
Abstract
In mammals, microbial colonization of the digestive tract (GIT) occurs right after birth by several bacterial phyla. Numerous human and mouse studies have reported the importance of early gut microbial inhabitants on host health. However, few attempts have been undertaken to directly interrogate the role of early gut/rumen microbial colonization on GIT development or host health in neonatal ruminants through artificial manipulation of the rumen microbiome. Thus, the molecular changes associated with bacterial colonization are largely unknown in cattle. In this study, we dosed young calves with exogenous rumen fluid obtained from an adult donor cow, starting at birth, and repeated every other week until six weeks of age. Eight Holstein bull calves were included in this study and were separated into two groups of four: the first group was treated with rumen content freshly extracted from an adult cow, and the second group was treated with sterilized rumen content. Using whole-transcriptome RNA-sequencing, we investigated the transcriptional changes in the host liver, which is a major metabolic organ and vital to the calf’s growth performance. Additionally, the comparison of rumen epimural microbial communities between the treatment groups was performed using the rRNA reads generated by sequencing. Liver transcriptome changes were enriched with genes involved in cell signaling and protein phosphorylation. Specifically, up-regulation of SGPL1 suggests a potential increase in the metabolism of sphingolipids, an essential molecular signal for bacterial survival in digestive tracts. Notably, eight genera, belonging to four phyla, had significant increases in abundance in treated calves. Our study provides insight into host liver transcriptome changes associated with early colonization of the microbial communities in neonatal calves. Such knowledge provides a foundation for future probiotics-based research in microbial organism mediated rumen development and nutrition in ruminants.
Similar content being viewed by others


Introduction
Young calves are born with an under-developed reticulo-rumen1 and are forced to behave like monogastrics while fed milk-based diets, which are digested in the abomasum. Later in life, the calves transition to dry and forage based diets, which requires further development of the rumen from the esophageal groove and the colonization of an associated microbial community2,3. During this transition, the optimal early development of the rumen is critical to ensure animal health, productivity, and related economic benefits to the producers. The development of the rumen during this transition involves three simultaneous components. (1) Firstly, there must be an anatomical development of the rumen, including growth in rumen volume and rumen papillation4. There are three phases involved in the anatomical development of rumen, including non-rumination (0–3 weeks); transitional phase (3–8 weeks) and rumination (from 8 weeks on)5. (2) Simultaneously, there is a colonization and establishment of the microbial community in the developing rumen6,7. (3) Finally, there is a functional development of fermentation capacity and enzyme activity in the rumen lumen and epimural layers8. These developments lead to a major change in nutrients delivered to the intestines and liver, and subsequent peripheral tissue of the animal9.
Gut microbial colonization is an important process that accompanies the rumen development process10. In ruminant livestock, rumen microbes provide 70% of the daily energy requirement of ruminants11. Studies have shown that different feeding management regimes may be associated with different microbial populations establishing in the rumen of young ruminants7,12,13,14. In the rumen of young lambs fed with forage, a different composition of microbial community was observed in comparison to the ones fed with concentrate, and this difference in microbial composition persisted over months. Artificially reared young ruminants have different protozoa and microbial populations as compared to those reared with their mothers, raising the possibility of a beneficial effect by direct microbial inoculation by the dam12,15. Moreover, despite a reported host-specificity in the rumen microbiome community, pre-weaning diet and feeding methods have been reported to have pronounced and long-lasting impacts on rumen microbial composition12,13. In comparison to calves weaned conventionally (at 6 weeks), calves introduced with solid feed early (at 3 weeks) showed much greater microbial abundance in the rumen16.
In addition to the effect on microbial communities observed using nutritional manipulation, direct feeding of live microorganisms to ruminants has also been reported to be advantageous17,18. Inoculation with fresh rumen fluid into rumens in early weaned lambs improved average daily gain and digestibility19. By adding a fungal probiotic, Theodorou et al.20 observed increased intake and live weight gain in calves at weaning. In adult ruminants, such artificial manipulation is short-lived and there is strong evidence of host specificity after mature rumen microbial colonization21. Early dietary experience contributes to a more pronounced and lasting effect on the rumen microbial composition22 and as a result various processes may be involved during early life adaptation, including neuroendocrine, morphological and physiological changes23. The most critical is the maturation of the immune system in the host modulated by early colonization events of microbes24. Collectively, these studies led to a hypothesis of microbial programming in early life.
Microbial colonization has an effect on the host’s innate immune response25,26. Tracheal antimicrobial peptide (TAP) gene, a β-Defensin gene27, is a front-line protectant against pathogens, and TAP expression has been associated with inflammation28. Elevated expression of TAP in tracheal epithelial cells has been observed after introduction of bacteria in cattle29. Additional roles of TAP include providing a link between innate and adaptive immune responses against microbial invasions30. TAP is believed to play a critical role in microbicidal activity against bacterial pathogens causing bovine respiratory disease. Notably, the β-defensin gene family in cattle was reported to have the most diverse repertoire so far identified31. In sheep, peak expression of β-defensin 1(also referred to as oBD1) from tongue to colon, and β-defensin 2 (oBD2) in the distal ileum were identified in the first 6–8 weeks of life32, with substantial expression in the digestive tract in pre-natal lambs33. Toll-like receptors (TLRs) are another class of host proteins with important roles in recognizing commensal and pathogen associated molecular patterns (PAMPs) from the gut flora. The utility of TLR signaling has been demonstrated in the maintenance of tight junctions between epithelial cells, and antimicrobial peptide expression34. These studies suggest that the genes involved in host innate immunity are critical players in managing a complex interface between host immune surveillance and the newly colonized rumen microbial community.
