs were C-tagged by the V5 epitope. Catalytically inactive hA3A mutants, UGI expression plasmid and other mammalian A3A expression plasmids, sheep, tamarin, rabbit, rhesus monkeys, dog, cow and horse were already described. All plasmids were verified by sequencing. A fatal case of encephalomyocarditis virus involving an African elephant (Loxodonta africana) occurred in November 2013 at the Réserve Africaine de Sigean in France32. Naturally infected samples were collected as part of routine veterinary investigation carried out by qualified veterinarians in the area of origin. All methods were carried out in accordance with relevant guidelines and regulations.
Cell transfection
Approximately 800,000 HeLa, HEK-293T, HEK-293T-UGI and QT6 cells were seeded into 6-well plates and transfected with 2 µg of A3 plasmid using the jetPRIME transfection kit (Polypus Transfection™) according to manufacturer’s instructions. For cotransfections, a plasmid ratio of 1:1 was used.
Deamination assay
At 48 hours post-transfection, A3-transfected 293 T cells were extensively washed with PBS and mechanically harvested. Total proteins were extracted using specific lysis buffer (25 mM HEPES pH 7.4, 10% glycerol, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, 1 mM MgCl2, 1 mM ZnCl2) supplemented with protease inhibitors. Deaminase activity was assessed by incubating whole cell lysates with 1 pmole DNA oligonucleotide 5′FAM-AAATTCTAATAGAT AATGTGA-TAMRA in the presence of 0.4 unit of uracil-DNA-glycosylase (UDG) (New England Biolabs) in a 20 mM Tris-HCl, 1 mM dithiothreitol, and 1 mM EDTA reaction buffer. After 2 hours of incubation at 37 °C, abasic sites were cleaved by heating for 2 min at 95 °C and end point fluorescence was measured using a RealPlex2 Mastercycler (Bio-Rad) with FAM setting and background fluorescence obtained with mock-transfected cells as negative control. Results are normalized to the quantity of protein using Pierce BCA protein assay kit (Thermo Scientific).
RNA and DNA extraction, 3D-PCR amplification and cloning
Total RNA from the elephant liver and DNA from transfected cells were extracted using the MasterPureTM complete DNA and RNA purification kit (Epicentre) and suspended in 35 µL of sterile water. cDNAs were synthetized using QuantiTect reverse transcription kit (Qiagen). For eA3Z1 amplification, a semi nested PCR was performed. For PCR1 and PCR2, first round primers were 1MYfwd: 5′CTGATGGATCAAAACATATTCCGCTTCA and 2MYrev: 5′TCAGTTGTTTCCATTCTGGAGAATAC. Second round primers for PCR1 were, 1MYfwd: 5′CTGATGGATCAAAACATATTCCGCTTCA and 1MYrev: 5′TGGGCACAGTTACGGCAGGGACTC. And for PCR2, second round primers were: 2MYfwd: 5′GGCCAGAAACAGACCTACCTGTGC and 2MYrev: 5′TCAGTTGTTTCCATTCTGGAGAATAC. First and second round reaction parameters were 95 °C for 5 min, followed by 35 cycles of 95 °C for 30 sec., 60 °C for 30 sec. and 72 °C for 1 min., with final extension for 10 min. at 72 °C. PCR1 and 2 products direct sequencing were outsourced to Eurofins and performed using amplification primers: 1MYfwd, 1MYrev, 2MYfwd and 2MYrev (accession number: MK156802).
For TP53 amplification, primers were: TP53out 5′GAGCTGGACCTTAGGCTCCAGAAAGGACAA TP53outrev 5′GCTGGTGTTG TTGGGCAGTGCTAGGAA, amplification was performed using first-round standard PCR with 5 µL of DNA extract followed by nested 3D-PCR9,33 with 5 µL of 1/50 dilution of the first PCR round. Primers for nested 3D-PCR were: TP53in 5′TTCTCTTTTCCTATCCTGAGTAGTGGTAA and TP53inrev 5′AAAGGTGATAAAAGTGAATCTGAGGCATAA. 3D-PCR was performed in 50 µL with 1 U Taq DNA polymerase (Eurobio) per reaction. PCR conditions for the first round of amplification were 5 min. of denaturation at 95 °C then 40 cycles of amplification (30 sec. 95 °C, 30 sec. 58 °C, 30 sec. 72 °C), followed by 7 min. at 72 °C. The condition of 3D-PCR were 5 min of denaturation temperature gradient of 84–92 °C then 40 cycles of amplification (1 min. 84–92 °C, 30 sec. 58 °C, 2 min. 72 °C), followed by 10 min. at 72 °C.