Several studies showed that initial microbial colonization occurs immediately after birth, with substantial colonization by critical bacterial phyla and genera in the first few days of life7,12,35. One of these studies found that, at birth, Methanogens abd fibrolytic bacteri were already present in the rumen35. At day two, the rumen microbial community was mainly composed of Proteobacteria, Bacteroidetes and Prevotella7. And an abrupt change was observed in the ruminal bacterial between days 2 and 3 and until day 12, with dominant genera being Bacteroides, Prevotella, Fusobacterium and Streptococcus7. Numerous human and mouse studies have reported the importance of early gut microbiota on host health, energy intake and storage36. However, few attempts have been undertaken to understand the role of early gut/rumen colonization on GIT development or host health in neonatal ruminants37. Thus, molecular mechanistic changes associated with bacterial colonization are largely unknown. In this study, we aim to characterize the host physiological changes resulting from the dosing of exogenous rumen content to post-natal calves. Specifically, we used a tissue transcriptome approach to study the host transcriptome changes in the liver that are vital to nutrient absorption. Additionally, we studied the meta-transcriptomics of the rumen microbial community using rRNA reads generated by RNA-sequencing.
Results
Bacterial community diversity of administered rumen inoculum
A total of 16 administered inoculum samples (collected at four times points immediately prior to the dosing of each treated calf) were subjected to bacterial composition analysis using next-generation sequencing. After sequence cleanup in mothur38, a total of 669,108 high quality reads were obtained for these samples (average per sample 51,469 ± 4,823 SEM). Good’s coverage was greater than 97% for all samples. Following normalization to 10,000 sequences per sample, observed bacterial community composition in all the rumen inoculum was comparable to each other. In Fig. 1, inoculum samples were grouped by the calf ID to which they were administered to. Within each calf ID, administered inoculum samples were labeled by the time they were collected (0, 2, 4 and 6 weeks from birth). In all samples, Bacteroidetes was the most abundant phylum (53.31 ± 1.85% SEM) followed by Firmicutes (34.32 ± 1.82% SEM) (Fig. 1).

Microbial community composition analysis using targeted 16S rRNA genomic sequencing. Sequencing reads were generated by next-generation sequencing of targeted, genomic amplicon 16S rRNA gene v4 region.
RNA quality, sequencing reads and total number of expressed genes
For liver samples, the average RIN (RNA integrity number) for extracted RNAs was 8.43 ± 0.09 (s.e.). The average RIN for RNAs extracted from rumen papillae was 9.08 ± 0.16 (s.e.). An average of five million rRNA reads, 5,072,494 ± 983,485 (s.e.), were obtained for microbial classification analysis for each rumen epimural microbial community. Total number of RNA sequencing raw reads for liver samples ranged from 69 M to 81 M, with an average of 76.8 M ± 1.29 M (s.e.). Total number of expressed genes ranges from 13,669 to 14,287 (fpkm cutoff > = 5) (Supplemental Table 1).
Differentially expressed genes between treated and untreated calves
In liver tissue, a total of 338 genes showed significant differential expression (fold change > = 1.5, and adjusted p-value < = 0.05). In the treated group (in comparison to the control group) 194 were up-regulated and 144 of them were down-regulated. (Supplemental Table 2). Gene ontology (GO) term enrichment analysis was performed using DAVID39, with Bos taurus as the reference. Top GO terms enriched by differentially expressed genes (DEGs) identified in liver transcriptomes include the following (Table 1): cellular component organization and biogenesis (GO:0071840, p-value = 0.02), single-organism metabolic process (GO:0044710, p-value = 0.01), regulation of signaling (GO:0023051, p-value = 0.04), positive regulation of nitrogen compound metabolic process (GO:0051173, p-value = 0.04), peptidyl-tyrosine phosphorylation (GO:0018108, p-value = 0.01), and innate immune response (GO:0045087, p-value = 0.05).
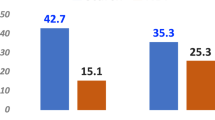
Annotation of the top 25 most upregulated genes include those associated with immune response and sphingolipid metabolism. Eight genes were involved in the innate immune response (Supplemental Table 3). The expression profile of homologs of these genes were investigated using tissue-specific gene expression data in cattle (Bos taurus) from the Gene Expression Atlas databases40 (https://www.ebi.ac.uk/gxa/home). And due to the comprehensive list of tissues included in the database, human tissue expression database (GTEx project, (https://www.gtexportal.org/home/) was also checked. Among these, SMPDL3B showed predominant expression in human GI tissue types (Supplemental Fig. 1a) and high expression in cattle liver (Supplemental Fig. 1b). Fibrinogen beta chain (FGB) and fibrinogen gamma chain (FGG) showed exclusive expression in human liver (Supplemental Fig. 2a) and high expression in cattle liver (Supplemental Fig. 2b). The rest of the genes showed nearly universal expression across investigated tissues. For RT-qPCR results, all of four of the chosen genes were confirmed by our RT-qPCR results, with significant up-regulation in treated animals (Fig. 2).

Fold-change (Treated vs. control) of four genes determined by both RT-qPCR and RNA-seqencing methods.
Results of taxonomic classification of rumen wall microbial community
The average classification rate is 97.9 ± 1.4 (s.e.). Among the top 10% most abundant taxa at genus level, 29 of them are shared between the calves from control and treated groups; four of them are unique to the control group (Aeromonas, Alistipes, Desulfotomaculum and Candidatus Arthromitus), and three of them are unique to the treated group (Corynebacterium, Thermoanaerobacterium and Algoriphagus). The abundance of 8 genera are significantly higher in the treated group (p-value < 0.05). These are: Acidiphilium, Jeotgalibaca, Polaribacter, Pseudodesulfovibrio, Bdellovibrio, Microbacterium, Eubacterium, Sporosarcina. And they belong to four phyla: Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria (Fig. 3). PCA analysis revealed that the calves treated with rumen-inoculum from high-efficiency donor (HE) separated from control animals along PC2 and PC4 (Fig. 4), counting for 31.52% of the overall differences between control and treated groups.

The abundance of eight genera are significantly higher in treated group (p-value < 0.05), in comparison to control group. rRNA sequencing reads mapped to each genera by Kraken were used to calculate the normalized read counts.

PCA plot using normalized, genus level read counts. Control and treated animals separate along PC2 and PC4, accounting 31.52% of the overall differences. Rumen microbial rRNA reads were obtained by rumen papillae tissue RNA-seq. Genus classification was done using Kraken.
Discussion
Genes with elevated expression changes and GO term annotation in liver
GO term annotation for the genes with most significant fold change between treated and control groups mainly fell into two groups: host-level molecular change in response to infection, including phosphorylation and cell signaling transduction; and mechanisms that facilitate bacterial establishment. Host protein phosphorylation during bacterial infection has been reported previously41,42. As the most widespread mechanism of post-translational modification, protein phosphorylation is reversible, and is commonly used to activate or deactivate cellular processes by switching enzyme activities “on” or “off”. During microbial colonization in treated calves, it is likely that phosphorylation-based cellular signaling sent from the host, contributes to immunity and to restricting foreign microbial invasion. Future protein phosphorylation based studies will most likely help identify the key protein phosphorylation events that co-occur with microbial invasion and establishment.
Among the major groups of microbial inhabitants in the rumen, bacteria are the most responsible for fermenting feed into an absorbable energy source by cattle21. In dairy cattle, Bacteroidetes is a dominant bacteria phylum in the rumen43,44, with a predominant presence in pre-weaned calves45. Several beneficial roles of Bacteroides have been reported and they include: contributing to the normal development of the gastrointestinal tract (GIT)46, interacting with the immune system to induce T-cell mediated responses47,48, and limiting the GIT colonization by potential pathogenic bacteria49.
Notably, we observed significant up-regulation of SGPL1 in liver transcripts. SGPL1 encodes Sphingosine 1-phosphate (S1P), which is a polar sphingolipid metabolite that regulates cell migration, differentiation, survival and complex physiological processes50. Sphingolipids entail diverse signal transduction, and are involved in stress response pathways with profound physiological impacts as demonstrated in a variety of eukaryotic cell types51. Quite interestingly, sphingolipid is reported to be essential for Bacteroides physiology, enabling them to perform functions related to symbiosis in the intestines52, and to play an important role in the development of immunological tolerance to commensal microbiota49. Given its diverse function in dealing with stress, sphingolipid-mediated signaling is an ideal mechanism for both the host and foreign bacteria to adapt and survive. Thus, we hypothesize that artificial dosing of rumen contents introduced a rich influx of Bacteroidetes into the GIT of newborn calves. These newly introduced bacteria imposed a threat to the host while, in the meantime, experienced intensive stress from the GIT of the host. As a metabolic organ, the metabolic activity of liver is controlled by many signals produced by the host. When the host produces excess amount of sphingolipid as an adaptive response to stress, increased metabolism of sphingolipid is observed in the liver.
Early dosing and host immune response
At the early stage of microbial invasion, the host immune response provides the frontline protection against newly introduced microbial species. Gut microbiota has a critical role in the development of both innate and adaptive components of the mucosal immune system10. Our RNA sequencing work provided evidence that immunity related genes responded to the alterations in gut microbiota with elevated expression levels in the liver. This finding suggests that the calves are coping to the exogenous bacterial introduction by turning on their immune and defense responses. Accordingly, we observed seven genes with substantial expression changes that are each involved in the adaptive and innate immune responses. Notably, SMPDL3B, encoding a membrane-modulating enzyme showed 2.5 fold change in expression in our experiment. Heinz et al.53 indicated that this gene may function at the interface of membrane biology and innate immunity. Interestingly, the human homolog of this gene shows predominant expression in an array of GI tissues, indicating a potentially significant role of this gene in the molecular mechanisms of GI physiology. Two genes, FGG and FGB, showed exclusive expression in liver, reflected by the human tissue-specific gene expression data. Aside from their roles in the adaptive immune response, these two genes were also shown to be involved in inflammatory pathways, with greatly increased expression in hepatocytes during inflammatory stress54.
Our study suggests that genes involved in the immune response, anti-inflammatory response and signaling are poised to provide both innate and adaptive immune responses and ‘tolerance’ to the first colonizers of the rumen. Immune responses consume a great deal of energy from the host with the desired outcome of destroying foreign intruders. Further studies may focus on targeted regulation of immunity- and anti-inflammation-associated genes during the neo-natal stage following artificially dosing rumen contents. For example, comparing the colonization rates of an exogenously introduced bacterial community in controls that do not demonstrate host gene regulation to those individuals comprising a treated group that does exhibit gene regulation. Such studies will undoubtedly provide more insights into the molecular mechanisms responsible for early microbial establishment in cattle.
In our experiment, we observed that innate immune response related genes showed significant up-regulation in calves dosed with the rumen inocula obtained from the high-milk production cow compared to the control calves. And yet, we did not observe clinical signs associated with pathological infection, e.g., no obvious increase in body temperature in treated calves in comparison to the control calves. Additionally, calves dosed with adult cow rumen content did not appear to have clinical signs consistent with enteric infections. Quite interestingly, studies that administered probiotics to calves have reported several beneficial effects, e.g., decreased incidence of diarrhea, and overall weight gain and increased efficiency in feed conversion18, which likely occurs due to the effect of promoting colonization of beneficial bacteria, while decreasing the occurrence of detrimental bacteria55. Thus, there might be a beneficial effect on calf health through early artificial dosing using adult cattle rumen content, which warrants further in-depth investigation.
Successful graft of exogenous inoculation of rumen content
Rumen content used in our dosing experiment was from an adult cow, within which the rumen microbial population has adapted to the host GIT environment and reached a homeostasis. When introduced to the newborn calf, this microbial population likely faced a new GIT environment, which undoubtedly elicited immune responses from the host. The first question regarding artificial dosing is whether these newly introduced rumen microbiota can survive in the new environment. Using the microbial rRNA reads extracted from our RNA-sequencing experiments, we identified that eight genera, belonging to four phyla, had significant increases in abundance in treated calves in comparison to control calves. Notably, three of these phyla, Bacteroidetes, Firmicutes and Proteobacteria have been reported as the most abundant microbial phyla in the rumens of adult dairy cattle56, suggesting that this inoculation may be hastening the development of a mature rumen microbial community in treated calves. Since the inoculum samples used for the treatment group were extracted from an adult cow, our meta-transcriptomic analysis provided strong evidence of successful graft of these adult cow-originated microbial phyla. Additionally, we observed significant up-regulation of genes involved in sphingolipid metabolism. These data indicate that the newly introduced microbiota have colonized the nascent rumen of the calf and actively interacted with the host GIT epithelial cells. Given existing evidence suggesting the host-specificity of cattle rumen microbial content21, follow-up studies may focus on determining the exact microbial species that successfully colonize within the calves. Gained knowledge might provide guidance into better management of feed during artificial dosing, e.g., variety of nutrients that facilitate establishment of beneficial microbial species, and selective dosing of a combination of microbial species to increase retention time and final colonization.