For the 5MeC deamination assay, a 679 bp fragment of HIV-I LAI env gene was amplified using total substitution of dCTP by 5Me-dCTP (Trilink) using the primer pair MC1, 5′TTGATGATCTGTAGTGCTACAGCA and MC2, 5′GCCTAATTCCATGTGTACATTGTA. The 5MeC containing DNA was heat denatured and chilled on ice and 200 ng of synthesized DNA was transfected using jetPRIME 24 hours following initial transfection of A3 coding plasmids in QT6 cells as described earlier16. The second round PCR was classical PCR, primers were: MC3 5′TGTACCCACAGACCCCAACCCACAA and MC4 5′TTCCATTGAACGTCTTATTATTACA, and PCR parameters were: 95 °C for 5 min., followed by 30 cycles of amplification (45 sec. 95 °C, 45 sec. 54 °C, 90 sec. 72 °C) followed by 20 min. at 72 °C. The 3D-PCR reaction parameters were 75–88 °C for 5 min., followed by 35 cycles of amplification (45 sec. 75–88 °C, 45 sec. 56 °C, 90 sec. 72 °C) followed by 20 min. at 72 °C, primers were MC5, 5′ATCAAAGCCTAAAGCCATGTGTAA and MC6, 5′CAATAATGTATGGGAATTGGCTCAA. 3DPCR products were cloned into the pCR2.1 TOPO cloning vector (Invitrogen) and sequenced (Eurofins).
Immunofluorescence
Approximately 50,000 HeLa cells were seeded in Nunc™ Lab-Tek™ II Chamber Slide™ System Thermo Scientific™ and transfected 24 hours later with 1 µg of plasmid DNA according to the Fugene® protocol. Two days after transfection, coverslip grown transfected HeLa cells were washed three times with PBS and fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 15 min. Cells were then washed two times and permeabilized with a 50% methanol/acetone mix for 10 min. After two PBS washings, permeabilized cells were incubated for 1 hour at room temperature, first with 0.5% bovine serum albumin (BSA) PBS 1/200 mouse monoclonal anti-V5 antibody (Invitrogen) and then with 0.5% bovine serum albumin PBS 1/500 anti mouse Alexa Fluor 488 conjugated antibody (ThermoFisher). After several PBS washings, coverslips were mounted with Vectashield mounting medium for immunofluorescence (Interchim). Imaging was performed using a Leica SP5 confocal microscope.
FACS analysis, double strand-breaks and apoptosis
Transfected cells were trypsinized, washed with PBS, fixed in 2% ice-cold paraformaldehyde (Electron Microscopy Sciences) for 10–20 min. on ice. After one PBS washing, cells were permeabilized in 90% ice-cold methanol (Sigma) for 30 min. For DSBs experiments, fixed and permeabilized cells were incubated 1 hour on ice with 1:200 PBS-0.5% BSA diluted mouse monoclonal anti V5-Tag Alexa Fluor® 488 antibody (AbD Serotec) and 1:100 diluted Alexa Fluor® 647 Mouse anti-H2AX (BD Pharmingen). For apoptosis, transfected HeLa cells were collected and washed with PBS, then incubated with complete DMEM medium at 37 °C for 2 hours. After washing with cold PBS, cells were resuspended in 1X Binding Buffer (BD Pharmingen) and then counterstained with 1 μg/ml FITC Annexin V antibody (BD Pharmingen) and 5 μg/ml Propidium Iodide (PI) (BD Pharmingen) to distinguish between early apoptotic and late apoptotic or necrotic events. Treatment by 100 mM etoposide in dimethylsulfoxide was used as positive control. The labelled samples were analyzed on a MACSQuant® analyzer harboring violet, blue, and either a red laser (measure of dsDNA breaks and apoptosis). The data were analyzed using the FlowJo® software (Tree Star Inc., version 10.1r5 for Mac).