Future perspectives
Our study represents a snap-shot of the host liver transcriptome changes in response to artificial dosing of rumen microbiota. Results from this study indicated that significant host liver transcriptome changes are triggered by the newly introduced microbial community. For future follow-up studies, comparative analyses at different time points during the artificial dosing experiments will provide more quantitative measures regarding how and when certain transcripts in the host are affected by exogenous, artificially dosed microbes. Knowledge obtained through these studies will most likely facilitate consistent and successful probiotic treatment of exogenous microbial species that are beneficial to the host. Additionally, further investigation into artificial dosing induced host immune responses may be leveraged as a means to promote health and productivity of newborn calves.
Methods
Experimental Design
This experiment was approved by University of Wisconsin-Madison, Institutional Animal Care and Use committee (IACUC). All animals involved in this study were fed and watered according to the herd standard practices used at the USDA Dairy Forage Research Center farm throughout the experiment. Animal care and use, and all methods involved in this study were performed in accordance with the relevant guidelines and regualtions by the Animal Wellfare Act from US Department of Agriculture and by the Federation of Animal Science Societies.
This study covered only a subset of a larger experiment. For the larger experimental design, two adult cows, previously identified as high milk production efficiency (HE) cow, and low milk production efficiency (LE) cow, as described in Jewell et al.56, were used as donors. The main objective of this study was to discern the impact of artificially dosed, live-microbes on the rumen epimural microbial community and liver transcriptome. For this purpose, one HE cow was used as the donor in this study. The diet composition of this donor cow was described in detail in Jewell et al.56. For the treated groups, the calves were dosed with the inocula obtained from this one donor cow’s rumen content (HE group). For the control group, they were dosed with sterilized inocula (control group).
Rumen inocula preparation and dosing
The inocula obtained from the donor cow were prepared as follows: rumen liquids and lightly squeezed solids were collected from the medio-ventral region of the rumen in a 3:1 ratio by volume. The mixture was immediately blended under CO2 for 1 minute, then squeezed through four layers of cheesecloth to remove large particles. Inoculum samples were taken right after blending and placed in a 15 mL conical tube (Corning, Oneonta, NY). For treatment cohorts, fresh inoculum was used same-day. The control inocula were prepared by autoclaving the combined inocula (50% from each) from the HE donor cow and the LE donor cow. For both treatments, doses were administered to bull calves within 3 days after birth, then 2, 4, and 6 weeks following the initial dosing. 50 mL of inoculum was delivered by oral gavage, then followed with 50 mL of sterile McDougall’s buffer to ensure complete clearance of the inoculum from the dosing tube.
DNA extraction and 16S Metagenomics analysis of HE rumen inoculum
Immediately prior to dosing, a portion of freshly prepared HE inoculum samples was collected and stored in −80 °C, at days 0, 2, 4 and 6. A total of 16 inoculum samples were collected, as four biological replicates were retrieved at each time point. To prepare for DNA extraction, frozen inoculum samples were thawed in a room temperature water bath. DNA was extracted using an adaptation of the methods described previously56,57. In short, cells were pelleted at 5000 g for one hour, then resuspended in approximately 2 mL of chilled DNA extraction buffer. 1 mL of this resuspension was transferred to a 2 mL screw-cap tube with 0.5 g for 0.1 mm zirconium beads, 50 uL 20% SDS, and 700 uL cold equilibrated phenol. This mixture was subjected to bead beating for 2 minutes on a tabletop bead beater (Mini Bead Beater, Biospec Products, Bartlesville, OK), then heated in a 60 °C water bath for 10 minutes before another 2 minutes of bead beating. The mixture was then centrifuged for 10 minutes at 4 °C on a tabletop centrifuge at max speed (~15,000 rpm, Microfuge 20 R, Beckman Coulter, Brea, CA). The aqueous layer was washed 2–4 times with cold equilibrated phenol until the white lipid layer disappeared before precipitation of DNA in a mixture of 0.1 vol 2 M Na acetate and 0.6 vol isopropanol overnight. DNA was pelleted in a tabletop centrifuge for 20 minutes at 4 °C with max speed. The pellet was washed with 70% ethanol, then dried overnight. Pellet was resuspended in 100 μL of elution buffer (Invitrogen, Thermo Fisher Scientific, Waltham, MA).
To amplify the 16S rRNA gene, universal primers flanking the variable 4 (V4) region were used58. For one reaction per sample, 25 ng of template DNA, 5 pmol of each of the forward and reverse primers, and 12.5 μL of 2X HotStart ReadyMix (KAPA Biosystems, Wilmington, MA), and water to a total volume of 25 μL were used. Cycling conditions were as follows: initial denaturation of 95 °C for 3 minutes, 25 cycles of 95 °C for 30 seconds, 55 °C for 30 seconds, and 72 °C for 30 seconds, and a final extension at 72 °C for 5 minutes. Size exclusion was performed by gel electrophoresis on a 1.0% low-melt agarose gel (National Diagnositcs, Atlanta, GA), followed by gel extraction of amplified DNA using a ZR-96 Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, CA). Extracted DNA was quantified via a 96-well protocol using manufacturer’s instructions with the Quant-iT™ dsDNA High-Sensitivity Assay Kit, using reagents from a Qubit® dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA). Reactions were read on a Synergy 2 Multi-Mode Reader (BioTek, Winooski, VT) following a programmed 3 second shaking period and a 2 minute incubation at 22 °C. Amplified DNA was equimolar pooled, combined with 10% PhiX control DNA, and sequenced using the MiSeq. 2 × 250 v2 kit (Illumina, San Diego, CA) with custom sequencing primers as described by Kozich et al.58.
The program mothur v.1.38.1 was used for further sequence processing38, following a protocol developed from Kozich et al.58, as described in Weimer et al.59. In short, paired-end sequences were assembled into continuous segments and poor-quality sequences were removed. The SILVA 16S rRNA gene reference alignment database v12860 was used to screen for alignment to the v4 region. Preclustering was performed (diffs = 2) to reduce error and computational load, and chimeric sequences were removed (UCHIME61). The 2013 release of the GreenGenes database62 was used to classify sequences with a bootstrap value cutoff of 80. Sequences classifying to cyanobacteria, mitochondria, Eukarya, or Archaea were removed. Bacterial sequences were grouped into operational taxonomic units (OTUs) with 97% sequence similarity. Good’s coverage63 was calculated in mothur. OTU counts were normalized to 10,000 sequences per sample, and normalized OTU counts were used for further analysis. Stacked bar plots were generated in R version 3.4.3 using the package phyloseq v1.22.364.
Calf tissue collection
Liver and rumen papillae were included in this study. These tissues were collected immediately after animal sacrifice. Upon collection, tissues were rinsed in PBS to remove feed particles if present, and cut with sterilized scalpels into 4–5 mm2 fragments and put into Eppendorf safe-lock tubes (Eppendorf North America, Hauppauge, NY). Collected tissues were flash frozen in liquid nitrogen and stored at −80 °C for long-term storage.
RNA extraction, quantification and whole transcriptome sequencing
Tissues were homogenized into fine powders in liquid nitrogen using a mortar and pestle. RNAs were extracted using miRNeasy kit (Qiagen, US) following manufacturer’s protocol. Quality of extracted RNAs were assessed using Bioanalyzer RNA 6000 nano kit (Agilent Technologies, US). RNA samples with RIN value > = 8 were pursued for RNA quantification using Qbit (Thermo Fisher, US). RNA-sequencing library preparation was done using Illumina TruSseq ribo-zero gold kit following manufacturer’s instructions. For each sample, 1 μg of total RNA was used for sequencing library preparation. After libraries were prepared for each sample, quantification of library was performed using Kapa quantification kit (Kapa systems) using ABI7300 instrument. Libraries were further normalized to ensure equal quantity before sequencing. 2 × 150 bp paired-end reads were obtained using Illumina NextSeq 500 instrument with high-output kit.
Mapping of RNA sequencing raw reads and differential gene expression analysis
Quality of raw reads were checked using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). For sequence alignment, NCBI UMD3.1, Bos taurus reference genome was used. Raw reads from all whole transcriptome RNA-seq libraries were aligned using a two-step alignment approach. First, Tophat265 was used with the following settings: ‘-r 70–mate-std-dec 90′ for paired-end reads from Illumina RNA-seq. Second, unmapped reads from step one were realigned with Bowtie266 using the “–very-sensitive-local” method. The genome annotation file (NCBI, UMD3.1) downloaded from Tophat website (http://ccb.jhu.edu/software/tophat/igenomes.shtml) was used as reference. Genes shorter than 150 bp were excluded from the GTF file. Raw reads shorter than 50 bps were excluded from the alignment process. Raw read counts for each gene were obtained using HTSeq (v0.6) HTseq.67. Combined (Tophat + bowtie2) sequence alignment generated by the two-step alignment approach served as input file for HTSeq. The expression level of mRNAs in each sample were normalized to fpkm using cufflinks68. Normalized fpkm values were used to assess gene expression profiles for each sample. Total number of expressed genes were calculated using fpkm cutoff value of 5.
DEG analysis was performed using the R/Bioconductor package DESeq 269 with raw read counts from RNA-Seq. Genes with less than ten normalized read counts were excluded from further analysis. Read count normalization was performed using the regularized logarithm (rlog) method provided in DESeq2. DEGs were determined by adjusted p-value (cutoff of 0.05) and the fold change (cutoff of 1.5) by DESeq2. Gene function annotation and pathway analysis were performed using DAVID39 and stringDB70,71.
Taxonomic classification of rumen wall microbial community using rRNA-sequencing reads
RNA-sequencing reads used for rumen wall microbial community classification were obtained using the following steps. Total RNA from rumen papilla samples were extracted using miRNeasy kit (Qiagen, US) following manufacturer’s instruction. Extracted total RNAs were further treated with DNase I (Qiagen, US). RNA qualities were assessed using Bioanalyzer RNA 6000 nano kit (Agilent Technologies, US). RNA samples with RIN value > = 8 were pursued for RNA quantification using Qbit (Thermo Fisher, US). RNA-sequencing library preparation was done using Illumina TruSseq ribozero gold kit following manufacturer’s instruction using 1ug of total RNA. Using STAR as the alignment tool72, RNA-seq reads mapped to the genome of Bos taurus (UMD 3.1) were filtered out. To enrich reads coming from microbial rRNA, the remaining, non-cattle RNA-seq raw reads were mapped to rRNA reference databases provided by SortMeRNA73 using STAR72. Mapped reads were used for downstream microbial taxonomic classification using Kraken74, following the protocol here (http://ccb.jhu.edu/software/kraken/MANUAL.html).
To compare the microbial community differences between control and treated groups, taxonomic classifications at the genus level were considered. For each sample, the total number of reads mapped to the each genus level is normalized by the total number of classified reads by Kraken. To do this, the total number of reads mapped to genus level was first divided to 1,000,000, which yields the “per million” factor. Then, the mapped reads at each genus was divided by the “per million factor”, yielding a normalized read count. The statistical significance of differences in genus level abundance between control and treated groups was carried out using non-parametric test, Kruskal-Wallis, by SciPY with p-value cutoff of 0.05. For each treatment group, the abundance of each taxon is ranked using averaged, normalized read counts at the genus level. The top 10% most common taxa were compared between treated and control groups. Principal component analysis (PCA) was performed using normalized read count at genus level prcomp in R (version 3.2).