Mitochondrial cytochrome c release
At 48 hours post-transfection, HeLa cells were trypsinized and investigated for cytochrome c release by using the FlowCellect cytochrome c kit from Millipore according to manufacturer’s instructions. Cells treated with 200 µM etoposide for 16 hours were used as a positive control of cytochrome c release. Stained samples were acquired on a MACSQuant Analyzer (Miltenyi Biotec) and the data was analyzed with FlowJo software (Tree Star Inc. version 10.0.8). For each sample 10,000 cells were counted.
Western blotting
Cells were recovered 48 hours after transfection. Protein extraction and Western blot analysis were carried out according to standard procedures. After blocking, membranes were probed with either a 1:5000 dilution of anti V5-tag horseradish peroxidase-coupled antibody (Invitrogen), or a 1:15000 dilution of anti β-actin (Sigma). The membrane was subjected to detection by SuperSignal™ West Pico chemiluminescent substrate (ThermoFisher Scientific).
References
Knisbacher, B. A., Gerber, D. & Levanon, E. Y. DNA Editing by APOBECs: A Genomic Preserver and Transformer. Trends Genet 32, 16–28 (2016).
Conticello, S. G., Thomas, C. J., Petersen-Mahrt, S. K. & Neuberger, M. S. Evolution of the AID/APOBEC family of polynucleotide (deoxy) cytidine deaminases. Mol Biol Evol 22, 367–377 (2005).
Larue, R. S. et al. Guidelines for Naming Non-Primate APOBEC3 Genes and Proteins. J Virol 83, 494–497 (2008).
LaRue, R. S. et al. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol 9, 104 (2008).
Munk, C., Willemsen, A. & Bravo, I. G. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol Biol 12, 71 (2012).
Caval, V. et al. Molecular basis of the attenuated phenotype of human APOBEC3B DNA mutator enzyme. Nucleic Acids Res 43, 9340–9349 (2015).
Caval, V., Suspène, R., Shapira, M., Vartanian, J. P. & Wain-Hobson, S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3′UTR enhances chromosomal DNA damage. Nat Commun 5, 5129 (2014).
Roberts, S. A. et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45, 970–976 (2013).
Suspène, R. et al. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci USA 108, 4858–4863 (2011).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Nik-Zainal, S. et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 149, 979–993 (2012).
Pleasance, E. D. et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463, 184–190 (2010).
Stephens, P. J. et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404 (2012).
Carpenter, M. A. et al. Methylcytosine and Normal Cytosine Deamination by the Foreign DNA Restriction Enzyme APOBEC3A. J Biol Chem 287, 34801–34808 (2012).
Denissenko, M. F., Chen, J. X., Tang, M. S. & Pfeifer, G. P. Cytosine methylation determines hot spots of DNA damage in the human P53 gene. Proc Natl Acad Sci USA 94, 3893–3898 (1997).
Suspène, R., Aynaud, M. M., Vartanian, J. P. & Wain-Hobson, S. Efficient deamination of 5-methylcytidine and 5-substituted cytidine residues in DNA by human APOBEC3A cytidine deaminase. PLoS ONE 8, e63461 (2013).
Burns, M. B. et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 (2013).
Jaguva Vasudevan, A. A. et al. APOBEC3B Activity Is Prevalent in Urothelial Carcinoma Cells and Only Slightly Affected by LINE-1 Expression. Front Microbiol 9, 2088 (2018).
Landry, S., Narvaiza, I., Linfesty, D. C. & Weitzman, M. D. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Reports 12, 444–450 (2011).
Komatsu, A., Nagasaki, K., Fujimori, M., Amano, J. & Miki, Y. Identification of novel deletion polymorphisms in breast cancer. Int J Oncol 33, 261–270 (2008).