RT-qPCR verification of target genes
Four genes were analyzed using real time quantitative PCR to assess their differential expression between treated and control groups. These genes are: SGPL1, TMEM175, SMPDL3B and CHST13. SMPDL3B encodes a lipid modifying enzyme, with a reported role in regulating innate immune signaling53 and in anti-inflammatory processes75. SGPL1 encodes a sphingolipid signaling molecule. TMEM17 encodes a transmembrane protein, with a reported role in regulating luminal pH stability76. CHST13 encodes a protein, belonging to sulfotransferase 2 family, and has highly exclusive expression in both fetal and adult liver77.
cDNA synthesis was performed using 2000 μg of RNA with High Capacity cDNA master mix (Life technologies). Gene-specific, Taqman assay probes were ordered from Thermo Fisher (Thermo Fisher, USA). All qPCR reactions were performed using the ABI7500 fast system (Applied Biosystems). The thermocycler steps were as follows: one step of uracil-N-glycosylase (UNG)78,79 treatment at 50 °C for 2 min, followed by an initial denaturation/activation step at 95 °C for 10 min, then 40 cycles at 95 °C for 15 s and 60 °C for 60 s. The experiments were carried out in triplicate for each data point. The fold change in gene expression was obtained following normalization to two reference genes, Beta-actin (ACTB) and hydroxymethylbilane synthase (HMBS). These two reference genes were found to be very consistent in the rumen epithelium80. The relative quantification of gene expression was determined using the 2−ΔΔCt method81.
Data Availability
Gene raw read-counts of liver samples were included in the supplemental dataset. rRNA raw reads obtained from rumen papilla tissues were submitted to NCBI with project accession number of PRJNA478608.
Change history
28 May 2019
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Warner, R. G. Dietary factors influencing the development of the ruminant stomach. Agric.Food Chem. 4, 788–801 (1956).
Shabat, S. K. et al. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J, https://doi.org/10.1038/ismej.2016.62 (2016).
Jami, E., White, B. A. & Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS One 9, e85423, https://doi.org/10.1371/journal.pone.0085423 (2014).
Reynolds, C. K., Durst, B., Lupoli, B., Humphries, D. J. & Beever, D. E. Visceral tissue mass and rumen volume in dairy cows during the transition from late gestation to early lactation. J Dairy Sci 87, 961–971, https://doi.org/10.3168/jds.S0022-0302(04)73240-3 (2004).
Lane, M. A., Baldwin, R. Lt & Jesse, B. W. Developmental changes in ketogenic enzyme gene expression during sheep rumen development. J Anim Sci 80, 1538–1544 (2002).
Fouts, D. E. et al. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen. PLoS One 7, e48289, https://doi.org/10.1371/journal.pone.0048289 (2012).
Rey, M. et al. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J Appl Microbiol 116, 245–257, https://doi.org/10.1111/jam.12405 (2014).
Rey, M., Enjalbert, F. & Monteils, V. Establishment of ruminal enzyme activities and fermentation capacity in dairy calves from birth through weaning. J Dairy Sci 95, 1500–1512, https://doi.org/10.3168/jds.2011-4902 (2012).
Baldwin, R., McLeod, K. R., Klotz, J. L. & Heitmann, R. N. Rumen Development, Intestinal Growth and Hepatic Metabolism In The Pre- and Postweaning Ruminant. Journal of Dairy Science 87, E55–E65 (2004).
Sommer, F. & Backhed, F. The gut microbiota—masters of host development and physiology. Nat Rev Microbiol 11, 227–238, https://doi.org/10.1038/nrmicro2974 (2013).
Yeoman, C. J. & White, B. A. Gastrointestinal tract microbiota and probiotics in production animals. Annu Rev Anim Biosci 2, 469–486, https://doi.org/10.1146/annurev-animal-022513-114149 (2014).
Abecia, L. et al. An antimethanogenic nutritional intervention in early life of ruminants modifies ruminal colonization by Archaea. Archaea 2014, 841463, https://doi.org/10.1155/2014/841463 (2014).
Abecia, L., Martin-Garcia, A. I., Martinez, G., Newbold, C. J. & Yanez-Ruiz, D. R. Nutritional intervention in early life to manipulate rumen microbial colonization and methane output by kid goats postweaning. J Anim Sci 91, 4832–4840, https://doi.org/10.2527/jas.2012-6142 (2013).
Jami, E., Israel, A., Kotser, A. & Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J 7, 1069–1079, https://doi.org/10.1038/ismej.2013.2 (2013).
Belanche, A., de la Fuente, G. & Newbold, C. J. Study of methanogen communities associated with different rumen protozoal populations. FEMS Microbiol Ecol 90, 663–677, https://doi.org/10.1111/1574-6941.12423 (2014).
Anderson, K. L., Nagaraja, T. G. & Morrill, J. L. Ruminal metabolic development in calves weaned conventionally or early. J Dairy Sci 70, 1000–1005, https://doi.org/10.3168/jds.S0022-0302(87)80105-4 (1987).
Jeyanathan, J., Martin, C. & Morgavi, D. P. The use of direct-fed microbials for mitigation of ruminant methane emissions: a review. Animal 8, 250–261, https://doi.org/10.1017/S1751731113002085 (2014).
Abe, F., Ishibashi, N. & Shimamura, S. Effect of administration of bifidobacteria and lactic acid bacteria to newborn calves and piglets. J Dairy Sci 78, 2838–2846, https://doi.org/10.3168/jds.S0022-0302(95)76914-4 (1995).
Zhong, R. Z., Sun, H. X., Li, G. D., Liu, H. W. & Zho, D. W. Effects of inoculation with rumen fluid on nutrient digestibility, growth performance and rumen fermentation of early weaned lambs. Livest. Sci. 162, 154–158 (2014)
Theodorou, M., Beever, D. E., Haines, M. J. & Brooks, A. The effect of a fungal probiotic on intake and performance in early weaned lambs. Anim. Prod. 53, 577 (1990).
Weimer, P. J., Stevenson, D. M., Mantovani, H. C. & Man, S. L. Host specificity of the ruminal bacterial community in the dairy cow following near-total exchange of ruminal contents. J Dairy Sci 93, 5902–5912, https://doi.org/10.3168/jds.2010-3500 (2010).
Distel, R. A., Villalba, J. J. & Laborde, H. E. Effects of early experience on voluntary intake of low-quality roughage by sheep. J Anim Sci 72, 1191–1195 (1994).
Yanez-Ruiz, D. R., Abecia, L. & Newbold, C. J. Manipulating rumen microbiome and fermentation through interventions during early life: a review. Front Microbiol 6, 1133, https://doi.org/10.3389/fmicb.2015.01133 (2015).
Schachtschneider, K. M. et al. Modulation of systemic immune responses through commensal gastrointestinal microbiota. PLoS One 8, e53969, https://doi.org/10.1371/journal.pone.0053969 (2013).
Mogensen, T. H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22, 240–273, Table of Contents, https://doi.org/10.1128/CMR.00046-08 (2009).
Janeway, C. A. Jr. & Medzhitov, R. Innate immune recognition. Annu Rev Immunol 20, 197–216, https://doi.org/10.1146/annurev.immunol.20.083001.084359 (2002).
Diamond, G. et al. Tracheal antimicrobial peptide, a cysteine-rich peptide from mammalian tracheal mucosa: peptide isolation and cloning of a cDNA. Proc Natl Acad Sci USA 88, 3952–3956 (1991).
Caverly, J. M. et al. Coordinated expression of tracheal antimicrobial peptide and inflammatory-response elements in the lungs of neonatal calves with acute bacterial pneumonia. Infect Immun 71, 2950–2955 (2003).
Diamond, G., Kaiser, V., Rhodes, J., Russell, J. P. & Bevins, C. L. Transcriptional regulation of beta-defensin gene expression in tracheal epithelial cells. Infect Immun 68, 113–119 (2000).
Yang, D., Biragyn, A., Kwak, L. W. & Oppenheim, J. J. Mammalian defensins in immunity: more than just microbicidal. Trends Immunol 23, 291–296 (2002).
Meade, K. G., Cormican, P., Narciandi, F., Lloyd, A. & O’Farrelly, C. Bovine beta-defensin gene family: opportunities to improve animal health? Physiol Genomics 46, 17–28, https://doi.org/10.1152/physiolgenomics.00085.2013 (2014).
Huttner, K. M., Brezinski-Caliguri, D. J., Mahoney, M. M. & Diamond, G. Antimicrobial peptide expression is developmentally regulated in the ovine gastrointestinal tract. J Nutr 128, 297S–299S (1998).
Meyerholz, D. K., Kawashima, K., Gallup, J. M., Grubor, B. & Ackermann, M. R. Expression of select immune genes (surfactant proteins A and D, sheep beta defensin 1, and toll-like receptor 4) by respiratory epithelia is developmentally regulated in the preterm neonatal lamb. Dev Comp Immunol 30, 1060–1069, https://doi.org/10.1016/j.dci.2006.01.001 (2006).
Abreu, M. T. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol 10, 131–144, https://doi.org/10.1038/nri2707 (2010).
Guzman, C. E., Bereza-Malcolm, L. T., De Groef, B. & Franks, A. E. Presence of Selected Methanogens, Fibrolytic Bacteria, and Proteobacteria in the Gastrointestinal Tract of Neonatal Dairy Calves from Birth to 72 Hours. PLoS One 10, e0133048, https://doi.org/10.1371/journal.pone.0133048 (2015).
Dave, M., Higgins, P. D., Middha, S. & Rioux, K. P. The human gut microbiome: current knowledge, challenges, and future directions. Transl Res 160, 246–257, https://doi.org/10.1016/j.trsl.2012.05.003 (2012).
Malmuthuge, N., Griebel, P. J. & Guan le, L. The Gut Microbiome and Its Potential Role in the Development and Function of Newborn Calf Gastrointestinal Tract. Front Vet Sci 2, 36, https://doi.org/10.3389/fvets.2015.00036 (2015).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75, 7537–7541, https://doi.org/10.1128/AEM.01541-09 (2009).
Dennis, G. Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3 (2003).
Petryszak, R. et al. Expression Atlas update–an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res 44, D746–752, https://doi.org/10.1093/nar/gkv1045 (2016).
Schmutz, C. et al. Systems-level overview of host protein phosphorylation during Shigella flexneri infection revealed by phosphoproteomics. Mol Cell Proteomics 12, 2952–2968, https://doi.org/10.1074/mcp.M113.029918 (2013).
Knodler, L. A., Finlay, B. B. & Steele-Mortimer, O. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem 280, 9058–9064, https://doi.org/10.1074/jbc.M412588200 (2005).
Mao, S., Zhang, M., Liu, J. & Zhu, W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci Rep 5, 16116, https://doi.org/10.1038/srep16116 (2015).
Malmuthuge, N., Griebel, P. J. & Guan le, L. Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl Environ Microbiol 80, 2021–2028, https://doi.org/10.1128/AEM.03864-13 (2014).
Li, R. W., Connor, E. E., Li, C., Baldwin Vi, R. L. & Sparks, M. E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ Microbiol 14, 129–139, https://doi.org/10.1111/j.1462-2920.2011.02543.x (2012).
D., A. G. Impact of the intestinal microflora on intestinal structure and function. Human Intestinal Microflora in Health and Disease, ed. Hentges D. J., editor. (New York, NY: Academic Press), 292–310 (1983).
Mazmanian, S. K. Capsular polysaccharides of symbiotic bacteria modulate immune responses during experimental colitis. J Pediatr Gastroenterol Nutr 46(Suppl 1), E11–12, https://doi.org/10.1097/01.mpg.0000313824.70971.a7 (2008).
Wen, L. et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455, 1109–1113, https://doi.org/10.1038/nature07336 (2008).
Mazmanian, S. K., Round, J. L. & Kasper, D. L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625, https://doi.org/10.1038/nature07008 (2008).
Bartke, N. & Hannun, Y. A. Bioactive sphingolipids: metabolism and function. J Lipid Res 50(Suppl), S91–96, https://doi.org/10.1194/jlr.R800080-JLR200 (2009).
Cowart, L. A., Gandy, J. L., Tholanikunnel, B. & Hannun, Y. A. Sphingolipids mediate formation of mRNA processing bodies during the heat-stress response of Saccharomyces cerevisiae. Biochem J 431, 31–38, https://doi.org/10.1042/BJ20100307 (2010).