Long, J. et al. A Common Deletion in the APOBEC3 Genes and Breast Cancer Risk. J Natl Cancer Inst 105, 573–579 (2013).
Xuan, D. et al. APOBEC3 deletion polymorphism is associated with breast cancer risk among women of European ancestry. Carcinogenesis 34, 2240–2243 (2013).
Zhang, T. et al. Evidence of associations of APOBEC3B gene deletion with susceptibility to persistent HBV infection and hepatocellular carcinoma. Hum Mol Genet 22, 1262–1269 (2012).
Nik-Zainal, S. et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet 46, 487–491 (2014).
Lamy, P. et al. Paired Exome Analysis Reveals Clonal Evolution and Potential Therapeutic Targets in Urothelial Carcinoma. Cancer Res 76, 5894–5906 (2016).
Caval, V., Suspène, R., Vartanian, J. P. & Wain-Hobson, S. Orthologous mammalian APOBEC3A cytidine deaminases hypermutate nuclear DNA. Mol Biol Evol 31, 330–340 (2014).
Peto, R., Roe, F. J., Lee, P. N., Levy, L. & Clack, J. Cancer and ageing in mice and men. Br J Cancer 32, 411–426 (1975).
Abegglen, L. M. et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA 314, 1850–1860 (2015).
Sulak, M. et al. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. Elife 5 (2016).
Henry, M., Terzian, C., Peeters, M., Wain-Hobson, S. & Vartanian, J. P. Evolution of the primate APOBEC3A cytidine deaminase gene and identification of related coding regions. PLoS ONE 7, e30036 (2012).
Thielen, B. K. et al. Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. J Biol Chem 285, 27753–27766 (2010).
Lamglait, B., Joris, A., Romey, A., Bakkali-Kassimi, L. & Lemberger, K. Fatal Encephalomyocarditis Virus Infection in an African Savanna Elephant (Loxodonta Africana) in a French Zoo. J Zoo Wildl Med 46, 393–396 (2015).
Suspène, R., Henry, M., Guillot, S., Wain-Hobson, S. & Vartanian, J. P. Recovery of APOBEC3-edited human immunodeficiency virus G- > A hypermutants by differential DNA denaturation PCR. J Gen Virol 86, 125–129 (2005).
Aynaud, M. M. et al. Human Tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J Biol Chem 287, 39182–39192 (2012).
Mussil, B. et al. Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to DNA damage response and apoptosis. PLoS ONE 8, e73641 (2013).
Berger, G. et al. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog 7, e1002221 (2011).
Vartanian, J. P., Guétard, D., Henry, M. & Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320, 230–233 (2008).
Suspène, R. et al. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci USA 102, 8321–8326 (2005).
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S. & Bonner, W. M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273, 5858–5868 (1998).
Westrich, J. A. et al. Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J Virol 92, e01318–17 (2018).
Acknowledgements
This work was supported by funds from the Institut Pasteur, the Centre National de la Recherche Scientifique (CNRS) and the Association pour la Recherche sur le Cancer (ARC, S-CR16140). XXL was supported by a stipend from Lanzhou Institute of Biological Products Co., Ltd (LIBP), subsidiary company of China National Biotec Group Company Limited (CNBG) and by a stipend from the Pasteur-Paris University (PPU) International PhD program. We would like to thank Drs Aurore Romey, Sandra Blaise-Boisseau and Labib Bakkali Kassimi for providing elephant tissues and for RNA extraction.
Author information
Authors and Affiliations
Contributions
X.X.L. and V.C. performed the experiments and analyzed the data. S.W.H. and J.P.V. designed the experiments, analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Caval, V., Wain-Hobson, S. et al. Elephant APOBEC3A cytidine deaminase induces massive double-stranded DNA breaks and apoptosis. Sci Rep 9, 728 (2019). https://doi.org/10.1038/s41598-018-37305-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37305-z
This article is cited by
-
Beyond tradition and convention: benefits of non-traditional model organisms in cancer research
Cancer and Metastasis Reviews (2021)
-
Mouse APOBEC1 cytidine deaminase can induce somatic mutations in chromosomal DNA
BMC Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