An, D., Na, C., Bielawski, J., Hannun, Y. A. & Kasper, D. L. Membrane sphingolipids as essential molecular signals for Bacteroides survival in the intestine. Proc Natl Acad Sci USA 108(Suppl 1), 4666–4671, https://doi.org/10.1073/pnas.1001501107 (2011).
Heinz, L. X. et al. The Lipid-Modifying Enzyme SMPDL3B Negatively RegulatesInnate Immunity. Cell Reports 11, 1919–1928, https://doi.org/10.1016/j.celrep.2015.05.006 (2015).
Fuller, G. M. & Zhang, Z. Transcriptional control mechanism of fibrinogen gene expression. Ann N Y Acad Sci 936, 469–479 (2001).
Ozawa, K. et al. Effect of Streptococcus faecalis BIO-4R on intestinal flora of weanling piglets and calves. Appl Environ Microbiol 45, 1513–1518 (1983).
Jewell, K. A., McCormick, C. A., Odt, C. L., Weimer, P. J. & Suen, G. Ruminal Bacterial Community Composition in Dairy Cows Is Dynamic over the Course of Two Lactations and Correlates with Feed Efficiency. Appl Environ Microbiol 81, 4697–4710, https://doi.org/10.1128/AEM.00720-15 (2015).
Stevenson, D. M. & Weimer, P. J. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl Microbiol Biotechnol 75, 165–174, https://doi.org/10.1007/s00253-006-0802-y (2007).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79, 5112–5120, https://doi.org/10.1128/AEM.01043-13 (2013).
Weimer, P. J. et al. Transient changes in milk production efficiency and bacterial community composition resulting from near-total exchange of ruminal contents between high- and low-efficiency Holstein cows. J Dairy Sci 100, 7165–7182, https://doi.org/10.3168/jds.2017-12746 (2017).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35, 7188–7196, https://doi.org/10.1093/nar/gkm864 (2007).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200, https://doi.org/10.1093/bioinformatics/btr381 (2011).
DeSantis, A. J. et al. Inpatient management of hyperglycemia: the Northwestern experience. Endocr Pract 12, 491–505, https://doi.org/10.4158/EP.12.5.491 (2006).
Good, I. J. The population frequencies of species and the estimation of population parameters. Biometrika 40, 237–264 (1953).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36, https://doi.org/10.1186/gb-2013-14-4-r36 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, https://doi.org/10.1093/bioinformatics/btu638 (2015).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578, https://doi.org/10.1038/nprot.2012.016 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Szklarczyk, D. et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39, D561–568, https://doi.org/10.1093/nar/gkq973 (2011).
Szklarczyk, D. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45, D362–D368, https://doi.org/10.1093/nar/gkw937 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Kopylova, E., Noe, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217, https://doi.org/10.1093/bioinformatics/bts611 (2012).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15, R46, https://doi.org/10.1186/gb-2014-15-3-r46 (2014).
Traini, M. et al. Sphingomyelin Phosphodiesterase Acid-like 3A (SMPDL3A) Is a Novel Nucleotide Phosphodiesterase Regulated by Cholesterol in Human Macrophages. Journal of Biological Chemistry 289, https://doi.org/10.1074/jbc.M114.612341 (2014).
Cang, C. L., Aranda, K., Seo, Y. J., Gasnier, B. & Ren, D. J. TMEM175 Is an Organelle K+ Channel Regulating Lysosomal Function. Cell 162, 1101–1112, https://doi.org/10.1016/j.cell.2015.08.002 (2015).
Kang, H. G., Evers, M. R., Xia, G., Baenziger, J. U. & Schachner, M. Molecular cloning and characterization of chondroitin-4-O-sulfotransferase-3. A novel member of the HNK-1 family of sulfotransferases. J Biol Chem 277, 34766–34772, https://doi.org/10.1074/jbc.M204907200 (2002).
Schormann, N., Ricciardi, R. & Chattopadhyay, D. Uracil-DNA glycosylases-Structural and functional perspectives on an essential family of DNA repair enzymes. Protein Science 23, 1667–1685, https://doi.org/10.1002/pro.2554 (2014).
Tetzner, R., Dietrich, D. & Distler, J. Control of carry-over contamination for PCR-based DNA methylation quantification using bisulfite treated DNA. Nucleic Acids Research 35, https://doi.org/10.1093/nar/gkl955 (2007).
Die, J. V. et al. Selection of internal reference genes for normalization of reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis in the rumen epithelium. PLoS One 12, e0172674, https://doi.org/10.1371/journal.pone.0172674 (2017).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Acknowledgements
We thank Laura Cersosimo and Rafael Oliveira for their generous help on calf tissue collection. WL and DB were supported by appropriated project 5090-31000-026-00-D from the USDA Agriculture Research Service (Dairy Forage Research Center). JW was supported by appropriated project 5090-43440-006-00-D from the USDA Agricultural Research Service (Cereal Crops Research Laboratory). GS was supported by, in part, by a USDA National Institute of Food and Agriculture HATCH grant WIS02007 and a USDA Agriculture and Food Research Initiative Competitive Grant no. 2015-67015-23246. MSC was supported by a USDA Agriculture and Food Research Initiative Education and Literacy Initiative predoctoral fellowship no. 2018-67011-27997. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation by the US Department of Agriculture. The USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Contributions
W.L. conceived the RNA sequencing experiment design, performed RNA sequencing data analysis, prepared the figures and wrote the manuscript. W.L. and D.B. contributed to the data analysis in microbial classification using rRNA reads. A.E. and C.R. contributed to animal tissue collection, RNA extraction and RNA sequencing efforts. A.E., C.R. and J.W. contributed to RT-qPCR experimentation and data analysis. M.S.C., S.R., J.H.S., A.J.S. and G.S. provided project management, logistical support and maintenance of experimental animals. All authors reviewes and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, W., Edwards, A., Riehle, C. et al. Transcriptomics analysis of host liver and meta-transcriptome analysis of rumen epimural microbial community in young calves treated with artificial dosing of rumen content from adult donor cow. Sci Rep 9, 790 (2019). https://doi.org/10.1038/s41598-018-37033-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37033-4
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
